Intracellular Peptides in Cell Biology and Pharmacology

 , ,
, ,
Abstract
1. Introduction
2. Intracellular Peptides—A Brief Historical Retrospective
3. Intracellular Peptides Generation
4. Intracellular Peptides Acting on G-protein Coupled Receptors
5. Pharmacological and Biochemical Analyses of Intracellular Peptides Suggest to Function within Cells
5.1. EL28
5.2. PepH
5.3. Pep5
6. Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Fricker, L.D. Neuropeptide-processing enzymes: Applications for drug discovery. AAPS J. 2005, 7, E449–E455. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Dolan, B.P.; Bennink, J.R.; Yewdell, J.W. Translating DRiPs: Progress in understanding viral and cellular sources of MHC class I peptide ligands. Cell. Mol. Life Sci. 2011, 68, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef]
- Rist, M.J.; Theodossis, A.; Croft, N.P.; Neller, M.A.; Welland, A.; Chen, Z.; Sullivan, L.C.; Burrows, J.M.; Miles, J.J.; Brennan, R.M.; et al. HLA peptide length preferences control CD8+ T cell responses. J. Immunol. 2013, 191, 561–571. [Google Scholar] [CrossRef]
- Burrows, J.M.; Bell, M.J.; Brennan, R.; Miles, J.J.; Khanna, R.; Burrows, S.R. Preferential binding of unusually long peptides to MHC class I and its influence on the selection of target peptides for T cell recognition. Mol. Immunol. 2008, 45, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Kloverpris, H.N.; Stryhn, A.; Harndahl, M.; Payne, R.; Towers, G.J.; Chen, F.; Riddell, L.; Walker, B.D.; Ndung’u, T.; Leslie, A.; et al. HLA-specific intracellular epitope processing shapes an immunodominance pattern for HLA-B*57 that is distinct from HLA-B*58:01. J. Virol. 2013, 87, 10889–10894. [Google Scholar] [CrossRef] [PubMed]
- Caron, E.; Kowalewski, D.J.; Chiek Koh, C.; Sturm, T.; Schuster, H.; Aebersold, R. Analysis of Major Histocompatibility Complex (MHC) Immunopeptidomes Using Mass Spectrometry. Mol. Cell. Proteom. 2015, 14, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Connell, G.E.; Watson, R.W. Intracellular peptides of Pseudomonas hydrophila. Biochim. Biophys. Acta 1957, 24, 226–227. [Google Scholar] [CrossRef]
- McManus, I. Synthesis of intracellular peptides in Torula utilis. J. Biol. Chem. 1958, 231, 777–785. [Google Scholar]
- Guidotti, A.; Forchetti, C.M.; Corda, M.G.; Konkel, D.; Bennett, C.D.; Costa, E. Isolation, characterization, and purification to homogeneity of an endogenous polypeptide with agonistic action on benzodiazepine receptors. Proc. Natl. Acad. Sci. USA 1983, 80, 3531–3535. [Google Scholar] [CrossRef]
- Alho, H.; Costa, E.; Ferrero, P.; Fujimoto, M.; Cosenza-Murphy, D.; Guidotti, A. Diazepam-binding inhibitor: A neuropeptide located in selected neuronal populations of rat brain. Science 1985, 229, 179–182. [Google Scholar] [CrossRef]
- Huyer, G.; Kistler, A.; Nouvet, F.J.; George, C.M.; Boyle, M.L.; Michaelis, S. Saccharomyces cerevisiae a-factor mutants reveal residues critical for processing, activity, and export. Eukaryot. Cell 2006, 5, 1560–1570. [Google Scholar] [CrossRef]
- Rioli, V.; Gozzo, F.C.; Heimann, A.S.; Linardi, A.; Krieger, J.E.; Shida, C.S.; Almeida, P.C.; Hyslop, S.; Eberlin, M.N.; Ferro, E.S. Novel natural peptide substrates for endopeptidase 24.15, neurolysin, and angiotensin-converting enzyme. J. Biol. Chem. 2003, 278, 8547–8555. [Google Scholar] [CrossRef]
- Rioli, V.; Ferro, E.S. Substrate Capture Assay Using Inactive Oligopeptidases to Identify Novel Peptides. Methods Mol. Biol. 2018, 1719, 97–105. [Google Scholar] [CrossRef]
- Dale, C.S.; Pagano Rde, L.; Rioli, V. Hemopressin: A novel bioactive peptide derived from the alpha1-chain of hemoglobin. Mem Inst. Oswaldo Cruz 2005, 100 (Suppl. 1), 105–106. [Google Scholar] [CrossRef]
- Heimann, A.S.; Favarato, M.H.; Gozzo, F.C.; Rioli, V.; Carreno, F.R.; Eberlin, M.N.; Ferro, E.S.; Krege, J.H.; Krieger, J.E. ACE gene titration in mice uncovers a new mechanism for ACE on the control of body weight. Physiol. Genom. 2005, 20, 173–182. [Google Scholar] [CrossRef]
