Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease



Abstract
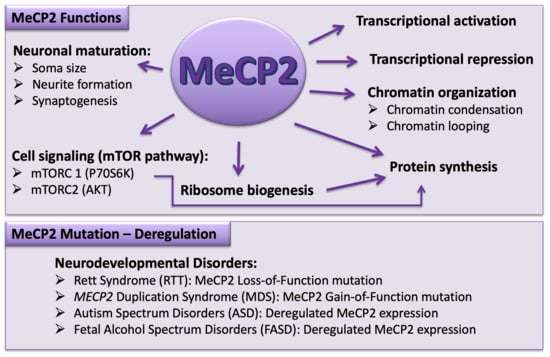
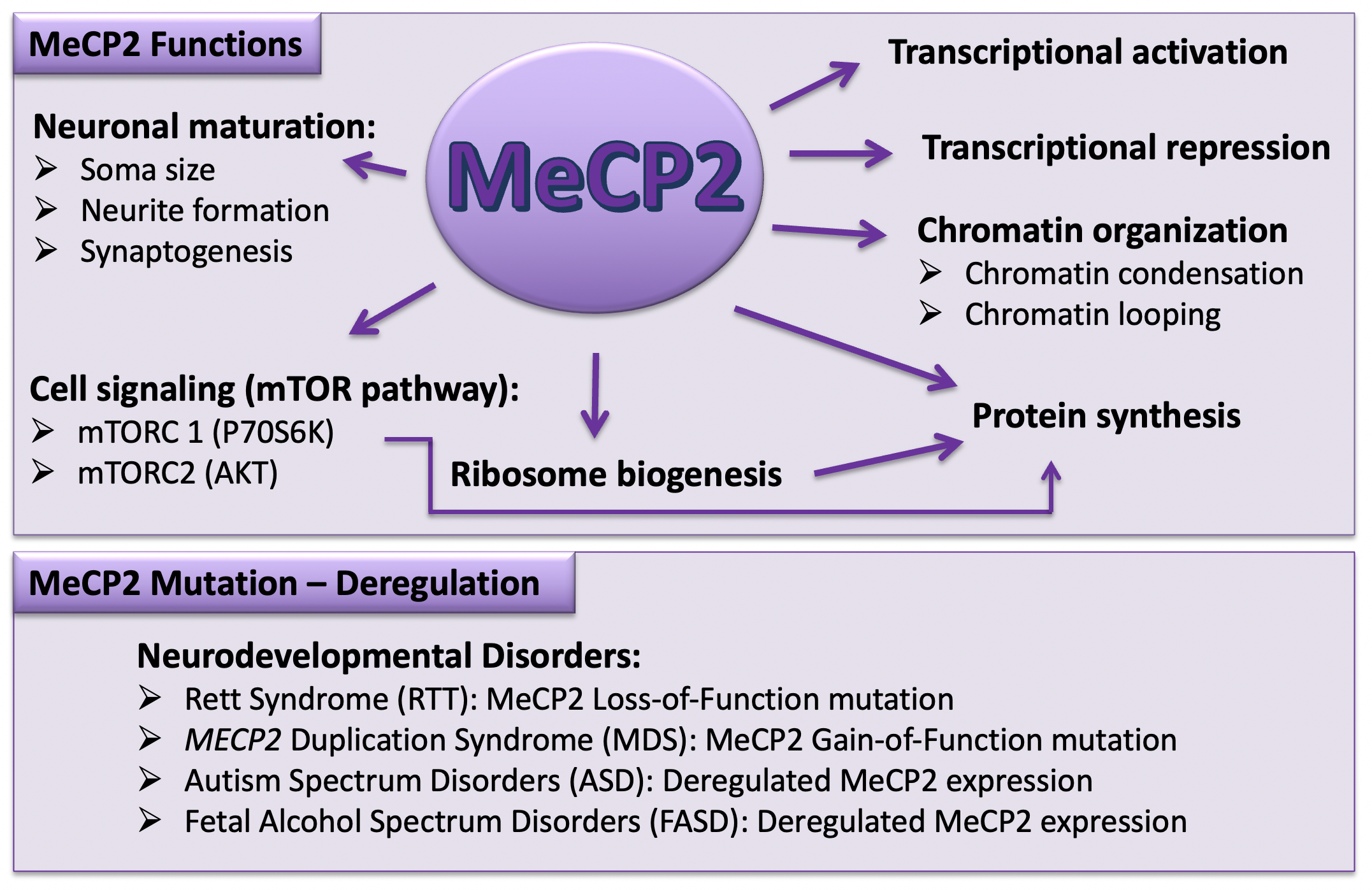{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction to Rett Syndrome
2. History of Rett Syndrome
3. Clinical Features, Diagnosis, and Histopathology of RTT
Gross and Microscopic Features of RTT
4. Genetic Basis of Rett Syndrome: Link between Genotype and Phenotype
4.1. Mutations in the N-Terminal Domain
4.2. Mutations in the Methyl-CpG-Binding Domain
4.3. Mutations in the Intervening Domain
4.4. Mutations in the Transcriptional Repression Domain
4.5. Mutations in the C-Terminal Domain
5. Biological Systems to Study Rett Syndrome
6. Epigenetic Regulation Mechanisms and Role in Controlling MeCP2 Homeostasis Network
6.1. Chromatin Remodelling
6.2. Histone Post-Translational Modifications
6.3. Noncoding RNAs
6.4. DNA Methylation
6.5. Writers of DNA Methylation
6.6. Erasers of DNA Methylation
6.7. Methyl-CpG-Binding Protein Family
6.8. MECP2/Mecp2 Gene Structure and MeCP2 Protein
6.9. MeCP2 Expression and Regulation
7. MeCP2 Target Genes: A Focus on BDNF and Its Related Signaling Cascades
7.1. Brain-Derived Neurotrophic Factor
7.1.1. BDNF Signaling
7.1.2. BDNF/Bdnf Regulation
7.1.3. Role of MeCP2 in BDNF Regulation
7.1.4. BDNF and Pathophysiology of RTT
7.1.5. BDNF and Cellular Origin of Detection in the Human Brain
7.2. MicroRNAs
7.2.1. Role of miRNAs in Central Nervous System Development
7.2.2. The Role of miR132 and its Effects on Neural Structure and Function
7.2.3. Homeostatic Regulation of MeCP2 by miR132
8. Lessons Learned from the Human Brain on MeCP2-BDNF-miR132 homeostasis Regulatory Components
9. Therapeutic Strategies for RTT
9.1. Molecular Treatments and Gene Dosage Concerns
9.2. Activating MECP2 on the Inactive X-Chromosome
9.3. Gene-Editing Strategies
9.4. Challenges of Protein Replacement
9.5. Targeting Downstream Signaling Pathways of MeCP2
9.6. Clinical Trials
10. Closing Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Leonard, H.; Cobb, S.; Downs, J. Clinical and biological progress over 50 years in Rett syndrome. Nat. Rev. Neurol. 2017, 13, 37–51. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Chahil, G.; Bollu, P.C. Rett Syndrome; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Takahashi, S.; Takeguchi, R.; Kuroda, M.; Tanaka, R. Atypical Rett syndrome in a girl with mosaic triple X and MECP2 variant. Mol. Genet. Genomic Med. 2020, 8, e1122. [Google Scholar] [CrossRef]
- Nomura, Y. Early behavior characteristics and sleep disturbance in Rett syndrome. Brain Dev. 2005, 27 (Suppl. S1), S35–S42. [Google Scholar] [CrossRef]
- Segawa, M. Early motor disturbances in Rett syndrome and its pathophysiological importance. Brain Dev. 2005, 27 (Suppl. S1), S54–S58. [Google Scholar] [CrossRef]
- Weese-Mayer, D.E.; Lieske, S.P.; Boothby, C.M.; Kenny, A.S.; Bennett, H.L.; Ramirez, J.-M. Autonomic dysregulation in young girls with Rett Syndrome during nighttime in-home recordings. Pediatr. Pulmonol. 2008, 43, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Weese-Mayer, D.E.; Lieske, S.P.; Boothby, C.M.; Kenny, A.S.; Bennett, H.L.; Silvestri, J.M.; Ramirez, J.-M. Autonomic nervous system dysregulation: Breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatric Res. 2006, 60, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B. Rett syndrome: Long-term clinical follow-up experiences over four decades. J. Child Neurol. 2005, 20, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Jian, L.; Nagarajan, L.; de Klerk, N.; Ravine, D.; Bower, C.; Anderson, A.; Williamson, S.; Christodoulou, J.; Leonard, H. Predictors of seizure onset in Rett syndrome. J. Pediatrics 2006, 149, 542–547. [Google Scholar] [CrossRef]
- Tarquinio, D.C.; Hou, W.; Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Motil, K.J.; Skinner, S.A.; Lee, H.-S.; Percy, A.K. The Changing Face of Survival in Rett Syndrome and MECP2-Related Disorders. Pediatric Neurol. 2015, 53, 402–411. [Google Scholar] [CrossRef]
- Percy, A.K.; Neul, J.L.; Glaze, D.G.; Motil, K.J.; Skinner, S.A.; Khwaja, O.; Lee, H.-S.; Lane, J.B.; Barrish, J.O.; Annese, F.; et al. Rett syndrome diagnostic criteria: Lessons from the Natural History Study. Ann. Neurol. 2010, 68, 951–955. [Google Scholar] [CrossRef]
- Duncan Armstrong, D. Neuropathology of Rett Syndrome. Neurobiol. Dis. Child. 2005, 20, 747–753. [Google Scholar] [CrossRef]
- Reiss, A.L.; Faruque, F.; Naidu, S.; Abrams, M.; Beaty, T.; Bryan, R.N.; Moser, H. Neuroanatomy of Rett syndrome: A volumetric imaging study. Ann. Neurol. 1993, 34, 227–234. [Google Scholar] [CrossRef]
- Subramaniam, B.; Naidu, S.; Reiss, A.L. Neuroanatomy in Rett syndrome: Cerebral cortex and posterior fossa. Neurology 1997, 48, 399–407. [Google Scholar] [CrossRef]
- Armstrong, D.; Dunn, J.K.; Antalffy, B.; Trivedi, R. Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 1995, 54, 195–201. [Google Scholar] [CrossRef]
- Jellinger, K.; Seitelberger, F. Neuropathology of Rett syndrome. Am. J. Med. Genet. Suppl. 1986, 1, 259–288. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Hagberg, B.; Dahlström, A. Morphological study of neocortical areas in Rett syndrome. Acta Neuropathol. 1997, 93, 50–61. [Google Scholar] [CrossRef]
- O’Connor, T.M.; O’Connell, J.; O’Brien, D.I.; Goode, T.; Bredin, C.P.; Shanahan, F. The role of substance P in inflammatory disease. J. Cell. Physiol. 2004, 201, 167–180. [Google Scholar] [CrossRef]
- Géranton, S.M.; Morenilla-Palao, C.; Hunt, S.P. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J. Neurosci. 2007, 27, 6163–6173. [Google Scholar] [CrossRef]
- Downs, J.; Géranton, S.M.; Bebbington, A.; Jacoby, P.; Bahi-Buisson, N.; Ravine, D.; Leonard, H. Linking MECP2 and pain sensitivity: The example of Rett syndrome. Am. J. Med. Genet. A 2010, 152A, 1197–1205. [Google Scholar] [CrossRef]
- Barney, C.C.; Feyma, T.; Beisang, A.; Symons, F.J. Pain experience and expression in Rett syndrome: Subjective and objective measurement approaches. J. Dev. Phys. Disabil. 2015, 27, 417–429. [Google Scholar] [CrossRef]
