Perspective: Potential Impact and Therapeutic Implications of Oncogenic PI3K Activation on Chromosomal Instability

 , ,
, ,
Abstract
1. Introduction
1.1. Class I PI3Ks–PIK3CA Mutation and Amplification
1.2. Chromosomal Instability, Whole-Genome Doubling and Aneuploidy-A Cellular Stress
1.2.1. Chromosomal Instability (CIN)
1.2.2. Whole Genome Doubling (WGD)
1.2.3. Aneuploidy, Cellular Stress and the CIN Paradox
1.3. Microtubules and the Mitotic Spindle
1.4. Centrosomes
2. PI3K Pathway Activation and CIN/WGD
2.1. PIK3CA/Akt
2.2. PTEN
3. Potential Molecular Mechanisms Underlying PIK3CA-Related CIN
3.1. Cell-Cycle Block by Constitutive PI3K (Over) Activation?
3.2. Impact of PI3K on Microtubules and the Mitotic Spindle
3.3. Impact of PI3K on Centrosomes (and Vice Versa)
4. Potential Therapeutic Exploitation in Cancer Prevention and Dampening of Tumour Evolution
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. Pi3k isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The pi3k pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the pten tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Madsen, R.R.; Knox, R.G.; Pearce, W.; Lopez, S.; Mahler-Araujo, B.; McGranahan, N.; Vanhaesebroeck, B.; Semple, R.K. Oncogenic pik3ca promotes cellular stemness in an allele dose-dependent manner. Proc. Natl. Acad. Sci. USA 2019, 116, 8380–8389. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Samuels, Y. Pik3ca in Cancer: The Past 30 Years. Semin Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Perisic, O.; Masson, G.R.; Vadas, O.; Williams, R.L. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (pik3ca). Proc. Natl. Acad. Sci. USA 2012, 109, 15259–15264. [Google Scholar] [CrossRef]
- Burke, J.E. Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol. Cell 2018, 71, 653–673. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Engelman, J.A.; Irie, H.Y.; Luo, J.; Brachmann, S.M.; Pearline, R.V.; Cantley, L.C.; Brugge, J.S. Breast cancer-associated pik3ca mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005, 65, 10992–11000. [Google Scholar] [CrossRef] [PubMed]
- Hutti, J.E.; Pfefferle, A.D.; Russell, S.C.; Sircar, M.; Perou, C.M.; Baldwin, A.S. Oncogenic pi3k mutations lead to nf-kappab-dependent cytokine expression following growth factor deprivation. Cancer Res. 2012, 72, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Denley, A.; Vanhaesebroeck, B.; Vogt, P.K. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class i phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2006, 103, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.H.; Pipinikas, C.P.; Pennycuick, A.; Lee-Six, H.; Chandrasekharan, D.; Beane, J.; Morris, T.J.; Karpathakis, A.; Feber, A.; Breeze, C.E.; et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat. Med. 2019, 25, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of pik3ca, pten, and akt mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Okada, J.; Timmerman, L.; Rodriguez-Viciana, P.; Stokoe, D.; Shoji, K.; Taketani, Y.; Kuramoto, H.; Knight, Z.A.; Shokat, K.M.; et al. Pik3ca cooperates with other phosphatidylinositol 3’-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 2008, 68, 8127–8136. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Donoghue, M.T.A.; Gadiya, M.; Hanrahan, A.J.; Won, H.H.; Chang, M.T.; Jonsson, P.; Penson, A.V.; Gorelick, A.; Harris, C.; et al. Widespread selection for oncogenic mutant allele imbalance in cancer. Cancer Cell 2018, 34, 852–862. [Google Scholar] [CrossRef]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Ollinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and kras dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. 2012, 13, 189–203. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Compton, D.A. A double-edged sword: How oncogenes and tumor suppressor genes can contribute to chromosomal instability. Front. Oncol. 2013, 3, 164. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [PubMed]
- Laughney, A.M.; Elizalde, S.; Genovese, G.; Bakhoum, S.F. Dynamics of tumor heterogeneity derived from clonal karyotypic evolution. Cell Rep. 2015, 12, 809–820. [Google Scholar] [CrossRef] [PubMed]