- Machado, M.F.; Cunha, F.M.; Berti, D.A.; Heimann, A.S.; Klitzke, C.F.; Rioli, V.; Oliveira, V.; Ferro, E.S. Substrate phosphorylation affects degradation and interaction to endopeptidase 24.15, neurolysin, and angiotensin-converting enzyme. Biochem. Biophys. Res. Commun. 2006, 339, 520–525. [Google Scholar] [CrossRef]
- Ferro, E.S.; Hyslop, S.; Camargo, A.C. Intracellullar peptides as putative natural regulators of protein interactions. J. Neurochem. 2004, 91, 769–777. [Google Scholar] [CrossRef]
- Fesenko, I.A.; Arapidi, G.P.; Skripnikov, A.Y.; Alexeev, D.G.; Kostryukova, E.S.; Manolov, A.I.; Altukhov, I.A.; Khazigaleeva, R.A.; Seredina, A.V.; Kovalchuk, S.I.; et al. Specific pools of endogenous peptides are present in gametophore, protonema, and protoplast cells of the moss Physcomitrella patens. BMC Plant. Biol. 2015, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Fesenko, I.; Khazigaleeva, R.; Govorun, V.; Ivanov, V. Analysis of Endogenous Peptide Pools of Physcomitrella patens Moss. Methods Mol. Biol. 2018, 1719, 395–405. [Google Scholar] [CrossRef]
- Dasgupta, S.; Yang, C.; Castro, L.M.; Tashima, A.K.; Ferro, E.S.; Moir, R.D.; Willis, I.M.; Fricker, L.D. Analysis of the Yeast Peptidome and Comparison with the Human Peptidome. PLoS ONE 2016, 11, e0163312. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.M.M.; Correa, C.N.; Iwai, L.K.; Ferro, E.S.; Castro, L.M. Characterization of intracellular peptides from zebrafish Danio rerio brain”. Zebrafish 2019. [Google Scholar]
- Fricker, L.D. Analysis of mouse brain peptides using mass spectrometry-based peptidomics: Implications for novel functions ranging from non-classical neuropeptides to microproteins. Mol. Biosyst. 2010, 6, 1355–1365. [Google Scholar] [CrossRef]
- Gelman, J.S.; Sironi, J.; Castro, L.M.; Ferro, E.S.; Fricker, L.D. Hemopressins and other hemoglobin-derived peptides in mouse brain: Comparison between brain, blood, and heart peptidome and regulation in Cpefat/fat mice. J. Neurochem. 2010, 113, 871–880. [Google Scholar] [CrossRef]
- Berezniuk, I.; Sironi, J.; Callaway, M.B.; Castro, L.M.; Hirata, I.Y.; Ferro, E.S.; Fricker, L.D. CCP1/Nna1 functions in protein turnover in mouse brain: Implications for cell death in Purkinje cell degeneration mice. FASEB J. 2010, 24, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Berti, D.A.; Morano, C.; Russo, L.C.; Castro, L.M.; Cunha, F.M.; Zhang, X.; Sironi, J.; Klitzke, C.F.; Ferro, E.S.; Fricker, L.D. Analysis of intracellular substrates and products of thimet oligopeptidase in human embryonic kidney 293 cells. J. Biol. Chem. 2009, 284, 14105–14116. [Google Scholar] [CrossRef]
- Gelman, J.S.; Sironi, J.; Castro, L.M.; Ferro, E.S.; Fricker, L.D. Peptidomic analysis of human cell lines. J. Proteom. Res. 2011, 10, 1583–1592. [Google Scholar] [CrossRef]
- Dasgupta, S.; Castro, L.M.; Dulman, R.; Yang, C.; Schmidt, M.; Ferro, E.S.; Fricker, L.D. Proteasome inhibitors alter levels of intracellular peptides in HEK293T and SH-SY5Y cells. PLoS ONE 2014, 9, e103604. [Google Scholar] [CrossRef]
- Cafe-Mendes, C.C.; Ferro, E.S.; Torrao, A.S.; Crunfli, F.; Rioli, V.; Schmitt, A.; Falkai, P.; Britto, L.R.; Turck, C.W.; Martins-de-Souza, D. Peptidomic analysis of the anterior temporal lobe and corpus callosum from schizophrenia patients. J. Proteom. 2017, 151, 97–105. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Wang, F.; You, L.; Xu, P.; Cao, Y.; Chen, L.; Wen, J.; Guo, X.; Cui, X.; et al. Identification of intracellular peptides associated with thermogenesis in human brown adipocytes. J. Cell. Physiol. 2018, 234, 7104–7114. [Google Scholar] [CrossRef]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef]
- Heimann, A.S.; Gomes, I.; Dale, C.S.; Pagano, R.L.; Gupta, A.; de Souza, L.L.; Luchessi, A.D.; Castro, L.M.; Giorgi, R.; Rioli, V.; et al. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 20588–20593. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Mancini, G.; Lutz, B.; Luckman, S.M. The peptide hemopressin acts through CB1 cannabinoid receptors to reduce food intake in rats and mice. J. Neurosci. 2010, 30, 7369–7376. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Grushko, J.S.; Golebiewska, U.; Hoogendoorn, S.; Gupta, A.; Heimann, A.S.; Ferro, E.S.; Scarlata, S.; Fricker, L.D.; Devi, L.A. Novel endogenous peptide agonists of cannabinoid receptors. FASEB J. 2009, 23, 3020–3029. [Google Scholar] [CrossRef]