- Takeguchi, R.; Takahashi, S.; Kuroda, M.; Tanaka, R.; Suzuki, N.; Tomonoh, Y.; Ihara, Y.; Sugiyama, N.; Itoh, M. MeCP2_e2 partially compensates for lack of MeCP2_e1: A male case of Rett syndrome. Mol. Genet. Genom. Med. 2020, 8, 1–5. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, X.; Wang, J.; Li, J.; Wu, Q.; Wen, Y.; Zhao, Y.; Zhang, X.; Yao, H.; Wu, X.; et al. Genomic mosaicism in the pathogenesis and inheritance of a Rett syndrome cohort. Genet. Med. 2019, 21, 1330–1338. [Google Scholar] [CrossRef]
- Reichow, B.; George-Puskar, A.; Lutz, T.; Smith, I.C.; Volkmar, F.R. Brief report: Systematic review of Rett syndrome in males. J. Autism Dev. Disord. 2015, 45, 3377–3383. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef]
- Girard, M.; Couvert, P.; Carrié, A.; Tardieu, M.; Chelly, J.; Beldjord, C.; Bienvenu, T. Parental origin of de novo MECP2 mutations in Rett syndrome. Eur. J. Hum. Genet. 2001, 9, 231–236. [Google Scholar] [CrossRef]
- Wan, M.; Lee, S.S.; Zhang, X.; Houwink-Manville, I.; Song, H.R.; Amir, R.E.; Budden, S.; Naidu, S.; Pereira, J.L.; Lo, I.F.; et al. Rett syndrome and beyond: Recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am. J. Hum. Genet. 1999, 65, 1520–1529. [Google Scholar] [CrossRef]
- Villard, L.; Kpebe, A.; Cardoso, C.; Chelly, P.J.; Tardieu, P.M.; Fontes, M. Two affected boys in a Rett syndrome family: Clinical and molecular findings. Neurology 2000, 55, 1188–1193. [Google Scholar] [CrossRef]
- Ortega-Alarcon, D.; Claveria-Gimeno, R.; Vega, S.; Jorge-Torres, O.C.; Esteller, M.; Abian, O.; Velazquez-Campoy, A. Molecular context-dependent effects induced by Rett syndrome-associated mutations in mecp2. Biomolecules 2020, 10, 1533. [Google Scholar] [CrossRef]
- Lamonica, J.M.; Kwon, D.Y.; Goffin, D.; Fenik, P.; Johnson, B.S.; Cui, Y.; Guo, H.; Veasey, S.; Zhou, Z. Elevating expression of MeCP2 T158M rescues DNA binding and Rett syndrome-like phenotypes. J. Clin. Investig. 2017, 127, 1889–1904. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, T.I.; de Paz, A.M.; Akhtar, S.; Ausió, J.; Vincent, J.B. MeCP2_E1 N-terminal modifications affect its degradation rate and are disrupted by the Ala2Val Rett mutation. Hum. Mol. Genet. 2017, 26, 4132–4141. [Google Scholar] [CrossRef] [PubMed]
- Krishnaraj, R.; Ho, G.; Christodoulou, J. RettBASE: Rett syndrome database update. Hum. Mutat. 2017, 38, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Archer, H.; Evans, J.; Leonard, H.; Colvin, L.; Ravine, D.; Christodoulou, J.; Williamson, S.; Charman, T.; Bailey, M.E.S.; Sampson, J.; et al. Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation. J. Med. Genet. 2007, 44, 148–152. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue-and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef]
- Renieri, A.; Meloni, I.; Longo, I.; Ariani, F.; Mari, F.; Pescucci, C.; Cambi, F. Rett syndrome: The complex nature of a monogenic disease. J. Mol. Med. 2003, 81, 346–354. [Google Scholar] [CrossRef]
- Bourdon, V.; Philippe, C.; Bienvenu, T.; Koenig, B.; Tardieu, M.; Chelly, J.; Jonveaux, P. Evidence of somatic mosaicism for a MECP2 mutation in females with Rett syndrome: Diagnostic implications. J. Med. Genet. 2001, 38, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Tate, P.; Li, E.; Bird, A. DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell. Biol. 1996, 16, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Smeets, E.; Terhal, P.; Casaer, P.; Peters, A.; Midro, A.; Schollen, E.; van Roozendaal, K.; Moog, U.; Matthijs, G.; Herbergs, J.; et al. Rett syndrome in females with CTS hot spot deletions: A disorder profile. Am. J. Med. Genet. A 2005, 132A, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Mnatzakanian, G.N.; Lohi, H.; Munteanu, I.; Alfred, S.E.; Yamada, T.; MacLeod, P.J.M.; Jones, J.R.; Scherer, S.W.; Schanen, N.C.; Friez, M.J.; et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 2004, 36, 339–341. [Google Scholar] [CrossRef]
- Bartholdi, D.; Klein, A.; Weissert, M.; Koenig, N.; Baumer, A.; Boltshauser, E.; Schinzel, A.; Berger, W.; Mátyás, G. Clinical profiles of four patients with Rett syndrome carrying a novel exon 1 mutation or genomic rearrangement in the MECP2 gene. Clin. Genet. 2006, 69, 319–326. [Google Scholar] [CrossRef]
- Nectoux, J.; Fichou, Y.; Rosas-Vargas, H.; Cagnard, N.; Bahi-Buisson, N.; Nusbaum, P.; Letourneur, F.; Chelly, J.; Bienvenu, T. Cell cloning-based transcriptome analysis in Rett patients: Relevance to the pathogenesis of Rett syndrome of new human MeCP2 target genes. J. Cell. Mol. Med. 2010, 14, 1962–1974. [Google Scholar] [CrossRef]
- Kerr, B.; Soto, C.J.; Saez, M.; Abrams, A.; Walz, K.; Young, J.I. Transgenic complementation of MeCP2 deficiency: Phenotypic rescue of Mecp2-null mice by isoform-specific transgenes. Eur. J. Hum. Genet. 2012, 20, 69–76. [Google Scholar] [CrossRef]
- Zhou, X.; Liao, Y.; Xu, M.; Ji, Z.; Xu, Y.; Zhou, L.; Wei, X.; Hu, P.; Han, P.; Yang, F.; et al. A novel mutation R190H in the AT-hook 1 domain of MeCP2 identified in an atypical Rett syndrome. Oncotarget 2017, 8, 82156–82164. [Google Scholar] [CrossRef]
- Sheikh, T.I.; Harripaul, R.; Ayub, M.; Vincent, J.B. MeCP2 AT-Hook1 mutations in patients with intellectual disability and/or schizophrenia disrupt DNA binding and chromatin compaction in vitro. Hum. Mutat. 2018, 39, 717–728. [Google Scholar] [CrossRef]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef]
- Hite, K.C.; Adams, V.H.; Hansen, J.C. Recent advances in MeCP2 structure and function. Biochem. Cell Biol. 2009, 87, 219–227. [Google Scholar] [CrossRef]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.-N.; Ho, H.-Y.H.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 2006, 52, 255–269. [Google Scholar] [CrossRef]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef]
- Ezeonwuka, C.D.; Rastegar, M. MeCP2-Related Diseases and Animal Models. Diseases 2014, 2, 45–70. [Google Scholar] [CrossRef]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef]
- Marchetto, M.C.N.M.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Kwok, S.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; Mitalipova, M.; et al. Global transcriptional and translational repression in human embryonic stem cells-derived Rett Syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef]