- López, S.; Lim, E.; Huebner, A.; Dietzen, M.; Mourikis, T.; Watkins, T.B.K.; Rowan, A.; Dewhurst, S.M.; Birkbak, N.J.; Wilson, G.A.; et al. Whole genome doubling mitigates muller’s ratchet in cancer evolution. bioRxiv 2019. [Google Scholar] [CrossRef]
- Oromendia, A.B.; Amon, A. Aneuploidy: Implications for protein homeostasis and disease. Dis. Mod. Mech. 2014, 7, 15–20. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Amon, A. The aneuploidy paradox: Costs and benefits of an incorrect karyotype. Trends Genet. 2011, 27, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.M.; van Deursen, J.M. Aneuploidy in cancer and aging. Annu. Rev. Genet. 2016, 50, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, S.; Amon, A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 2015, 16, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Stingele, S.; Stoehr, G.; Peplowska, K.; Cox, J.; Mann, M.; Storchova, Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 2012, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Stingele, S.; Stoehr, G.; Storchova, Z. Activation of autophagy in cells with abnormal karyotype. Autophagy 2013, 9, 246–248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, Q.; Au, B.; Kulkarni, M.; Shen, Y.; Lim, K.J.; Maimaiti, J.; Wong, C.K.; Luijten, M.N.H.; Chong, H.C.; Lim, E.H.; et al. Chromosomal instability-induced senescence potentiates cell non-autonomous tumourigenic effects. Oncogenesis 2018, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed]
- Andriani, G.A.; Almeida, V.P.; Faggioli, F.; Mauro, M.; Tsai, W.L.; Santambrogio, L.; Maslov, A.; Gadina, M.; Campisi, J.; Vijg, J.; et al. Whole chromosome instability induces senescence and promotes sasp. Sci. Rep. 2016, 6, 35218. [Google Scholar] [CrossRef] [PubMed]
- Oromendia, A.B.; Dodgson, S.E.; Amon, A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012, 26, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.J.; Nelliat, A.R.; Choudhury, M.I.; Kucharavy, A.; Bradford, W.D.; Cook, M.E.; Kim, J.; Mair, D.B.; Sun, S.X.; Schatz, M.C.; et al. Hypo-osmotic-like stress underlies general cellular defects of aneuploidy. Nature 2019, 570, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Nassour, J.; Radford, R.; Correia, A.; Fuste, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. P53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra254. [Google Scholar] [CrossRef]
- Hanel, W.; Moll, U.M. Links between mutant p53 and genomic instability. J. Cell. Biochem. 2012, 113, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Lansbergen, G.; Akhmanova, A. Microtubule plus end: A hub of cellular activities. Traffic 2006, 7, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Lyle, K.; Kumar, P.; Wittmann, T. Snapshot: Microtubule regulators ii. Cell 2009, 136, 566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lyle, K.; Kumar, P.; Wittmann, T. Snapshot: Microtubule regulators i. Cell 2009, 136, 380. [Google Scholar] [PubMed]
- Bakhoum, S.F.; Thompson, S.L.; Manning, A.L.; Compton, D.A. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. Cell Biol. 2009, 11, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Ertych, N.; Stolz, A.; Stenzinger, A.; Weichert, W.; Kaulfuss, S.; Burfeind, P.; Aigner, A.; Wordeman, L.; Bastians, H. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat. Cell Biol. 2014, 16, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.R.; Kramer, A. Centrosome amplification, chromosomal instability and cancer: Mechanistic, clinical and therapeutic issues. Chromosome Res. 2016, 24, 105–126. [Google Scholar] [CrossRef] [PubMed]
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130467. [Google Scholar] [CrossRef] [PubMed]
- Gonczy, P. Centrosomes and cancer: Revisiting a long-standing relationship. Nat. Rev. Cancer 2015, 15, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Maier, B.; Bartek, J. Centrosome clustering and chromosomal (in)stability: A matter of life and death. Mol. Oncol. 2011, 5, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Silk, A.D.; Montagna, C.; Verdier-Pinard, P.; Cleveland, D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef]