- Gomes, I.; Dale, C.S.; Casten, K.; Geigner, M.A.; Gozzo, F.C.; Ferro, E.S.; Heimann, A.S.; Devi, L.A. Hemoglobin-derived peptides as novel type of bioactive signaling molecules. AAPS J. 2010, 12, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Chicca, A.; Tamborrini, M.; Eisen, D.; Lerner, R.; Lutz, B.; Poetz, O.; Pluschke, G.; Gertsch, J. Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J. Biol. Chem. 2012, 287, 36944–36967. [Google Scholar] [CrossRef]
- Hofer, S.C.; Ralvenius, W.T.; Gachet, M.S.; Fritschy, J.M.; Zeilhofer, H.U.; Gertsch, J. Localization and production of peptide endocannabinoids in the rodent CNS and adrenal medulla. Neuropharmacology 2015, 98, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Xapelli, S.; Agasse, F.; Grade, S.; Bernardino, L.; Ribeiro, F.F.; Schitine, C.S.; Heimann, A.S.; Ferro, E.S.; Sebastiao, A.M.; De Melo Reis, R.A.; et al. Modulation of subventricular zone oligodendrogenesis: A role for hemopressin? Front. Cell. Neurosci. 2014, 8, 59. [Google Scholar] [CrossRef]
- Khilnani, G.; Khilnani, A.K. Inverse agonism and its therapeutic significance. Indian J. Pharmacol. 2011, 43, 492–501. [Google Scholar] [CrossRef]
- Bomar, M.G.; Samuelsson, S.J.; Kibler, P.; Kodukula, K.; Galande, A.K. Hemopressin forms self-assembled fibrillar nanostructures under physiologically relevant conditions. Biomacromolecules 2012, 13, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Dale, C.S.; Pagano Rde, L.; Rioli, V.; Hyslop, S.; Giorgi, R.; Ferro, E.S. Antinociceptive action of hemopressin in experimental hyperalgesia. Peptides 2005, 26, 431–436. [Google Scholar] [CrossRef]
- Blais, P.A.; Cote, J.; Morin, J.; Larouche, A.; Gendron, G.; Fortier, A.; Regoli, D.; Neugebauer, W.; Gobeil, F., Jr. Hypotensive effects of hemopressin and bradykinin in rabbits, rats and mice. A comparative study. Peptides 2005, 26, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Lippton, H.; Lin, B.; Gumusel, B.; Witriol, N.; Wasserman, A.; Knight, M. Hemopressin, a hemoglobin fragment, dilates the rat systemic vascular bed through release of nitric oxide. Peptides 2006, 27, 2284–2288. [Google Scholar] [CrossRef] [PubMed]
- Gelman, J.S.; Fricker, L.D. Hemopressin and other bioactive peptides from cytosolic proteins: Are these non-classical neuropeptides? AAPS J. 2010, 12, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Scrima, M.; Di Marino, S.; Grimaldi, M.; Mastrogiacomo, A.; Novellino, E.; Bifulco, M.; D’Ursi, A.M. Binding of the hemopressin peptide to the cannabinoid CB1 receptor: Structural insights. Biochemistry 2010, 49, 10449–10457. [Google Scholar] [CrossRef]
- Horvath, G.; Mecs, L. Antinociception by endogenous ligands at peripheral level. Ideggyogy Sz 2011, 64, 193–207. [Google Scholar] [PubMed]
- Hama, A.; Sagen, J. Activation of spinal and supraspinal cannabinoid-1 receptors leads to antinociception in a rat model of neuropathic spinal cord injury pain. Brain Res. 2011, 1412, 44–54. [Google Scholar] [CrossRef]
- Hama, A.; Sagen, J. Centrally mediated antinociceptive effects of cannabinoid receptor ligands in rat models of nociception. Pharmacol. Biochem. Behav. 2011, 100, 340–346. [Google Scholar] [CrossRef]
- Petrovszki, Z.; Kovacs, G.; Tomboly, C.; Benedek, G.; Horvath, G. The effects of peptide and lipid endocannabinoids on arthritic pain at the spinal level. Anest. Anal.g 2012, 114, 1346–1352. [Google Scholar] [CrossRef]
- Bomar, M.G.; Galande, A.K. Modulation of the cannabinoid receptors by hemopressin peptides. Life Sci. 2013, 92, 520–524. [Google Scholar] [CrossRef]
- Zhou, L.; Jin, Q.; Yang, Y.; Liu, Z.; Li, X.; Dong, S.; Zhao, L. Effects of endokinin A/B and endokinin C/D on the antinociception properties of hemopressin in mice. Peptides 2012, 38, 70–80. [Google Scholar] [CrossRef]
- Reddy, P.A.; Jones, S.T.; Lewin, A.H.; Carroll, F.I. Synthesis of hemopressin peptides by classical solution phase fragment condensation. Int. J. Pept. 2012, 2012, 186034. [Google Scholar] [CrossRef]