- Yazdani, M.; Deogracias, R.; Guy, J.; Poot, R.A.; Bird, A.; Barde, Y.A. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem Cells 2012. [Google Scholar] [CrossRef]
- Fyffe, S.L.; Neul, J.L.; Samaco, R.C.; Chao, H.-T.; Ben-Shachar, S.; Moretti, P.; McGill, B.E.; Goulding, E.H.; Sullivan, E.; Tecott, L.H.; et al. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 2008, 59, 947–958. [Google Scholar] [CrossRef]
- Samaco, R.C.; Mandel-Brehm, C.; Chao, H.-T.; Ward, C.S.; Fyffe-Maricich, S.L.; Ren, J.; Hyland, K.; Thaller, C.; Maricich, S.M.; Humphreys, P.; et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2009, 106, 21966–21971. [Google Scholar] [CrossRef] [PubMed]
- Lawson-Yuen, A.; Liu, D.; Han, L.; Jiang, Z.I.; Tsai, G.E.; Basu, A.C.; Picker, J.; Feng, J.; Coyle, J.T. Ube3a mRNA and protein expression are not decreased in Mecp2R168X mutant mice. Brain Res. 2007, 1180, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jentarra, G.M.; Olfers, S.L.; Rice, S.G.; Srivastava, N.; Homanics, G.E.; Blue, M.; Naidu, S.; Narayanan, V. Abnormalities of cell packing density and dendritic complexity in the MeCP2 A140V mouse model of Rett syndrome/X-linked mental retardation. BMC Neurosci. 2010, 11, 19. [Google Scholar] [CrossRef]
- Calfa, G.; Percy, A.K.; Pozzo-Miller, L. Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp. Biol. Med. 2011, 236, 3–19. [Google Scholar] [CrossRef]
- Lyst, M.J.; Bird, A. Rett syndrome: A complex disorder with simple roots. Nat. Rev. Genet. 2015, 16, 261–274. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, J.-S.; Spruyt, K. Sleep problems in Rett syndrome animal models: A systematic review. J. Neurosci. Res. 2021, 99, 529–544. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; MacDonald, J.L. Sex differences in Mecp2-mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development. Brain Res. 2020, 1729, 146644. [Google Scholar] [CrossRef]
- Vashi, N.; Justice, M.J. Treating Rett syndrome: From mouse models to human therapies. Mamm. Genome 2019, 30, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H.; Yi, S.A.; Jang, H.J.; Han, J.W.; Lee, J. In vitro modeling for inherited neurological diseases using induced pluripotent stem cells: From 2D to organoid. Arch. Pharm. Res. 2020, 43, 877–889. [Google Scholar] [CrossRef]
- Sandweiss, A.J.; Brandt, V.L.; Zoghbi, H.Y. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: Prospects for future therapies. Lancet Neurol. 2020, 19, 689–698. [Google Scholar] [CrossRef]
- Freitas, B.C.; Beltrão-Braga, P.C.B.; Marchetto, M.C. Modeling Inflammation on Neurodevelopmental Disorders Using Pluripotent Stem Cells. In Neurodevelopmental Disorders: Employing iPSC Technologies to Define and Treat Childhood Brain Diseases; DiCicco-Bloom, E., Millonig, J.H., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 207–218. ISBN 978-3-030-45493-7. [Google Scholar] [CrossRef]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar] [CrossRef]
- Lee, K.M.; Hawi, Z.H.; Parkington, H.C.; Parish, C.L.; Kumar, P.V.; Polo, J.M.; Bellgrove, M.A.; Tong, J. The application of human pluripotent stem cells to model the neuronal and glial components of neurodevelopmental disorders. Mol. Psychiatry 2020, 25, 368–378. [Google Scholar] [CrossRef]
- Smith, E.S.; Smith, D.R.; Eyring, C.; Braileanu, M.; Smith-Connor, K.S.; Ei Tan, Y.; Fowler, A.Y.; Hoffman, G.E.; Johnston, M.V.; Kannan, S.; et al. Altered trajectories of neurodevelopment and behavior in mouse models of Rett syndrome. Neurobiol. Learn. Mem. 2019, 165, 106962. [Google Scholar] [CrossRef]
- Fallah, M.S.; Eubanks, J.H. Seizures in Mouse Models of Rare Neurodevelopmental Disorders. Neuroscience 2020, 445, 50–68. [Google Scholar] [CrossRef]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett syndrome: A neurological disorder with metabolic components. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Ausió, J. Role of MeCP2 in neurological disorders: Current status and future perspectives. Epigenomics 2018, 10, 5–8. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Rastegar, M.; Davie, J.R. Epigenetic control. J. Cell. Physiol. 2009, 219, 243–250. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Rastegar, M. Rett syndrome and MeCP2. Neuromol. Med. 2014, 16, 231–264. [Google Scholar] [CrossRef]
- Kobrossy, L.; Rastegar, M.; Featherstone, M. Interplay between chromatin and trans-acting factors regulating the Hoxd4 promoter during neural differentiation. J. Biol. Chem. 2006, 281, 25926–25939. [Google Scholar] [CrossRef]
- Segal, E.; Widom, J. What controls nucleosome positions? Trends Genet. 2009, 25, 335–343. [Google Scholar] [CrossRef]
- Rastegar, M.; Kobrossy, L.; Kovacs, E.N.; Rambaldi, I.; Featherstone, M. Sequential Histone Modifications at Hoxd4 Regulatory Regions Distinguish Anterior from Posterior Embryonic Compartments. Mol. Cell. Biol. 2004. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Zachariah, R.M.; Delcuve, G.P.; Davie, J.R.; Rastegar, M. New Developments in Chromatin Research: An Epigenetic Perspective. In New Developments in Chromatin Research; Simpson, N.M., Stewart, V.J., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2012; pp. 29–58. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Barber, B.A.; Rastegar, M. Epigenetic control of Hox genes during neurogenesis, development, and disease. Ann. Anat. 2010, 192, 261–274. [Google Scholar] [CrossRef]
- Patel, D.J.; Wang, Z. Readout of epigenetic modifications. Annu. Rev. Biochem. 2013, 82, 81–118. [Google Scholar] [CrossRef]
- Huisinga, K.L.; Brower-Toland, B.; Elgin, S.C.R. The contradictory definitions of heterochromatin: Transcription and silencing. Chromosoma 2006, 115, 110–122. [Google Scholar] [CrossRef]
- Trojer, P.; Reinberg, D. Facultative heterochromatin: Is there a distinctive molecular signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Jarmasz, J.S.; Murugeshan, N.; Del Bigio, M.R.; Rastegar, M.; Davie, J.R. DNA modifications: Function and applications in normal and disease States. Biology 2014, 3, 670–723. [Google Scholar] [CrossRef]