- Brinkley, B.R. Managing the centrosome numbers game: From chaos to stability in cancer cell division. Trends Cell Biol. 2001, 11, 18–21. [Google Scholar] [CrossRef]
- Basto, R.; Brunk, K.; Vinadogrova, T.; Peel, N.; Franz, A.; Khodjakov, A.; Raff, J.W. Centrosome amplification can initiate tumorigenesis in flies. Cell 2008, 133, 1032–1042. [Google Scholar] [CrossRef]
- Marthiens, V.; Piel, M.; Basto, R. Never tear us apart--the importance of centrosome clustering. J. Cell Sci. 2012, 125, 3281–3292. [Google Scholar] [CrossRef]
- Sabino, D.; Gogendeau, D.; Gambarotto, D.; Nano, M.; Pennetier, C.; Dingli, F.; Arras, G.; Loew, D.; Basto, R. Moesin is a major regulator of centrosome behavior in epithelial cells with extra centrosomes. Curr. Biol. 2015, 25, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Rhys, A.D.; Monteiro, P.; Smith, C.; Vaghela, M.; Arnandis, T.; Kato, T.; Leitinger, B.; Sahai, E.; McAinsh, A.; Charras, G.; et al. Loss of e-cadherin provides tolerance to centrosome amplification in epithelial cancer cells. J. Cell Biol. 2018, 217, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Silkworth, W.T.; Nardi, I.K.; Scholl, L.M.; Cimini, D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS ONE 2009, 4, e6564. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev. Cell 2017, 40, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Raff, J.W.; Basto, R. Centrosome amplification and cancer: A question of sufficiency. Dev. Cell 2017, 40, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Coelho, P.A.; Bury, L.; Shahbazi, M.N.; Liakath-Ali, K.; Tate, P.H.; Wormald, S.; Hindley, C.J.; Huch, M.; Archer, J.; Skarnes, W.C.; et al. Over-expression of plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 2015, 5, 150209. [Google Scholar] [CrossRef] [PubMed]
- Sercin, O.; Larsimont, J.C.; Karambelas, A.E.; Marthiens, V.; Moers, V.; Boeckx, B.; Le Mercier, M.; Lambrechts, D.; Basto, R.; Blanpain, C. Transient plk4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat. Cell Biol. 2016, 18, 100–110. [Google Scholar] [CrossRef]
- Chen, J.H.; Segni, M.; Payne, F.; Huang-Doran, I.; Sleigh, A.; Adams, C.; Consortium, U.K.; Savage, D.B.; O’Rahilly, S.; Semple, R.K.; et al. Truncation of poc1a associated with short stature and extreme insulin resistance. J. Mol. Endocrinol. 2015, 55, 147–158. [Google Scholar] [CrossRef]
- Bettencourt-Dias, M.; Hildebrandt, F.; Pellman, D.; Woods, G.; Godinho, S.A. Centrosomes and cilia in human disease. Trends Genet. 2011, 27, 307–315. [Google Scholar] [CrossRef]
- Chavali, P.L.; Putz, M.; Gergely, F. Small organelle, big responsibility: The role of centrosomes in development and disease. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130468. [Google Scholar] [CrossRef]
- Nigg, E.A.; Raff, J.W. Centrioles, centrosomes, and cilia in health and disease. Cell 2009, 139, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Arquint, C.; Gabryjonczyk, A.M.; Nigg, E.A. Centrosomes as signalling centres. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130464. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Hong, W.J.; Majeti, R.; Stearns, T. Centrosome-kinase fusions promote oncogenic signaling and disrupt centrosome function in myeloproliferative neoplasms. PLoS ONE 2014, 9, e92641. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.; Alonso-Nunez, M.; Grallert, A.; Tanaka, K.; Connolly, Y.; Smith, D.L.; Hagan, I.M. Dialogue between centrosomal entrance and exit scaffold pathways regulates mitotic commitment. J. Cell Biol. 2017, 216, 2795–2812. [Google Scholar] [CrossRef] [PubMed]
- Kapeller, R.; Chakrabarti, R.; Cantley, L.; Fay, F.; Corvera, S. Internalization of activated platelet-derived growth factor receptor-phosphatidylinositol-3’ kinase complexes: Potential interactions with the microtubule cytoskeleton. Mol. Cell. Biol. 1993, 13, 6052–6063. [Google Scholar] [CrossRef] [PubMed]
- Kapeller, R.; Toker, A.; Cantley, L.C.; Carpenter, C.L. Phosphoinositide 3-kinase binds constitutively to alpha/beta-tubulin and binds to gamma-tubulin in response to insulin. J. Biol. Chem. 1995, 270, 25985–25991. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, J.G.; Stephens, D.J.; Tavare, J.M. A role for glycogen synthase kinase-3 in mitotic spindle dynamics and chromosome alignment. J. Cell Sci. 2003, 116, 637–646. [Google Scholar] [CrossRef]