- Gelman, J.S.; Dasgupta, S.; Berezniuk, I.; Fricker, L.D. Analysis of peptides secreted from cultured mouse brain tissue. Biochim. Biophys. Acta 2013, 1834, 2408–2417. [Google Scholar] [CrossRef]
- Dodd, G.T.; Worth, A.A.; Hodkinson, D.J.; Srivastava, R.K.; Lutz, B.; Williams, S.R.; Luckman, S.M. Central functional response to the novel peptide cannabinoid, hemopressin. Neuropharmacology 2013, 71, 27–36. [Google Scholar] [CrossRef]
- Tanaka, K.; Shimizu, T.; Yanagita, T.; Nemoto, T.; Nakamura, K.; Taniuchi, K.; Dimitriadis, F.; Yokotani, K.; Saito, M. Brain RVD-haemopressin, a haemoglobin-derived peptide, inhibits bombesin-induced central activation of adrenomedullary outflow in the rat. Br. J. Pharmacol. 2014, 171, 202–213. [Google Scholar] [CrossRef]
- Rashid, M.; Wangler, N.J.; Yang, L.; Shah, K.; Arumugam, T.V.; Abbruscato, T.J.; Karamyan, V.T. Functional up-regulation of endopeptidase neurolysin during post-acute and early recovery phases of experimental stroke in mouse brain. J. Neurochem. 2014, 129, 179–189. [Google Scholar] [CrossRef]
- Han, Z.L.; Fang, Q.; Wang, Z.L.; Li, X.H.; Li, N.; Chang, X.M.; Pan, J.X.; Tang, H.Z.; Wang, R. Antinociceptive effects of central administration of the endogenous cannabinoid receptor type 1 agonist VDPVNFKLLSH-OH [(m)VD-hemopressin(alpha)], an N-terminally extended hemopressin peptide. J. Pharmacol. Exp. Ther. 2014, 348, 316–323. [Google Scholar] [CrossRef]
- Li, X.H.; Li, N.; Wang, Z.L.; Pan, J.X.; Han, Z.L.; Chang, X.M.; Tang, H.H.; Wang, P.; Wang, R.; Fang, Q. The hypotensive effect of intrathecally injected (m)VD-hemopressin(alpha) in urethane-anesthetized rats. Peptides 2014, 56, 45–51. [Google Scholar] [CrossRef]
- Toniolo, E.F.; Maique, E.T.; Ferreira, W.A., Jr.; Heimann, A.S.; Ferro, E.S.; Ramos-Ortolaza, D.L.; Miller, L.; Devi, L.A.; Dale, C.S. Hemopressin, an inverse agonist of cannabinoid receptors, inhibits neuropathic pain in rats. Peptides 2014, 56, 125–131. [Google Scholar] [CrossRef]
- Pan, J.X.; Wang, Z.L.; Li, N.; Han, Z.L.; Li, X.H.; Tang, H.H.; Wang, P.; Zheng, T.; Fang, Q.; Wang, R. Analgesic tolerance and cross-tolerance to the cannabinoid receptors ligands hemopressin, VD-hemopressin(alpha) and WIN55,212-2 at the supraspinal level in mice. Neurosci. Lett. 2014, 578, 187–191. [Google Scholar] [CrossRef]
- Mahmoud, M.F.; Swefy, S.E.; Hasan, R.A.; Ibrahim, A. Role of cannabinoid receptors in hepatic fibrosis and apoptosis associated with bile duct ligation in rats. Eur. J. Pharmacol. 2014, 742, 118–124. [Google Scholar] [CrossRef]
- Fogaca, M.V.; Sonego, A.B.; Rioli, V.; Gozzo, F.C.; Dale, C.S.; Ferro, E.S.; Guimaraes, F.S. Anxiogenic-like effects induced by hemopressin in rats. Pharmacol. Biochem. Behav. 2015, 129, 7–13. [Google Scholar] [CrossRef]
- Song, B.; Kibler, P.D.; Endsley, A.N.; Nayak, S.K.; Galande, A.K.; Jambunathan, K. Site-specific Substitutions Eliminate Aggregation Properties of Hemopressin. Chem. Biol. Drug Des. 2015, 86, 1433–1437. [Google Scholar] [CrossRef]
- Straiker, A.; Mitjavila, J.; Yin, D.; Gibson, A.; Mackie, K. Aiming for allosterism: Evaluation of allosteric modulators of CB1 in a neuronal model. Pharmacol. Res. 2015, 99, 370–376. [Google Scholar] [CrossRef]
- Ma, L.; Jia, J.; Niu, W.; Jiang, T.; Zhai, Q.; Yang, L.; Bai, F.; Wang, Q.; Xiong, L. Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions. Sci. Rep. 2015, 5, 12440. [Google Scholar] [CrossRef]
- Szlavicz, E.; Perera, P.S.; Tomboly, C.; Helyes, Z.; Zador, F.; Benyhe, S.; Borsodi, A.; Bojnik, E. Further Characterization of Hemopressin Peptide Fragments in the Opioid and Cannabinoid Systems. Anest. Analg. 2015, 121, 1488–1494. [Google Scholar] [CrossRef]
- Zhang, L.; Kolaj, M.; Renaud, L.P. Intracellular postsynaptic cannabinoid receptors link thyrotropin-releasing hormone receptors to TRPC-like channels in thalamic paraventricular nucleus neurons. Neuroscience 2015, 311, 81–91. [Google Scholar] [CrossRef]
- Pan, J.X.; Wang, Z.L.; Li, N.; Zhang, N.; Wang, P.; Tang, H.H.; Zhang, T.; Yu, H.P.; Zhang, R.; Zheng, T.; et al. Effects of neuropeptide FF and related peptides on the antinociceptive activities of VD-hemopressin(alpha) in naive and cannabinoid-tolerant mice. Eur. J. Pharmacol. 2015, 767, 119–125. [Google Scholar] [CrossRef]