- Rastegar, M.; Delcuve, G.P.; Davie, J.R. Epigenetic Analysis of Pluripotent Cells. In Human Stem Cell Technology and Biology: A Research Guide and Laboratory Manual, 1st ed.; Wiley-Blackwell: New Jersey, NY, USA, 2011; pp. 273–288. [Google Scholar]
- Patil, V.S.; Zhou, R.; Rana, T.M. Gene regulation by non-coding RNAs. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 16–32. [Google Scholar] [CrossRef]
- Wu, H.; Tao, J.; Chen, P.J.; Shahab, A.; Ge, W.; Hart, R.P.; Ruan, X.; Ruan, Y.; Sun, Y.E. Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 18161–18166. [Google Scholar] [CrossRef]
- Beckedorff, F.C.; Ayupe, A.C.; Crocci-Souza, R.; Amaral, M.S.; Nakaya, H.I.; Soltys, D.T.; Menck, C.F.M.; Reis, E.M.; Verjovski-Almeida, S. The intronic long noncoding RNA ANRASSF1 recruits PRC2 to the RASSF1A promoter, reducing the expression of RASSF1A and increasing cell proliferation. PLoS Genet. 2013, 9, e1003705. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.B.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. Ethanol deregulates Mecp2/MeCP2 in differentiating neural stem cells via interplay between 5-methylcytosine and 5-hydroxymethylcytosine at the Mecp2 regulatory elements. Exp. Neurol. 2015, 265, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.; Song, H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle 2011, 10, 2662–2668. [Google Scholar] [CrossRef]
- Rastegar, M. Epigenetics and Cerebellar Neurodevelopmental Disorders. In Development of the Cerebellum from Molecular Aspects to Diseases; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Defossez, P.-A.; Stancheva, I. Biological functions of methyl-CpG-binding proteins. Prog. Mol. Biol. Transl. Sci. 2011, 101, 377–398. [Google Scholar] [CrossRef]
- Li, L.; Chen, B.-F.; Chan, W.-Y. An epigenetic regulator: Methyl-CpG-binding domain protein 1 (MBD1). Int. J. Mol. Sci. 2015, 16, 5125–5140. [Google Scholar] [CrossRef]
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef]
- Lewis, J.; Bird, A. DNA methylation and chromatin structure. FEBS Lett. 1991, 285, 155–159. [Google Scholar] [CrossRef]
- Boyes, J.; Bird, A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell 1991, 64, 1123–1134. [Google Scholar] [CrossRef]
- Reeves, R.; Nissen, M.S. The A.T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J. Biol. Chem. 1990, 265, 8573–8582. [Google Scholar] [CrossRef]
- Adams, V.H.; McBryant, S.J.; Wade, P.A.; Woodcock, C.L.; Hansen, J.C. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J. Biol. Chem. 2007, 282, 15057–15064. [Google Scholar] [CrossRef]
- Martínez De Paz, A.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Tom Dieck, S.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin 2019, 12, 1–16. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Bird, A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004, 32, 1818–1823. [Google Scholar] [CrossRef]
- Zachariah, R.M.; Olson, C.O.; Ezeonwuka, C.; Rastegar, M. Novel MeCP2 Isoform-Specific Antibody Reveals the Endogenous MeCP2E1 Expression in Murine Brain, Primary Neurons and Astrocytes. PLoS ONE 2012, 7, 24–28. [Google Scholar] [CrossRef]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.B.; Rastegar, M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef]
- Pejhan, S.; Del Bigio, M.R.; Rastegar, M. The MeCP2E1/E2-BDNF-miR132 Homeostasis Regulatory Network Is Region-Dependent in the Human Brain and Is Impaired in Rett Syndrome Patients. Front. Cell Dev. Biol. 2020, 8, 763. [Google Scholar] [CrossRef]
- Rastegar, M.; Hotta, A.; Pasceri, P.; Makarem, M.; Cheung, A.Y.L.; Elliott, S.; Park, K.J.; Adachi, M.; Jones, F.S.; Clarke, I.D.; et al. MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS ONE 2009, 4, e6810. [Google Scholar] [CrossRef]
- Ballas, N.; Lioy, D.T.; Grunseich, C.; Mandel, G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 2009, 12, 311–317. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Zachariah, R.M.; Rastegar, M. Decitabine alters the expression of Mecp2 isoforms via dynamic DNA methylation at the Mecp2 regulatory elements in neural stem cells. Mol. Autism 2013, 4, 46. [Google Scholar] [CrossRef]
- Maezawa, I.; Jin, L.-W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef]
- Yasui, D.H.; Xu, H.; Dunaway, K.W.; LaSalle, J.M.; Jin, L.-W.; Maezawa, I. MeCP2 modulates gene expression pathways in astrocytes. Mol. Autism 2013, 4, 3. [Google Scholar] [CrossRef]
- Derecki, N.C.; Cronk, J.C.; Lu, Z.; Xu, E.; Abbott, S.B.G.; Guyenet, P.G.; Kipnis, J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 2012, 484, 105–109. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Olson, C.O.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. DNA methylation contributes to the differential expression levels of Mecp2 in male mice neurons and astrocytes. Int. J. Mol. Sci. 2019, 20, 1845. [Google Scholar] [CrossRef]
- Xu, W.; Liyanage, V.R.B.; MacAulay, A.; Levy, R.D.; Curtis, K.; Olson, C.O.; Zachariah, R.M.; Amiri, S.; Buist, M.; Hicks, G.G.; et al. Genome-Wide Transcriptome Landscape of Embryonic Brain-Derived Neural Stem Cells Exposed to Alcohol with Strain-Specific Cross-Examination in BL6 and CD1 Mice. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]
- Amiri, S.; Davie, J.R.; Rastegar, M. Chronic Ethanol Exposure Alters DNA Methylation in Neural Stem Cells: Role of Mouse Strain and Sex. Mol. Neurobiol. 2020, 57, 650–667. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef]
- Mellén, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.W.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 Is Expressed at Near Histone-Octamer Levels and Globally Alters the Chromatin State. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef]
- Georgel, P.T.; Horowitz-Scherer, R.A.; Adkins, N.; Woodcock, C.L.; Wade, P.A.; Hansen, J.C. Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 2003, 278, 32181–32188. [Google Scholar] [CrossRef]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.-F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, R.M.; Rastegar, M. Linking epigenetics to human disease and Rett syndrome: The emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012, 2012, 415825. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Francke, U. Identification of cis-regulatory elements for MECP2 expression. Hum. Mol. Genet. 2006, 15, 1769–1782. [Google Scholar] [CrossRef] [PubMed]