- Zhu, D.; Shi, S.; Wang, H.; Liao, K. Growth arrest induces primary-cilium formation and sensitizes igf-1-receptor signaling during differentiation induction of 3t3-l1 preadipocytes. J. Cell Sci. 2009, 122, 2760–2768. [Google Scholar] [CrossRef]
- Suizu, F.; Hirata, N.; Kimura, K.; Edamura, T.; Tanaka, T.; Ishigaki, S.; Donia, T.; Noguchi, H.; Iwanaga, T.; Noguchi, M. Phosphorylation-dependent akt-inversin interaction at the basal body of primary cilia. EMBO J. 2016, 35, 1346–1363. [Google Scholar] [CrossRef]
- Leonard, M.K.; Hill, N.T.; Bubulya, P.A.; Kadakia, M.P. The pten-akt pathway impacts the integrity and composition of mitotic centrosomes. Cell Cycle 2013, 12, 1406–1415. [Google Scholar] [CrossRef]
- Astrinidis, A.; Senapedis, W.; Henske, E.P. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum. Mol. Genet. 2006, 15, 287–297. [Google Scholar] [CrossRef] [PubMed]
- van Ree, J.H.; Nam, H.J.; Jeganathan, K.B.; Kanakkanthara, A.; van Deursen, J.M. Pten regulates spindle pole movement through dlg1-mediated recruitment of eg5 to centrosomes. Nat. Cell Biol. 2016, 18, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zou, Y.; Liu, H.; Wang, H.; Zhang, H.; Hou, W.; Li, X.; Jia, X.; Zhang, J.; Hou, L.; et al. Teif associated centrosome activity is regulated by egf/pi3k/akt signaling. Biochim. Biophys. Acta 2014, 1843, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Murakami, H.; Asai, N.; Morone, N.; Watanabe, T.; Kawai, K.; Murakumo, Y.; Usukura, J.; Kaibuchi, K.; Takahashi, M. Akt/pkb regulates actin organization and cell motility via girdin/ape. Dev. Cell 2005, 9, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.Z.; Jiang, P.; Cui, S.P.; Ren, Y.L.; Zhao, J.; Yin, X.H.; Enomoto, A.; Liu, H.J.; Hou, L.; Takahashi, M.; et al. Girdin locates in centrosome and midbody and plays an important role in cell division. Cancer Sci. 2012, 103, 1780–1787. [Google Scholar] [CrossRef] [PubMed]
- Kamata, T.; Pritchard, C. Mechanisms of aneuploidy induction by ras and raf oncogenes. Am. J. Cancer Res. 2011, 1, 955–971. [Google Scholar] [PubMed]
- Cui, Y.; Borysova, M.K.; Johnson, J.O.; Guadagno, T.M. Oncogenic b-raf(v600e) induces spindle abnormalities, supernumerary centrosomes, and aneuploidy in human melanocytic cells. Cancer Res. 2010, 70, 675–684. [Google Scholar] [CrossRef]
- Denko, N.C.; Giaccia, A.J.; Stringer, J.R.; Stambrook, P.J. The human ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc. Natl. Acad. Sci. USA 1994, 91, 5124–5128. [Google Scholar] [CrossRef]
- Saavedra, H.I.; Fukasawa, K.; Conn, C.W.; Stambrook, P.J. Mapk mediates ras-induced chromosome instability. J. Biol. Chem. 1999, 274, 38083–38090. [Google Scholar] [CrossRef]
- Knauf, J.A.; Ouyang, B.; Knudsen, E.S.; Fukasawa, K.; Babcock, G.; Fagin, J.A. Oncogenic ras induces accelerated transition through g2/m and promotes defects in the g2 DNA damage and mitotic spindle checkpoints. J. Biol. Chem. 2006, 281, 3800–3809. [Google Scholar] [CrossRef]
- Abulaiti, A.; Fikaris, A.J.; Tsygankova, O.M.; Meinkoth, J.L. Ras induces chromosome instability and abrogation of the DNA damage response. Cancer Res. 2006, 66, 10505–10512. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Sambandam, V.; Vijayaraghavan, S.; Balaji, K.; Mukherjee, S. Molecular genetics and cellular events of k-ras-driven tumorigenesis. Oncogene 2018, 37, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Berenjeno, I.M.; Pineiro, R.; Castillo, S.D.; Pearce, W.; McGranahan, N.; Dewhurst, S.M.; Meniel, V.; Birkbak, N.J.; Lau, E.; Sansregret, L.; et al. Oncogenic pik3ca induces centrosome amplification and tolerance to genome doubling. Nat. Commun. 2017, 8, 1773. [Google Scholar] [CrossRef] [PubMed]