- El Swefy, S.; Hasan, R.A.; Ibrahim, A.; Mahmoud, M.F. Curcumin and hemopressin treatment attenuates cholestasis-induced liver fibrosis in rats: Role of CB1 receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 389, 103–116. [Google Scholar] [CrossRef]
- Cunha, F.M.; Berti, D.A.; Ferreira, Z.S.; Klitzke, C.F.; Markus, R.P.; Ferro, E.S. Intracellular peptides as natural regulators of cell signaling. J. Biol. Chem. 2008, 283, 24448–24459. [Google Scholar] [CrossRef]
- Russo, L.C.; Asega, A.F.; Castro, L.M.; Negraes, P.D.; Cruz, L.; Gozzo, F.C.; Ulrich, H.; Camargo, A.C.; Rioli, V.; Ferro, E.S. Natural intracellular peptides can modulate the interactions of mouse brain proteins and thimet oligopeptidase with 14-3-3epsilon and calmodulin. Proteomics 2012, 12, 2641–2655. [Google Scholar] [CrossRef]
- Berti, D.A.; Russo, L.C.; Castro, L.M.; Cruz, L.; Gozzo, F.C.; Heimann, J.C.; Lima, F.B.; Oliveira, A.C.; Andreotti, S.; Prada, P.O.; et al. Identification of intracellular peptides in rat adipose tissue: Insights into insulin resistance. Proteomics 2012, 12, 2668–2681. [Google Scholar] [CrossRef]
- Fricker, L.D.; Gelman, J.S.; Castro, L.M.; Gozzo, F.C.; Ferro, E.S. Peptidomic analysis of HEK293T cells: Effect of the proteasome inhibitor epoxomicin on intracellular peptides. J. Proteom. Res. 2012, 11, 1981–1990. [Google Scholar] [CrossRef]
- Ribeiro, N.M.; Toniolo, E.F.; Castro, L.M.; Russo, L.C.; Rioli, V.; Ferro, E.S.; Dale, C.S. AGH is a new hemoglobin alpha-chain fragment with antinociceptive activity. Peptides 2013, 48, 10–20. [Google Scholar] [CrossRef]
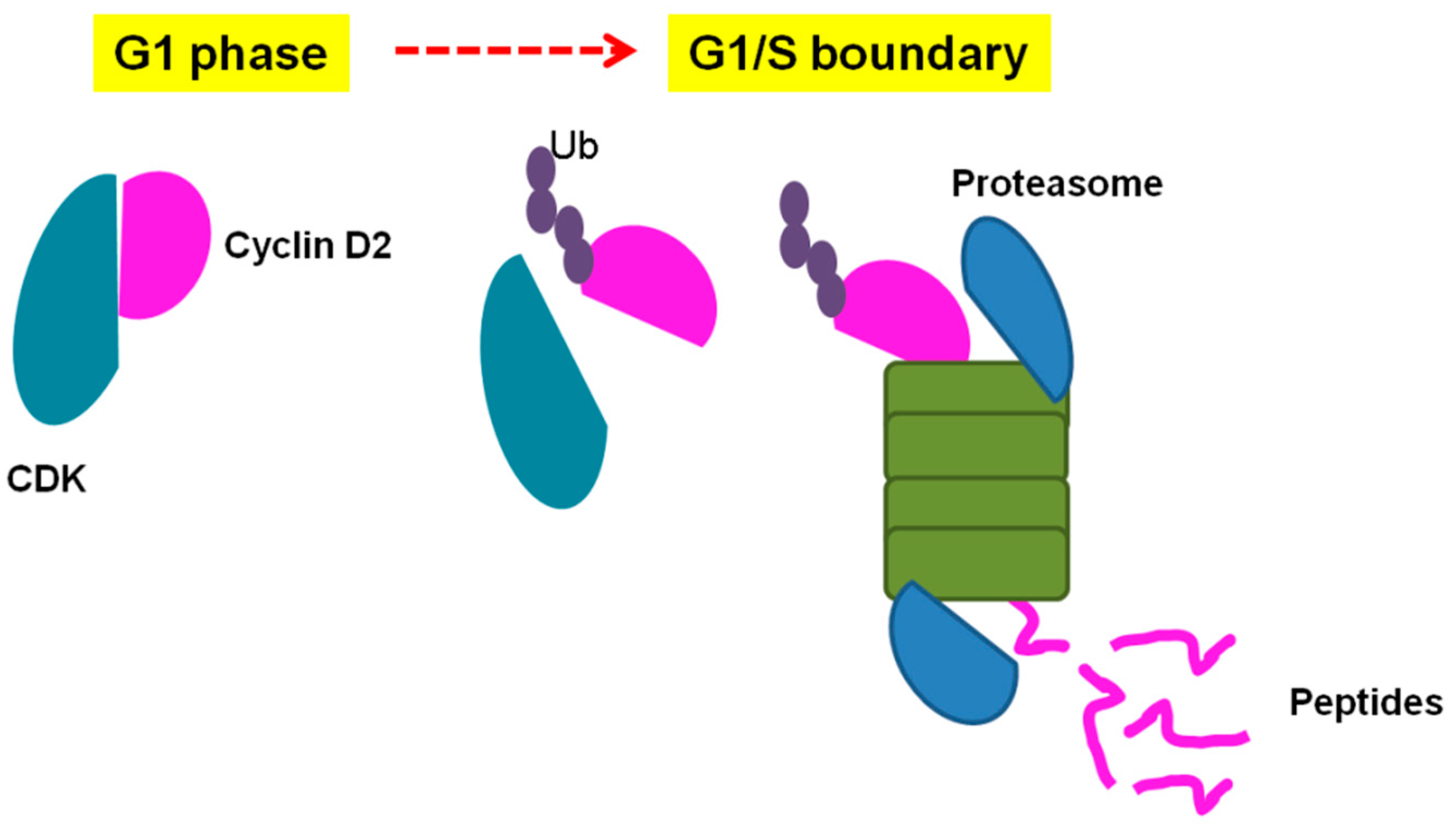
- de Araujo, C.B.; Russo, L.C.; Castro, L.M.; Forti, F.L.; do Monte, E.R.; Rioli, V.; Gozzo, F.C.; Colquhoun, A.; Ferro, E.S. A novel intracellular peptide derived from g1/s cyclin d2 induces cell death. J. Biol. Chem. 2014, 289, 16711–16726. [Google Scholar] [CrossRef]
- Russo, L.C.; Araujo, C.B.; Iwai, L.K.; Ferro, E.S.; Forti, F.L. A Cyclin D2-derived peptide acts on specific cell cycle phases by activating ERK1/2 to cause the death of breast cancer cells. J. Proteom. 2016, 151, 24–32. [Google Scholar] [CrossRef]
- de Araujo, C.B.; de Lima, L.P.; Calderano, S.G.; Damasceno, F.S.; Silber, A.M.; Elias, M.C. Pep5, a fragment of cyclin D2, shows antiparasitic effects in different stages of the Trypanosoma cruzi life cycle and blocks parasite infectivity. Antimicrob. Agents Chemother. 2019. [Google Scholar] [CrossRef]
- Monte-Silva ERC, R.C.; Russo, L.C.; Castro, L.M.; Gozzo, F.C.; de Araujo, C.B.; Peron, J.P.S.; Rioli, V.; Ferro, E.S. EL28 is a novel intracellular peptide that activates immune proteasome and CD8+ T-cell response. J. Proteom. 2016, 16, S1874–3919. [Google Scholar]
- Gelman, J.S.; Sironi, J.; Berezniuk, I.; Dasgupta, S.; Castro, L.M.; Gozzo, F.C.; Ferro, E.S.; Fricker, L.D. Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib. PLoS ONE 2013, 8, e53263. [Google Scholar] [CrossRef]
- Dasgupta, S.; Fishman, M.A.; Mahallati, H.; Castro, L.M.; Tashima, A.K.; Ferro, E.S.; Fricker, L.D. Reduced Levels of Proteasome Products in a Mouse Striatal Cell Model of Huntington’s Disease. PLoS ONE 2015, 10, e0145333. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.V.; Karpov, V.L. Biological consequences of structural and functional proteasome diversity. Heliyon 2018, 4, e00894. [Google Scholar] [CrossRef]
- Russo, L.C.; Castro, L.M.; Gozzo, F.C.; Ferro, E.S. Inhibition of thimet oligopeptidase by siRNA alters specific intracellular peptides and potentiates isoproterenol signal transduction. FEBS Lett 2012, 586, 3287–3292. [Google Scholar] [CrossRef]
- Cavalcanti, D.M.; Castro, L.M.; Rosa Neto, J.C.; Seelaender, M.; Neves, R.X.; Oliveira, V.; Forti, F.L.; Iwai, L.K.; Gozzo, F.C.; Todiras, M.; et al. Neurolysin knockout mice generation and initial phenotype characterization. J. Biol. Chem. 2014, 289, 15426–15440. [Google Scholar] [CrossRef]
- Castro, L.M.; Cavalcanti, D.M.; Araujo, C.B.; Rioli, V.; Icimoto, M.Y.; Gozzo, F.C.; Juliano, M.; Juliano, L.; Oliveira, V.; Ferro, E.S. Peptidomic analysis of the neurolysin-knockout mouse brain. J. Proteom. 2014, 111, 238–248. [Google Scholar] [CrossRef]