- Newnham, C.M.; Hall-Pogar, T.; Liang, S.; Wu, J.; Tian, B.; Hu, J.; Lutz, C.S. Alternative polyadenylation of MeCP2: Influence of cis-acting elements and trans-acting factors. RNA Biol. 2010, 7, 361–372. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coy, J.F.; Sedlacek, Z.; Bächner, D.; Delius, H.; Poustka, A. A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3”-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum. Mol. Genet. 1999, 8, 1253–1262. [Google Scholar] [CrossRef]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Urdinguio, R.G.; Fernandez, A.F.; Lopez-Nieva, P.; Rossi, S.; Huertas, D.; Kulis, M.; Liu, C.-G.; Croce, C.M.; Calin, G.A.; Esteller, M. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 2010, 5, 656–663. [Google Scholar] [CrossRef]
- Bothwell, M. NGF, BDNF, NT3, and NT4. Handb. Exp. Pharmacol. 2014, 220, 3–15. [Google Scholar] [CrossRef]
- Binder, D.K.; Scharfman, H.E. Brain-derived neurotrophic factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef]
- Bronfman, F.C.; Lazo, O.M.; Flores, C.; Escudero, C.A. Spatiotemporal intracellular dynamics of neurotrophin and its receptors. Implications for neurotrophin signaling and neuronal function. Handb. Exp. Pharmacol. 2014, 220, 33–65. [Google Scholar] [CrossRef] [PubMed]
- West, A.E.; Pruunsild, P.; Timmusk, T. Neurotrophins: Transcription and translation. Handb. Exp. Pharmacol. 2014, 220, 67–100. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef] [PubMed]
- An, J.J.; Gharami, K.; Liao, G.-Y.; Woo, N.H.; Lau, A.G.; Vanevski, F.; Torre, E.R.; Jones, K.R.; Feng, Y.; Lu, B.; et al. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 2008, 134, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.G.; Irier, H.A.; Gu, J.; Tian, D.; Ku, L.; Liu, G.; Xia, M.; Fritsch, B.; Zheng, J.Q.; Dingledine, R.; et al. Distinct 3′UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor (BDNF). Proc. Natl. Acad. Sci. USA 2010, 107, 15945–15950. [Google Scholar] [CrossRef]
- Waterhouse, E.G.; An, J.J.; Orefice, L.L.; Baydyuk, M.; Liao, G.-Y.; Zheng, K.; Lu, B.; Xu, B. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J. Neurosci. 2012, 32, 14318–14330. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Lu, Y.; Christian, K.; Lu, B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 2008, 89, 312–323. [Google Scholar] [CrossRef]
- Begni, V.; Riva, M.A.; Cattaneo, A. Cellular and molecular mechanisms of the brain-derived neurotrophic factor in physiological and pathological conditions. Clin. Sci. 2017, 131, 123–138. [Google Scholar] [CrossRef]
- Bibel, M.; Hoppe, E.; Barde, Y.A. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 1999, 18, 616–622. [Google Scholar] [CrossRef]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.-Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef]
- Mizui, T.; Ishikawa, Y.; Kumanogoh, H.; Kojima, M. Neurobiological actions by three distinct subtypes of brain-derived neurotrophic factor: Multi-ligand model of growth factor signaling. Pharmacol. Res. 2016, 105, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef]
- Grewal, S.S.; York, R.D.; Stork, P.J. Extracellular-signal-regulated kinase signalling in neurons. Curr. Opin. Neurobiol. 1999, 9, 544–553. [Google Scholar] [CrossRef]
- Shaywitz, A.J.; Greenberg, M.E. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 1999, 68, 821–861. [Google Scholar] [CrossRef]
- Patapoutian, A.; Reichardt, L.F. Trk receptors: Mediators of neurotrophin action. Curr. Opin. Neurobiol. 2001, 11, 272–280. [Google Scholar] [CrossRef]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Lin, G.; Bella, A.J.; Lue, T.F.; Lin, C.-S. Brain-derived neurotrophic factor (BDNF) acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat: Part 2. J. Sex. Med. 2006, 3, 821–829. [Google Scholar] [CrossRef]
- Ashcroft, M.; Stephens, R.M.; Hallberg, B.; Downward, J.; Kaplan, D.R. The selective and inducible activation of endogenous PI 3-kinase in PC12 cells results in efficient NGF-mediated survival but defective neurite outgrowth. Oncogene 1999, 18, 4586–4597. [Google Scholar] [CrossRef][Green Version]
- Kang, H.; Schuman, E.M. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 1996, 273, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Kawamura, M.; Hara, K.; Yonezawa, K.; Nawa, H. Brain-derived neurotrophic factor enhances neuronal translation by activating multiple initiation processes: Comparison with the effects of insulin. J. Biol. Chem. 2001, 276, 42818–42825. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Ali Sher, A.; Del Bigio, M.R.; Sztainberg, Y.; Siu, V.M.; et al. MECP2 Mutation Interrupts Nucleolin–mTOR–P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Zafra, F.; Thoenen, H.; Lindholm, D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc. Natl. Acad. Sci. USA 1992, 89, 9444–9448. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, Y.; Pizzorusso, T.; Cremisi, F.; Rossi, F.M.; Barsacchi, G.; Maffei, L. Monocular deprivation decreases the expression of messenger RNA for brain-derived neurotrophic factor in the rat visual cortex. Neuroscience 1995, 69, 1133–1144. [Google Scholar] [CrossRef]
- Rocamora, N.; Welker, E.; Pascual, M.; Soriano, E. Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. J. Neurosci. 1996, 16, 4411–4419. [Google Scholar] [CrossRef]
- Zafra, F.; Hengerer, B.; Leibrock, J.; Thoenen, H.; Lindholm, D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990, 9, 3545–3550. [Google Scholar] [CrossRef]
- Zafra, F.; Castrén, E.; Thoenen, H.; Lindholm, D. Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1991, 88, 10037–10041. [Google Scholar] [CrossRef]
- Zafra, F.; Lindholm, D.; Castrén, E.; Hartikka, J.; Thoenen, H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J. Neurosci. 1992, 12, 4793–4799. [Google Scholar] [CrossRef]
- Isackson, P.J.; Huntsman, M.M.; Murray, K.D.; Gall, C.M. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: Temporal patterns of induction distinct from NGF. Neuron 1991, 6, 937–948. [Google Scholar] [CrossRef]