- Plo, I.; Lopez, B. Akt1 represses gene conversion induced by different genotoxic stresses and induces supernumerary centrosomes and aneuploidy in hamster ovary cells. Oncogene 2009, 28, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Woodgett, J.R. Chronic activation of protein kinase bbeta/akt2 leads to multinucleation and cell fusion in human epithelial kidney cells: Events associated with tumorigenesis. Oncogene 2005, 24, 5459–5470. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Van Veen, E.; Van Vugt, M.A.; Thoreen, C.C.; Sabatini, D.M. Mtorc1 regulates cytokinesis through activation of rho-rock signaling. arXiv 2012, arXiv:1506.04437v1. Available online: https://arxiv.org/abs/1506.04437 (accessed on 1 August 2019).
- Moniz, L.S.; Surinova, S.; Ghazaly, E.; Velasco, L.G.; Haider, S.; Rodriguez-Prados, J.C.; Berenjeno, I.M.; Chelala, C.; Vanhaesebroeck, B. Phosphoproteomic comparison of pik3ca and pten signalling identifies the nucleotidase nt5c as a novel akt substrate. Sci. Rep. 2017, 7, 39985. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, V.Y.; Juvin, V.; Malek, M.; Luscombe, N.; Hawkins, P.; Le Novere, N.; Stephens, L. Perturbations of pip3 signalling trigger a global remodelling of mrna landscape and reveal a transcriptional feedback loop. Nucleic Acids Res. 2015, 43, 9663–9679. [Google Scholar] [CrossRef] [PubMed]
- Vinayagam, A.; Kulkarni, M.M.; Sopko, R.; Sun, X.; Hu, Y.; Nand, A.; Villalta, C.; Moghimi, A.; Yang, X.; Mohr, S.E.; et al. An integrative analysis of the inr/pi3k/akt network identifies the dynamic response to insulin signaling. Cell Rep. 2016, 16, 3062–3074. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.J.; Chae, S.; Jang, S.H.; Cho, H.; Lee, J.H. The pi3k-akt mediates oncogenic met-induced centrosome amplification and chromosome instability. Carcinogenesis 2010, 31, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Celton-Morizur, S.; Merlen, G.; Couton, D.; Margall-Ducos, G.; Desdouets, C. The insulin/akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J. Clin. Investig. 2009, 119, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.Q.; Ouyang, M.; Brandmaier, A.; Hao, H.; Shen, W.H. Pten in the maintenance of genome integrity: From DNA replication to chromosome segregation. Bioessays 2017, 39, 1700082. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Martini, M.; De Santis, M.C.; Hirsch, E. How pi3k-derived lipids control cell division. Front. Cell Dev. Biol. 2015, 3, 61. [Google Scholar] [CrossRef] [PubMed]
- Garcia, Z.; Kumar, A.; Marques, M.; Cortes, I.; Carrera, A.C. Phosphoinositide 3-kinase controls early and late events in mammalian cell division. EMBO J. 2006, 25, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Klinghoffer, R.; Prestwich, G.D.; Toker, A.; Kazlauskas, A. Pdgf induces an early and a late wave of pi 3-kinase activity, and only the late wave is required for progression through g1. Curr. Biol. 1999, 9, 512–521. [Google Scholar] [CrossRef]
- Marques, M.; Kumar, A.; Cortes, I.; Gonzalez-Garcia, A.; Hernandez, C.; Moreno-Ortiz, M.C.; Carrera, A.C. Phosphoinositide 3-kinases p110alpha and p110beta regulate cell cycle entry, exhibiting distinct activation kinetics in g1 phase. Mol. Cell. Biol. 2008, 28, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Sussman, J.; Stokoe, D. A role for pi 3-kinase and pkb activity in the g2/m phase of the cell cycle. Curr. Biol. 2002, 12, 919–924. [Google Scholar] [CrossRef]
- Jones, S.M.; Kazlauskas, A. Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat. Cell Biol. 2001, 3, 165–172. [Google Scholar] [CrossRef]
- Klippel, A.; Escobedo, M.A.; Wachowicz, M.S.; Apell, G.; Brown, T.W.; Giedlin, M.A.; Kavanaugh, W.M.; Williams, L.T. Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol. Cell. Biol. 1998, 18, 5699–5711. [Google Scholar] [CrossRef]
- Alvarez, B.; Martinez, A.C.; Burgering, B.M.; Carrera, A.C. Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature 2001, 413, 744–747. [Google Scholar] [CrossRef]