- Ramachandran, K.V.; Margolis, S.S. A mammalian nervous-system-specific plasma membrane proteasome complex that modulates neuronal function. Nat. Struct. Mol. Biol. 2017, 24, 419–430. [Google Scholar] [CrossRef]
- Mechoulam, R.; Hanus, L.O.; Pertwee, R.; Howlett, A.C. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 2014, 15, 757–764. [Google Scholar] [CrossRef]
- Wang, P.; Zheng, T.; Zhang, M.; Xu, B.; Zhang, R.; Zhang, T.; Zhao, W.; Shi, X.; Zhang, Q.; Fang, Q. Antinociceptive effects of the endogenous cannabinoid peptide agonist VD-hemopressin(beta) in mice. Brain Res. Bull. 2018, 139, 48–55. [Google Scholar] [CrossRef]
- Zheng, T.; Zhang, R.; Zhang, T.; Zhang, M.N.; Xu, B.; Song, J.J.; Li, N.; Tang, H.H.; Wang, P.; Wang, R.; et al. CB1 cannabinoid receptor agonist mouse VD-hemopressin(alpha) produced supraspinal analgesic activity in the preclinical models of pain. Brain Res. 2017, 1680, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Recinella, L.; Chiavaroli, A.; Ferrante, C.; Mollica, A.; Macedonio, G.; Stefanucci, A.; Dimmito, M.P.; Dvoracsko, S.; Tomboly, C.; Brunetti, L.; et al. Effects of central RVD-hemopressin(alpha) administration on anxiety, feeding behavior and hypothalamic neuromodulators in the rat. Pharmacol. Rep. 2018, 70, 650–657. [Google Scholar] [CrossRef]
- Leone, S.; Recinella, L.; Chiavaroli, A.; Martinotti, S.; Ferrante, C.; Mollica, A.; Macedonio, G.; Stefanucci, A.; Dvoracsko, S.; Tomboly, C.; et al. Emotional disorders induced by Hemopressin and RVD-hemopressin(alpha) administration in rats. Pharmacol. Rep. 2017, 69, 1247–1253. [Google Scholar] [CrossRef]
- Mechoulam, R. Interview with Prof. Raphael Mechoulam, codiscoverer of THC. Einstein. Int. J. Addict. 1986, 21, 579–587. [Google Scholar] [CrossRef]
- Mechoulam, R. [Endocannabinoids and psychiatric disorders: The road ahead]. Braz J. Psychiatr. 2010, 32 (Suppl. 1), S5–S6. [Google Scholar]
- Mechoulam, R.; Shani, A.; Edery, H.; Grunfeld, Y. Chemical basis of hashish activity. Science 1970, 169, 611–612. [Google Scholar] [CrossRef]
- Blair, R.E.; Deshpande, L.S.; Sombati, S.; Falenski, K.W.; Martin, B.R.; DeLorenzo, R.J. Activation of the cannabinoid type-1 receptor mediates the anticonvulsant properties of cannabinoids in the hippocampal neuronal culture models of acquired epilepsy and status epilepticus. J. Pharmacol. Exp. Ther. 2006, 317, 1072–1078. [Google Scholar] [CrossRef]
- Jones, N.A.; Hill, A.J.; Smith, I.; Bevan, S.A.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J. Pharmacol. Exp. Ther. 2010, 332, 569–577. [Google Scholar] [CrossRef]
- Shafaroodi, H.; Samini, M.; Moezi, L.; Homayoun, H.; Sadeghipour, H.; Tavakoli, S.; Hajrasouliha, A.R.; Dehpour, A.R. The interaction of cannabinoids and opioids on pentylenetetrazole-induced seizure threshold in mice. Neuropharmacology 2004, 47, 390–400. [Google Scholar] [CrossRef]
- Wallace, M.J.; Martin, B.R.; DeLorenzo, R.J. Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. Eur. J. Pharmacol. 2002, 452, 295–301. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef]
- Hildebrandt, A.K.; Dietzen, M.; Lengauer, T.; Lenhof, H.P.; Althaus, E.; Hildebrandt, A. Efficient computation of root mean square deviations under rigid transformations. J. Comput. Chem. 2014, 35, 765–771. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef]
- Turski, W.A.; Cavalheiro, E.A.; Schwarz, M.; Czuczwar, S.J.; Kleinrok, Z.; Turski, L. Limbic seizures produced by pilocarpine in rats: Behavioural, electroencephalographic and neuropathological study. Behav. Brain Res. 1983, 9, 315–335. [Google Scholar] [CrossRef]
- Rioli, V.; Kato, A.; Portaro, F.C.; Cury, G.K.; te Kaat, K.; Vincent, B.; Checler, F.; Camargo, A.C.; Glucksman, M.J.; Roberts, J.L.; et al. Neuropeptide specificity and inhibition of recombinant isoforms of the endopeptidase 3.4.24.16 family: Comparison with the related recombinant endopeptidase 3.4.24.15. Biochem Biophys. Res. Commun. 1998, 250, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Heimann, A.S.; Gupta, A.; Gomes, I.; Rayees, R.; Schlessinger, A.; Ferro, E.S.; Unterwald, E.M.; Devi, L.A. Generation of G protein-coupled receptor antibodies differentially sensitive to conformational states. PLoS ONE 2018, 12, e0187306. [Google Scholar] [CrossRef]