- Pruunsild, P.; Sepp, M.; Orav, E.; Koppel, I.; Timmusk, T. Identification of cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF gene. J. Neurosci. 2011, 31, 3295–3308. [Google Scholar] [CrossRef] [PubMed]
- Mellios, N.; Huang, H.-S.; Grigorenko, A.; Rogaev, E.; Akbarian, S. A set of differentially expressed miRNAs, including miR-30a-5p, act as post-transcriptional inhibitors of BDNF in prefrontal cortex. Hum. Mol. Genet. 2008, 17, 3030–3042. [Google Scholar] [CrossRef] [PubMed]
- Varendi, K.; Mätlik, K.; Andressoo, J.-O. From microRNA target validation to therapy: Lessons learned from studies on BDNF. Cell. Mol. Life Sci. 2015, 72, 1779–1794. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Lioy, D.T.; Ma, L.; Impey, S.; Mandel, G.; Goodman, R.H. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 2007, 10, 1513–1514. [Google Scholar] [CrossRef] [PubMed]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Khare, G.; Dani, V.; Nelson, S.; Jaenisch, R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 2006, 49, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Calfa, G.; Larimore, J.; Pozzo-Miller, L. Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proc. Natl. Acad. Sci. USA 2012, 109, 17087–17092. [Google Scholar] [CrossRef]
- Li, W.; Pozzo-Miller, L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014, 76 Pt C, 737–746. [Google Scholar] [CrossRef]
- Gonzales, M.L.; Adams, S.; Dunaway, K.W.; LaSalle, J.M. Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol. Cell. Biol. 2012, 32, 2894–2903. [Google Scholar] [CrossRef]
- Katz, D.M. Brain-derived neurotrophic factor and Rett syndrome. In Neurotrophic Factors; Springer: Berlin/Heidelberg, Germany, 2014; pp. 481–495. ISBN 978-3-642-45105-8. [Google Scholar] [CrossRef]
- Vanhala, R.; Korhonen, L.; Mikelsaar, M.; Lindholm, D.; Riikonen, R. Neurotrophic factors in cerebrospinal fluid and serum of patients with Rett syndrome. J. Child Neurol. 1998, 13, 429–433. [Google Scholar] [CrossRef]
- Riikonen, R. Neurotrophic factors in the pathogenesis of Rett syndrome. J. Child Neurol. 2003, 18, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Abuhatzira, L.; Makedonski, K.; Kaufman, Y.; Razin, A.; Shemer, R. MeCP2 deficiency in the brain decreases BDNF levels by REST/CoREST-mediated repression and increases TRKB production. Epigenetics 2007, 2, 214–222. [Google Scholar] [CrossRef]
- Deng, V.; Matagne, V.; Banine, F.; Frerking, M.; Ohliger, P.; Budden, S.; Pevsner, J.; Dissen, G.A.; Sherman, L.S.; Ojeda, S.R. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum. Mol. Genet. 2007, 16, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Pejhan, S.; Siu, V.M.; Ang, L.C.; Del Bigio, M.R.; Rastegar, M. Differential brain region-specific expression of MeCP2 and BDNF in Rett Syndrome patients: A distinct grey-white matter variation. Neuropathol. Appl. Neurobiol. 2020, 1–16. [Google Scholar] [CrossRef]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.T.; Conover, J.C.; Borday, V.; Champagnat, J.; Barbacid, M.; Yancopoulos, G.; Katz, D.M. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J. Neurosci. 1996, 16, 5361–5371. [Google Scholar] [CrossRef]
- Conover, J.C.; Erickson, J.T.; Katz, D.M.; Bianchi, L.M.; Poueymirou, W.T.; McClain, J.; Pan, L.; Helgren, M.; Ip, N.Y.; Boland, P. Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT4. Nature 1995, 375, 235–238. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Alemán, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Tropea, D.; Kreiman, G.; Lyckman, A.; Mukherjee, S.; Yu, H.; Horng, S.; Sur, M. Gene expression changes and molecular pathways mediating activity-dependent plasticity in visual cortex. Nat. Neurosci. 2006, 9, 660–668. [Google Scholar] [CrossRef]
- Zheng, W.H.; Quirion, R. Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J. Neurochem. 2004, 89, 844–852. [Google Scholar] [CrossRef]
- Nishijima, T.; Piriz, J.; Duflot, S.; Fernandez, A.M.; Gaitan, G.; Gomez-Pinedo, U.; Verdugo, J.M.G.; Leroy, F.; Soya, H.; Nuñez, A.; et al. Neuronal Activity Drives Localized Blood-Brain-Barrier Transport of Serum Insulin-like Growth Factor-I into the CNS. Neuron 2010, 67, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Pini, G.; Scusa, M.F.; Congiu, L.; Benincasa, A.; Morescalchi, P.; Bottiglioni, I.; Di Marco, P.; Borelli, P.; Bonuccelli, U.; Della-Chiesa, A.; et al. IGF1 as a Potential Treatment for Rett Syndrome: Safety Assessment in Six Rett Patients. Autism Res. Treat. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science 2007, 315, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.; Guy, J.; McKay, L.; Brockett, E.; Spike, R.C.; Selfridge, J.; De Sousa, D.; Merusi, C.; Riedel, G.; Bird, A.; et al. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain 2012, 135, 2699–2710. [Google Scholar] [CrossRef]
- Tropea, D.; Giacometti, E.; Wilson, N.R.; Beard, C.; McCurry, C.; Dong, D.F.; Flannery, R.; Jaenisch, R.; Sur, M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2029–2034. [Google Scholar] [CrossRef]
- Castro, J.; Garcia, R.I.; Kwok, S.; Banerjee, A.; Petravicz, J.; Woodson, J.; Mellios, N.; Tropea, D.; Sur, M. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 9941–9946. [Google Scholar] [CrossRef] [PubMed]
- Keogh, C.; Pini, G.; Gemo, I.; Kaufmann, W.E.; Tropea, D. Functional network mapping reveals state-dependent response to IGF1 treatment in Rett syndrome. Brain Sci. 2020, 10, 515. [Google Scholar] [CrossRef]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Leventhal, C.; Rafii, S.; Rafii, D.; Shahar, A.; Goldman, S.A. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol. Cell. Neurosci. 1999, 13, 450–464. [Google Scholar] [CrossRef]
- Kallmann, B.A.; Wagner, S.; Hummel, V.; Buttmann, M.; Bayas, A.; Tonn, J.C.; Rieckmann, P. Characteristic gene expression profile of primary human cerebral endothelial cells. FASEB J. 2002, 16, 589–591. [Google Scholar] [CrossRef]
- Quirié, A.; Hervieu, M.; Garnier, P.; Demougeot, C.; Mossiat, C.; Bertrand, N.; Martin, A.; Marie, C.; Prigent-Tessier, A. Comparative Effect of Treadmill Exercise on Mature BDNF Production in Control versus Stroke Rats. PLoS ONE 2012, 7, e44218. [Google Scholar] [CrossRef]