- Yuan, T.L.; Wulf, G.; Burga, L.; Cantley, L.C. Cell-to-cell variability in pi3k protein level regulates pi3k-akt pathway activity in cell populations. Curr. Biol. 2011, 21, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Sizek, H.; Hamel, A.; Deritei, D.; Campbell, S.; Ravasz Regan, E. Boolean model of growth signaling, cell cycle and apoptosis predicts the molecular mechanism of aberrant cell cycle progression driven by hyperactive pi3k. PLoS Comput. Biol. 2019, 15, e1006402. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef] [PubMed]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. Akt induces senescence in human cells via mtorc1 and p53 in the absence of DNA damage: Implications for targeting mtor during malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, C.; Bonifant, C.L.; Ressom, H.; Waldman, T. Activation of p53-dependent growth suppression in human cells by mutations in pten or pik3ca. Mol. Cell. Biol. 2007, 27, 662–677. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Sandoval, M.; Beronja, S. Oncogenic activation of pi3k induces progenitor cell differentiation to suppress epidermal growth. Nat. Cell Biol. 2018, 20, 1256–1266. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Morton, J.P.; Manoharan, I.; Nelson, D.M.; Jamieson, N.B.; Pawlikowski, J.S.; McBryan, T.; Doyle, B.; McKay, C.; Oien, K.A.; et al. Activation of the pik3ca/akt pathway suppresses senescence induced by an activated ras oncogene to promote tumorigenesis. Mol. Cell 2011, 42, 36–49. [Google Scholar] [CrossRef]
- Onishi, K.; Higuchi, M.; Asakura, T.; Masuyama, N.; Gotoh, Y. The pi3k-akt pathway promotes microtubule stabilization in migrating fibroblasts. Genes Cells 2007, 12, 535–546. [Google Scholar] [CrossRef]
- Gasic, I.; Boswell, S.A.; Mitchison, T.J. Tubulin mrna stability is sensitive to change in microtubule dynamics caused by multiple physiological and toxic cues. PLoS Biol. 2019, 17, e3000225. [Google Scholar] [CrossRef]
- Akhmanova, A.; Hoogenraad, C.C.; Drabek, K.; Stepanova, T.; Dortland, B.; Verkerk, T.; Vermeulen, W.; Burgering, B.M.; De Zeeuw, C.I.; Grosveld, F.; et al. Clasps are clip-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 2001, 104, 923–935. [Google Scholar] [CrossRef]
- Di Paolo, G.; Antonsson, B.; Kassel, D.; Riederer, B.M.; Grenningloh, G. Phosphorylation regulates the microtubule-destabilizing activity of stathmin and its interaction with tubulin. FEBS Lett. 1997, 416, 149–152. [Google Scholar] [CrossRef]
- Wik, E.; Birkeland, E.; Trovik, J.; Werner, H.M.; Hoivik, E.A.; Mjos, S.; Krakstad, C.; Kusonmano, K.; Mauland, K.; Stefansson, I.M.; et al. High phospho-stathmin(serine38) expression identifies aggressive endometrial cancer and suggests an association with pi3k inhibition. Clin. Cancer Res. 2013, 19, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Sathyanarayanan, S.; Di Bacco, A.; Chi, A.; Zhang, T.; Chen, A.H.; Dolinski, B.; Kraus, M.; Roberts, B.; Arthur, W.; et al. Pathway-based identification of biomarkers for targeted therapeutics: Personalized oncology with pi3k pathway inhibitors. Sci. Transl. Med. 2010, 2, 43ra55. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase b. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Buttrick, G.J.; Beaumont, L.M.; Leitch, J.; Yau, C.; Hughes, J.R.; Wakefield, J.G. Akt regulates centrosome migration and spindle orientation in the early drosophila melanogaster embryo. J. Cell Biol. 2008, 180, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Buttrick, G.J.; Wakefield, J.G. Pi3-k and gsk-3: Akt-ing together with microtubules. Cell Cycle 2008, 7, 2621–2625. [Google Scholar] [CrossRef] [PubMed]
- Mitsushima, M.; Toyoshima, F.; Nishida, E. Dual role of cdc42 in spindle orientation control of adherent cells. Mol. Cell. Biol. 2009, 29, 2816–2827. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, F.; Matsumura, S.; Morimoto, H.; Mitsushima, M.; Nishida, E. Ptdins(3,4,5)p3 regulates spindle orientation in adherent cells. Dev. Cell 2007, 13, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Silio, V.; Redondo-Munoz, J.; Carrera, A.C. Phosphoinositide 3-kinase beta regulates chromosome segregation in mitosis. Mol. Biol. Cell 2012, 23, 4526–4542. [Google Scholar] [CrossRef]