- Gupta, A.; Decaillot, F.M.; Gomes, I.; Tkalych, O.; Heimann, A.S.; Ferro, E.S.; Devi, L.A. Conformation state-sensitive antibodies to G-protein-coupled receptors. J. Biol. Chem. 2007, 282, 5116–5124. [Google Scholar] [CrossRef]
- Reckziegel, P.; Festuccia, W.T.; Britto, L.R.G.; Jang, K.L.L.; Romão, C.M.; Heimann, J.C.; Fogaça, M.V.; Rodrigues, N.S.; Silva, N.R.; Guimarães, F.S.; et al. A novel peptide that improves metabolic parameters without adverse central nervous system effects. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Groll, M.; Ditzel, L.; Lowe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Reits, E.; Griekspoor, A.; Neijssen, J.; Groothuis, T.; Jalink, K.; van Veelen, P.; Janssen, H.; Calafat, J.; Drijfhout, J.W.; Neefjes, J. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity 2003, 18, 97–108. [Google Scholar] [CrossRef]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell. Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef]
- Kohler, A.; Bajorek, M.; Groll, M.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. The substrate translocation channel of the proteasome. Biochimie 2001, 83, 325–332. [Google Scholar] [CrossRef]
- Tian, G.; Park, S.; Lee, M.J.; Huck, B.; McAllister, F.; Hill, C.P.; Gygi, S.P.; Finley, D. An asymmetric interface between the regulatory and core particles of the proteasome. Nat. Struct. Mol. Biol. 2011, 18, 1259–1267. [Google Scholar] [CrossRef]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef]
- Sanders, A.R.; Goring, H.H.; Duan, J.; Drigalenko, E.I.; Moy, W.; Freda, J.; He, D.; Shi, J.; Gejman, P.V. Transcriptome study of differential expression in schizophrenia. Hum. Mol. Genet. 2013, 22, 5001–5014. [Google Scholar] [CrossRef]
- Fierz, B.; Chatterjee, C.; McGinty, R.K.; Bar-Dagan, M.; Raleigh, D.P.; Muir, T.W. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 2011, 7, 113–119. [Google Scholar] [CrossRef]
- Norbury, C.; Nurse, P. Animal cell cycles and their control. Annu. Rev. Biochem. 1992, 61, 441–470. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 2005, 30, 630–641. [Google Scholar] [CrossRef]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef]
- Hunter, T.; Pines, J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 1994, 79, 573–582. [Google Scholar] [CrossRef]
- Sherr, C.J. Mammalian G1 cyclins. Cell 1993, 73, 1059–1065. [Google Scholar] [CrossRef]
- Waclaw, R.R.; Chatot, C.L. Patterns of expression of cyclins A, B1, D, E and cdk 2 in preimplantation mouse embryos. Zygote 2004, 12, 19–30. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. (Berl) 2016, 94, 1313–1326. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Vodermaier, H.C. APC/C and SCF: Controlling each other and the cell cycle. Curr. Biol. 2004, 14, R787–R796. [Google Scholar] [CrossRef]
- Harper, J.W.; Burton, J.L.; Solomon, M.J. The anaphase-promoting complex: it’s not just for mitosis any more. Genes Dev. 2002, 16, 2179–2206. [Google Scholar] [CrossRef]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef]
- Pagano, M.; Theodoras, A.M.; Tam, S.W.; Draetta, G.F. Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev. 1994, 8, 1627–1639. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Caserta, T.M.; Smith, A.N.; Gultice, A.D.; Reedy, M.A.; Brown, T.L. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 2003, 8, 345–352. [Google Scholar] [CrossRef]
- Sodeoka, M.; Dodo, K. Development of selective inhibitors of necrosis. Chem. Rec. 2010, 10, 308–314. [Google Scholar] [CrossRef]
- Fernandez-Salas, E.; Suh, K.S.; Speransky, V.V.; Bowers, W.L.; Levy, J.M.; Adams, T.; Pathak, K.R.; Edwards, L.E.; Hayes, D.D.; Cheng, C.; et al. mtCLIC/CLIC4, an organellular chloride channel protein, is increased by DNA damage and participates in the apoptotic response to p53. Mol. Cell. Biol. 2002, 22, 3610–3620. [Google Scholar] [CrossRef]