- Navaratna, D.; Guo, S.Z.; Hayakawa, K.; Wang, X.; Gerhardinger, C.; Lo, E.H. Decreased cerebrovascular brain-derived neurotrophic factor-mediated neuroprotection in the diabetic brain. Diabetes 2011, 60, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Kim, W.J.; Lok, J.; Lee, S.-R.; Besancon, E.; Luo, B.-H.; Stins, M.F.; Wang, X.; Dedhar, S.; Lo, E.H. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 7582–7587. [Google Scholar] [CrossRef] [PubMed]
- Monnier, A.; Prigent-Tessier, A.; Quirié, A.; Bertrand, N.; Savary, S.; Gondcaille, C.; Garnier, P.; Demougeot, C.; Marie, C. Brain-derived neurotrophic factor of the cerebral microvasculature: A forgotten and nitric oxide-dependent contributor of brain-derived neurotrophic factor in the brain. Acta Physiol. 2017, 219, 790–802. [Google Scholar] [CrossRef]
- Bagayogo, I.P.; Dreyfus, C.F. Regulated Release of BDNF by Cortical Oligodendrocytes is Mediated Through Metabotropic Glutamate Receptors and the PLC Pathway. ASN Neuro 2009, 1, AN20090006. [Google Scholar] [CrossRef] [PubMed]
- Jean, Y.Y.; Lercher, L.D.; Dreyfus, C.F. Glutamate elicits release of BDNF from basal forebrain astrocytes in a process dependent on metabotropic receptors and the PLC pathway. Neuron Glia Biol. 2008, 4, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, A.L.; Babbitt, C.C. The predictive nature of transcript expression levels on protein expression in adult human brain. BMC Genom. 2017, 18, 322. [Google Scholar] [CrossRef] [PubMed]
- Satriotomo, I.; Nichols, N.L.; Dale, E.A.; Emery, A.T.; Dahlberg, J.M.; Mitchell, G.S. Repetitive acute intermittent hypoxia increases growth/neurotrophic factor expression in non-. Neuroscience 2016, 322, 479–488. [Google Scholar] [CrossRef]
- Hartman, W.; Helan, M.; Smelter, D.; Sathish, V.; Thompson, M.; Pabelick, C.M.; Johnson, B.; Prakash, Y.S. Role of Hypoxia-Induced Brain Derived Neurotrophic Factor in Human Pulmonary Artery Smooth Muscle. PLoS ONE 2015, 10, e0129489. [Google Scholar] [CrossRef]
- Rajman, M.; Schratt, G. MicroRNAs in neural development: From master regulators to fine-tuners. Development 2017, 144, 2310–2322. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Cochella, L.; Hobert, O. Diverse functions of microRNAs in nervous system development. Curr. Top. Dev. Biol. 2012, 99, 115–143. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-Y.M.; Papp, J.W.; Varlamova, O.; Dziema, H.; Russell, B.; Curfman, J.P.; Nakazawa, T.; Shimizu, K.; Okamura, H.; Impey, S.; et al. microRNA modulation of circadian-clock period and entrainment. Neuron 2007, 54, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Marler, K.J.; Suetterlin, P.; Dopplapudi, A.; Rubikaite, A.; Adnan, J.; Maiorano, N.A.; Lowe, A.S.; Thompson, I.D.; Pathania, M.; Bordey, A.; et al. BDNF promotes axon branching of retinal ganglion cells via miRNA-132 and p250GAP. J. Neurosci. 2014, 34, 969–979. [Google Scholar] [CrossRef]
- Magill, S.T.; Cambronne, X.A.; Luikart, B.W.; Lioy, D.T.; Leighton, B.H.; Westbrook, G.L.; Mandel, G.; Goodman, R.H. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 20382–20387. [Google Scholar] [CrossRef] [PubMed]
- Remenyi, J.; van den Bosch, M.W.M.; Palygin, O.; Mistry, R.B.; McKenzie, C.; Macdonald, A.; Hutvagner, G.; Arthur, J.S.C.; Frenguelli, B.G.; Pankratov, Y. miR-132/212 knockout mice reveal roles for these miRNAs in regulating cortical synaptic transmission and plasticity. PLoS ONE 2013, 8, e62509. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef]
- Jasińska, M.; Miłek, J.; Cymerman, I.A.; Łęski, S.; Kaczmarek, L.; Dziembowska, M. miR-132 Regulates Dendritic Spine Structure by Direct Targeting of Matrix Metalloproteinase 9 mRNA. Mol. Neurobiol. 2016, 53, 4701–4712. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-T.; Chu, K.; Im, W.-S.; Yoon, H.-J.; Im, J.-Y.; Park, J.-E.; Park, K.-H.; Jung, K.-H.; Lee, S.K.; Kim, M.; et al. Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 2011, 227, 172–179. [Google Scholar] [CrossRef]
- Miller, B.H.; Zeier, Z.; Xi, L.; Lanz, T.A.; Deng, S.; Strathmann, J.; Willoughby, D.; Kenny, P.J.; Elsworth, J.D.; Lawrence, M.S.; et al. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc. Natl. Acad. Sci. USA 2012, 109, 3125–3130. [Google Scholar] [CrossRef]
- Wanet, A.; Tacheny, A.; Arnould, T.; Renard, P. miR-212/132 expression and functions: Within and beyond the neuronal compartment. Nucleic Acids Res. 2012, 40, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hao, J.; Xie, F.; Hu, X.; Liu, C.; Tong, J.; Zhou, J.; Wu, J.; Shao, C. Downregulation of miR-132 by promoter methylation contributes to pancreatic cancer development. Carcinogenesis 2011, 32, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Liu, C.-G.; Sevignani, C.; Ferracin, M.; Felli, N.; Dumitru, C.D.; Shimizu, M.; Cimmino, A.; Zupo, S.; Dono, M.; et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. USA 2004, 101, 11755–11760. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.F.; Karelina, K.; Sakamoto, K.; Wayman, G.A.; Impey, S.; Obrietan, K. miRNA-132: A dynamic regulator of cognitive capacity. Brain Struct. Funct. 2013, 218, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.F.; Sakamoto, K.; Wayman, G.A.; Impey, S.; Obrietan, K. Transgenic miR132 alters neuronal spine density and impairs novel object recognition memory. PLoS ONE 2010, 5, e15497. [Google Scholar] [CrossRef]
- Han, K.; Gennarino, V.A.; Lee, Y.; Pang, K.; Hashimoto-Torii, K.; Choufani, S.; Raju, C.S.; Oldham, M.C.; Weksberg, R.; Rakic, P.; et al. Human-specific regulation of MeCP2 levels in fetal brains by microRNA miR-483-5p. Genes Dev. 2013, 27, 485–490. [Google Scholar] [CrossRef]
- Zhang, K.; Jing, X.; Wang, G. MicroRNAs as regulators of drug abuse and immunity. Cent. J. Immunol. 2016, 41, 426–434. [Google Scholar] [CrossRef]
- Zhang, R.; Huang, M.; Cao, Z.; Qi, J.; Qiu, Z.; Chiang, L.-Y. MeCP2 plays an analgesic role in pain transmission through regulating CREB/miR-132 pathway. Mol. Pain 2015, 11, 19. [Google Scholar] [CrossRef]
- Ausió, J.; Martínez de Paz, A.; Esteller, M. MeCP2: The long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 2014, 20, 487–498. [Google Scholar] [CrossRef]