- Paranavitane, V.; Coadwell, W.J.; Eguinoa, A.; Hawkins, P.T.; Stephens, L. Ll5beta is a phosphatidylinositol (3,4,5)-trisphosphate sensor that can bind the cytoskeletal adaptor, gamma-filamin. J. Biol. Chem. 2003, 278, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Lansbergen, G.; Grigoriev, I.; Mimori-Kiyosue, Y.; Ohtsuka, T.; Higa, S.; Kitajima, I.; Demmers, J.; Galjart, N.; Houtsmuller, A.B.; Grosveld, F.; et al. Clasps attach microtubule plus ends to the cell cortex through a complex with ll5beta. Dev. Cell 2006, 11, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Naguib, A.; Bencze, G.; Cho, H.; Zheng, W.; Tocilj, A.; Elkayam, E.; Faehnle, C.R.; Jaber, N.; Pratt, C.P.; Chen, M.; et al. Pten functions by recruitment to cytoplasmic vesicles. Mol. Cell 2015, 58, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Hosokawa, Y.; Watanabe, K.; Tanimura, S.; Ozaki, K.; Kohno, M. Blockade of the phosphatidylinositol-3-kinase-akt signaling pathway enhances the induction of apoptosis by microtubule-destabilizing agents in tumor cells in which the pathway is constitutively activated. Mol. Cancer Ther. 2007, 6, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Bohnacker, T.; Prota, A.E.; Beaufils, F.; Burke, J.E.; Melone, A.; Inglis, A.J.; Rageot, D.; Sele, A.M.; Cmiljanovic, V.; Cmiljanovic, N.; et al. Deconvolution of buparlisib’s mechanism of action defines specific pi3k and tubulin inhibitors for therapeutic intervention. Nat. Commun. 2017, 8, 14683. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, S.M.; Kleylein-Sohn, J.; Gaulis, S.; Kauffmann, A.; Blommers, M.J.; Kazic-Legueux, M.; Laborde, L.; Hattenberger, M.; Stauffer, F.; Vaxelaire, J.; et al. Characterization of the mechanism of action of the pan class i pi3k inhibitor nvp-bkm120 across a broad range of concentrations. Mol. Cancer Ther. 2012, 11, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Mardin, B.R.; Isokane, M.; Cosenza, M.R.; Kramer, A.; Ellenberg, J.; Fry, A.M.; Schiebel, E. Egf-induced centrosome separation promotes mitotic progression and cell survival. Dev. Cell 2013, 25, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A.; Rodriguez-Viciana, P.; Wennstrom, S.; Warne, P.H.; Downward, J. Matrix adhesion and ras transformation both activate a phosphoinositide 3-oh kinase and protein kinase b/akt cellular survival pathway. EMBO J. 1997, 16, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic instability is induced by persistent proliferation of cells undergoing epithelial-to-mesenchymal transition. Cell Rep. 2016, 17, 2632–2647. [Google Scholar] [CrossRef] [PubMed]
- Gergely, F.; Karlsson, C.; Still, I.; Cowell, J.; Kilmartin, J.; Raff, J.W. The tacc domain identifies a family of centrosomal proteins that can interact with microtubules. Proc. Natl. Acad. Sci. USA 2000, 97, 14352–14357. [Google Scholar] [CrossRef] [PubMed]
- Peset, I.; Vernos, I. The tacc proteins: Tacc-ling microtubule dynamics and centrosome function. Trends Cell Biol. 2008, 18, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Zwang, Y.; Jonas, O.; Chen, C.; Rinne, M.L.; Doench, J.G.; Piccioni, F.; Tan, L.; Huang, H.T.; Wang, J.; Ham, Y.J.; et al. Synergistic interactions with pi3k inhibition that induce apoptosis. eLife 2017, 6, e24523. [Google Scholar] [CrossRef] [PubMed]
- Cully, M.; Shiu, J.; Piekorz, R.P.; Muller, W.J.; Done, S.J.; Mak, T.W. Transforming acidic coiled coil 1 promotes transformation and mammary tumorigenesis. Cancer Res. 2005, 65, 10363–10370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gomez-Baldo, L.; Schmidt, S.; Maxwell, C.A.; Bonifaci, N.; Gabaldon, T.; Vidalain, P.O.; Senapedis, W.; Kletke, A.; Rosing, M.; Barnekow, A.; et al. Tacc3-tsc2 maintains nuclear envelope structure and controls cell division. Cell Cycle 2010, 9, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Kumar, S.; Zaman, N.; Pan, C.C.; Bloodworth, J.C.; Lei, W.; Streicher, J.M.; Hempel, N.; Mythreye, K.; Lee, N.Y. Tak1 activation of alpha-tat1 and microtubule hyperacetylation control akt signaling and cell growth. Nat. Commun. 2018, 9, 1696. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. bioRxiv 2018, 161562. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Shaw, J.A.; Guttery, D.S.; Hills, A.; Fernandez-Garcia, D.; Page, K.; Rosales, B.M.; Goddard, K.S.; Hastings, R.K.; Luo, J.; Ogle, O.; et al. Mutation analysis of cell-free DNA and single circulating tumor cells in metastatic breast cancer patients with high circulating tumor cell counts. Clin. Cancer Res. 2017, 23, 88–96. [Google Scholar] [CrossRef]
- Semple, R.K.; Vanhaesebroeck, B. Lessons for cancer drug treatment from tackling a non-cancerous overgrowth syndrome. Nature 2018, 558, 523–525. [Google Scholar] [CrossRef]
- Fisher, R.; Horswell, S.; Rowan, A.; Salm, M.P.; de Bruin, E.C.; Gulati, S.; McGranahan, N.; Stares, M.; Gerlinger, M.; Varela, I.; et al. Development of synchronous vhl syndrome tumors reveals contingencies and constraints to tumor evolution. Genome Biol. 2014, 15, 433. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Birkbak, N.J.; Swanton, C. Constraints in cancer evolution. Biochem. Soc. Trans. 2017, 45, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Madsen, R.R.; Vanhaesebroeck, B.; Semple, R.K. Cancer-associated pik3ca mutations in overgrowth disorders. Trends Mol. Med. 2018, 24, 856–870. [Google Scholar] [CrossRef] [PubMed]
- Venot, Q.; Blanc, T.; Rabia, S.H.; Berteloot, L.; Ladraa, S.; Duong, J.P.; Blanc, E.; Johnson, S.C.; Hoguin, C.; Boccara, O.; et al. Targeted therapy in patients with pik3ca-related overgrowth syndrome. Nature 2018, 558, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Knouse, K.A.; Lopez, K.E.; Bachofner, M.; Amon, A. Chromosome segregation fidelity in epithelia requires tissue architecture. Cell 2018, 175, 200–211. [Google Scholar] [CrossRef] [PubMed]



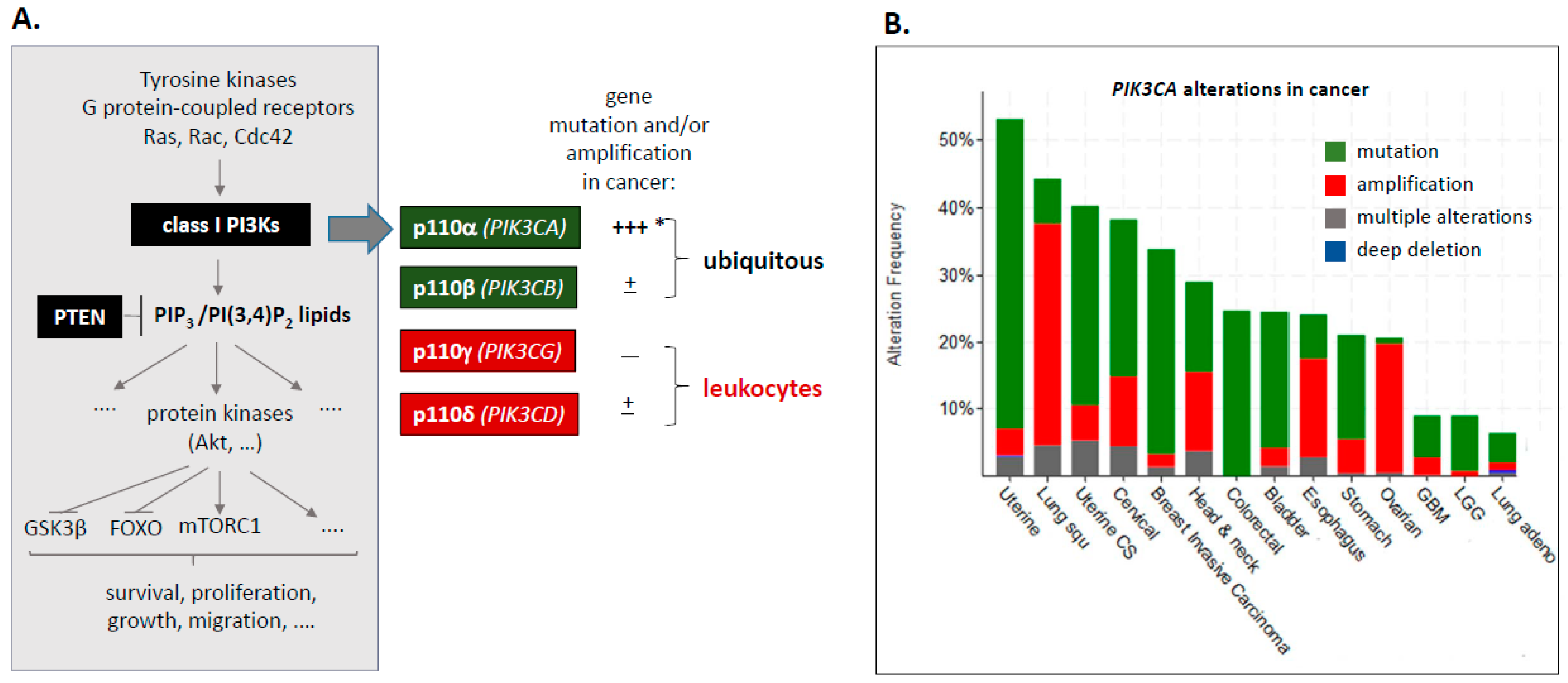{kind=link}
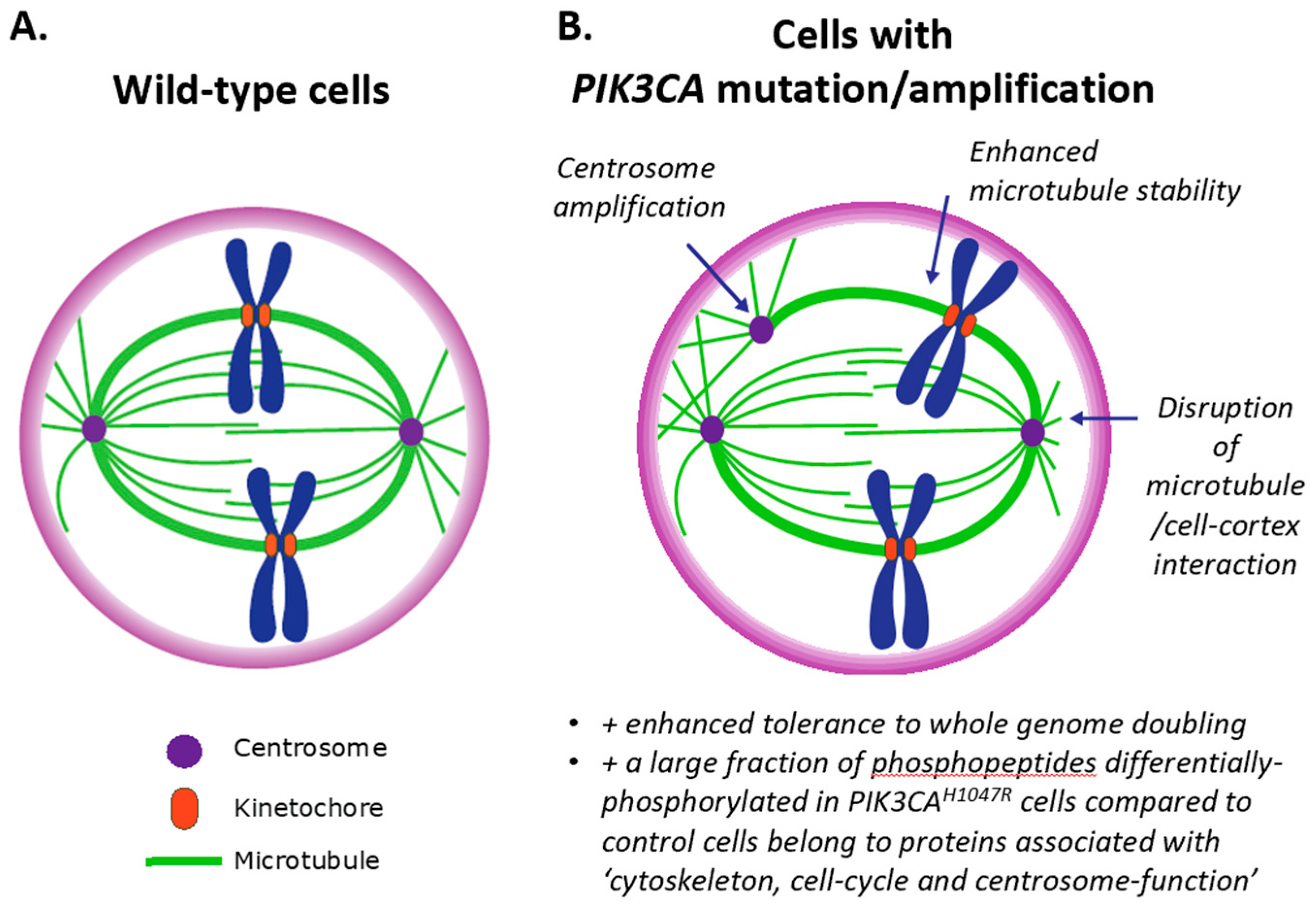{kind=link}
| PI3K Pathway Component | References | Additional Information |
|---|---|---|
| p85 regulatory subunit of PI3K | [75,76] | p85 can associate with the centrosome in an insulin-dependent manner |
| Akt/PKB | [77] | Akt/PKB is phosphorylated during mitosis and is present in the centrosome |
| [78] | T308-phosphorylated Akt/PKB is present in basal body of primary cilia | |
| [79] | S473-phosphorylated Akt/PKB is present in basal body of primary cilia. Akt/PKB interacts with and phosphorylates the ciliary protein Inversin. | |
| [80] | Akt/PKB-inhibition prevents recruitment of PTEN to mitotic centrosomes | |
| TSC1/TSC2 (TSC2 is an Akt/PKB substrate) | [81] | TSC1 is present in centrosome. Phosphorylated TSC1 and phosphorylated TSC2 co-immunoprecipitate with Plk1 |
| GSK3β (Akt/PKB substrate) | [77] | Phospho-GSK-3 at the centrosomes upon entry into mitosis |
| PTEN | [82] | Phosphatase-independent (scaffold function) of PTEN is recruited to pre-mitotic centrosomes in a Plk1-dependent fashion |
| Akt/PKB Substrate | References | Additional Information |
|---|---|---|
| TSC1/TSC2 (TSC2 is an Akt/PKB substrate) | [81] | TSC1 is present in centrosome. Phosphorylated TSC1 and phosphorylated TSC2 co-immunoprecipitate with Plk1 |
| GSK3β | [77] | Phospho-GSK3 at the centrosomes upon entry into mitosis |
| Inversin | [79] | Akt/PKB interacts with and phosphorylates the ciliary protein Inversin—dimerisation. Co-localisation of Inversin and phosphorylated-Akt/PKB at the basal body is augmented by PDGF-AA. |
| TEIF | [83] | TIEF is a potential centrosome component |
| Girdin | [84,85] | Girdin may localise to centrosomes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanhaesebroeck, B.; Bilanges, B.; Madsen, R.R.; Dale, K.L.; Lau, E.; Vladimirou, E. Perspective: Potential Impact and Therapeutic Implications of Oncogenic PI3K Activation on Chromosomal Instability. Biomolecules 2019, 9, 331. https://doi.org/10.3390/biom9080331
Vanhaesebroeck B, Bilanges B, Madsen RR, Dale KL, Lau E, Vladimirou E. Perspective: Potential Impact and Therapeutic Implications of Oncogenic PI3K Activation on Chromosomal Instability. Biomolecules. 2019; 9(8):331. https://doi.org/10.3390/biom9080331
Chicago/Turabian StyleVanhaesebroeck, Bart, Benoit Bilanges, Ralitsa R. Madsen, Katie L. Dale, Evelyn Lau, and Elina Vladimirou. 2019. "Perspective: Potential Impact and Therapeutic Implications of Oncogenic PI3K Activation on Chromosomal Instability" Biomolecules 9, no. 8: 331. https://doi.org/10.3390/biom9080331
APA StyleVanhaesebroeck, B., Bilanges, B., Madsen, R. R., Dale, K. L., Lau, E., & Vladimirou, E. (2019). Perspective: Potential Impact and Therapeutic Implications of Oncogenic PI3K Activation on Chromosomal Instability. Biomolecules, 9(8), 331. https://doi.org/10.3390/biom9080331