- Fichou, Y.; Nectoux, J.; Bahi-Buisson, N.; Rosas-Vargas, H.; Girard, B.; Chelly, J.; Bienvenu, T. The first missense mutation causing Rett syndrome specifically affecting the MeCP2_e1 isoform. Neurogenetics 2009, 10, 127–133. [Google Scholar] [CrossRef]
- Quenard, A.; Yilmaz, S.; Fontaine, H.; Bienvenu, T.; Moncla, A.; des Portes, V.; Rivier, F.; Mathieu, M.; Raux, G.; Jonveaux, P.; et al. Deleterious mutations in exon 1 of MECP2 in Rett syndrome. Eur. J. Med. Genet. 2006, 49, 313–322. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M.; Goldstine, J.; Balmer, D.; Greco, C.M. Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum. Mol. Genet. 2001, 10, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.D. Neuropathology of Rett syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Petel-Galil, Y.; Benteer, B.; Galil, Y.P.; Zeev, B.B.; Greenbaum, I.; Vecsler, M.; Goldman, B.; Lohi, H.; Minassian, B.A.; Gak, E. Comprehensive diagnosis of Rett’s syndrome relying on genetic, epigenetic and expression evidence of deficiency of the methyl-CpG-binding protein 2 gene: Study of a cohort of Israeli patients. J. Med. Genet. 2006, 43, e56. [Google Scholar] [CrossRef][Green Version]
- Katz, D.M.; Bird, A.; Coenraads, M.; Gray, S.J.; Menon, D.U.; Philpot, B.D.; Tarquinio, D.C. Rett Syndrome: Crossing the Threshold to Clinical Translation. Trends Neurosci. 2016, 39, 100–113. [Google Scholar] [CrossRef]
- Chapleau, C.A.; Lane, J.; Larimore, J.; Li, W.; Pozzo-Miller, L.; Percy, A.K. Recent Progress in Rett Syndrome and MeCP2 Dysfunction: Assessment of Potential Treatment Options. Future Neurol. 2013, 8. [Google Scholar] [CrossRef]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Gecz, J.; et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef]
- del Gaudio, D.; Fang, P.; Scaglia, F.; Ward, P.A.; Craigen, W.J.; Glaze, D.G.; Neul, J.L.; Patel, A.; Lee, J.A.; Irons, M.; et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 2006, 8, 784–792. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Belichenko, P.V.; Oldfors, A.; Hagberg, B.; Dahlström, A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport 1994, 5, 1509–1513. [Google Scholar] [CrossRef]
- Stearns, N.A.; Schaevitz, L.R.; Bowling, H.; Nag, N.; Berger, U.V.; Berger-Sweeney, J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: A model for Rett syndrome. Neuroscience 2007, 146, 907–921. [Google Scholar] [CrossRef]
- Chapleau, C.A.; Calfa, G.D.; Lane, M.C.; Albertson, A.J.; Larimore, J.L.; Kudo, S.; Armstrong, D.L.; Percy, A.K.; Pozzo-Miller, L. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 2009, 35, 2019–2233. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, G.; Angelucci, A.; Morando, L.; Boggio, E.M.; Giustetto, M.; Pizzorusso, T. Early Environmental Enrichment Moderates the Behavioral and Synaptic Phenotype of MeCP2 Null Mice. Biol. Psychiatry 2010, 67, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Landi, S.; Putignano, E.; Boggio, E.M.; Giustetto, M.; Pizzorusso, T.; Ratto, G.M. The short-time structural plasticity of dendritic spines is altered in a model of Rett syndrome. Sci. Rep. 2011, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Rikhye, R.V.; Breton-Provencher, V.; Tangb, X.; Li, C.; Li, K.; Runyan, C.A.; Fu, Z.; Jaenisch, R.; Sur, M. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E7287–E7296. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Takumi, T.; Nakai, J.; Sato, M. Common defects of spine dynamics and circuit function in neurodevelopmental disorders: A systematic review of findings from in vivo optical imaging of mouse models. Front. Neurosci. 2018, 12, 1–18. [Google Scholar] [CrossRef]
- Zoghbi, H.Y. Postnatal Neurodevelopmental Disorders: Meeting at the Synapse? Science 2003, 302, 826–830. [Google Scholar] [CrossRef]
- Bodda, C.; Tantra, M.; Mollajew, R.; Arunachalam, J.P.; Laccone, F.A.; Can, K.; Rosenberger, A.; Mironov, S.L.; Ehrenreich, H.; Mannan, A.U. Mild overexpression of Mecp2 in mice causes a higher susceptibility toward seizures. Am. J. Pathol. 2013, 183, 195–210. [Google Scholar] [CrossRef]
- Nagarajan, R.P.; Hogart, A.R.; Gwye, Y.; Martin, M.R.; LaSalle, J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006, 1, 172–182. [Google Scholar] [CrossRef]
- Huang, H.-S.; Allen, J.A.; Mabb, A.M.; King, I.F.; Miriyala, J.; Taylor-Blake, B.; Sciaky, N.; Dutton, J.W.; Lee, H.-M.; Chen, X.; et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011, 481, 185–189. [Google Scholar] [CrossRef]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef]
- Garg, S.K.; Lioy, D.T.; Cheval, H.; McGann, J.C.; Bissonnette, J.M.; Murtha, M.J.; Foust, K.D.; Kaspar, B.K.; Bird, A.; Mandel, G. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J. Neurosci. 2013, 33, 13612–13620. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics based on stop codon readthrough. Annu. Rev. Genom. Hum. Genet. 2014, 15, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Belakhov, V.; Werner, H.; Wegener, E.; Gärtner, J.; Nudelman, I.; Baasov, T.; Huppke, P. Readthrough of nonsense mutations in Rett syndrome: Evaluation of novel aminoglycosides and generation of a new mouse model. J. Mol. Med. 2011, 89, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, K.K.E.; Bailey, M.E.S.; Cobb, S.R. MeCP2 and Rett syndrome: Reversibility and potential avenues for therapy. Biochem. J. 2011, 439, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.M.; Baker, S.A.; Zoghbi, H.Y. MECP2 disorders: From the clinic to mice and back. J. Clin. Investig. 2015, 125, 2914–2923. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pejhan, S.; Rastegar, M. Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules 2021, 11, 75. https://doi.org/10.3390/biom11010075
Pejhan S, Rastegar M. Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules. 2021; 11(1):75. https://doi.org/10.3390/biom11010075
Chicago/Turabian StylePejhan, Shervin, and Mojgan Rastegar. 2021. "Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease" Biomolecules 11, no. 1: 75. https://doi.org/10.3390/biom11010075
APA StylePejhan, S., & Rastegar, M. (2021). Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules, 11(1), 75. https://doi.org/10.3390/biom11010075