- Valenzuela, S.M.; Mazzanti, M.; Tonini, R.; Qiu, M.R.; Warton, K.; Musgrove, E.A.; Campbell, T.J.; Breit, S.N. The nuclear chloride ion channel NCC27 is involved in regulation of the cell cycle. J. Physiol. 2000, 529 Pt 3, 541–552. [Google Scholar] [CrossRef]
- Argenzio, E.; Moolenaar, W.H. Emerging biological roles of Cl- intracellular channel proteins. J. Cell Sci. 2016, 129, 4165–4174. [Google Scholar] [CrossRef]
- Wang, P.; Zeng, Y.; Liu, T.; Zhang, C.; Yu, P.W.; Hao, Y.X.; Luo, H.X.; Liu, G. Chloride intracellular channel 1 regulates colon cancer cell migration and invasion through ROS/ERK pathway. World J. Gastroenterol. 2014, 20, 2071–2078. [Google Scholar] [CrossRef]
- Tian, Y.; Guan, Y.; Jia, Y.; Meng, Q.; Yang, J. Chloride intracellular channel 1 regulates prostate cancer cell proliferation and migration through the MAPK/ERK pathway. Cancer Biother. Radiopharm. 2014, 29, 339–344. [Google Scholar] [CrossRef]
- Manso, J.A.; Garcia Rubio, I.; Gomez-Hernandez, M.; Ortega, E.; Buey, R.M.; Carballido, A.M.; Carabias, A.; Alonso-Garcia, N.; de Pereda, J.M. Purification and Structural Analysis of Plectin and BPAG1e. Methods Enzymol. 2016, 569, 177–196. [Google Scholar]
- Kazerounian, S.; Uitto, J.; Aho, S. Unique role for the periplakin tail in intermediate filament association: Specific binding to keratin 8 and vimentin. Exp. Dermatol. 2002, 11, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Bausch, D.; Mino-Kenudson, M.; Fernandez-Del Castillo, C.; Warshaw, A.L.; Kelly, K.A.; Thayer, S.P. Plectin-1 is a biomarker of malignant pancreatic intraductal papillary mucinous neoplasms. J. Gastrointest. Surg. 2009, 13, 1948–1954. [Google Scholar] [CrossRef]
- Bausch, D.; Thomas, S.; Mino-Kenudson, M.; Fernandez-del, C.C.; Bauer, T.W.; Williams, M.; Warshaw, A.L.; Thayer, S.P.; Kelly, K.A. Plectin-1 as a novel biomarker for pancreatic cancer. Clin. Cancer Res. 2011, 17, 302–309. [Google Scholar] [CrossRef]
- Katada, K.; Tomonaga, T.; Satoh, M.; Matsushita, K.; Tonoike, Y.; Kodera, Y.; Hanazawa, T.; Nomura, F.; Okamoto, Y. Plectin promotes migration and invasion of cancer cells and is a novel prognostic marker for head and neck squamous cell carcinoma. J. Proteom. 2012, 75, 1803–1815. [Google Scholar] [CrossRef]
- Pawar, H.; Kashyap, M.K.; Sahasrabuddhe, N.A.; Renuse, S.; Harsha, H.C.; Kumar, P.; Sharma, J.; Kandasamy, K.; Marimuthu, A.; Nair, B.; et al. Quantitative tissue proteomics of esophageal squamous cell carcinoma for novel biomarker discovery. Cancer Biol. Ther. 2011, 12, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Vallelian, F.; Deuel, J.W.; Opitz, L.; Schaer, C.A.; Puglia, M.; Lonn, M.; Engelsberger, W.; Schauer, S.; Karnaukhova, E.; Spahn, D.R.; et al. Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ. 2015, 22, 597–611. [Google Scholar] [CrossRef]
- Fortes, G.B.; Alves, L.S.; de Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; De Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Amino Acid Sequence | Protein Precursor | Pharmacological Activity | Level of Evidence | Reference(s) |
|---|---|---|---|---|---|
| Hemopressin | PVNFKFLSH | Hemoglobin alpha-chain | First intracellular peptide identified using the substrate-capture assay. Has hypotensive action in anesthetized rats if administered intravenously or intra arterially. Was found to bind CB1R receptor as an inverse agonist and to have oral activity in rats and mice with antinociceptive action in hyperalgesia models. Also, orally administrated is capable to reduce appetite in experimental rat and mouse models. It has potent activity inducing myelination. | Bind CB1R receptor as inverse agonist (EC50 = 0.35 nM); increases adenylyl cyclase activity in rat striatal membranes. The short hemopressin sequence PVNFKF was shown to have inverse agonist activity on CB1R receptors. | [14,25,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70] |
| VD and RVD-hemopressin | RVDPVNFKFLSH | Hemoglobin alpha-chain | Found as the endogenous hemopressins, have CB1R receptor agonist activity in opposition to inverse agonist activity of hemopressin. Also described as negative allosteric modulator of CB1R receptors. | Found in mouse blood. Increase cannabinoid 1 and 2 receptor-mediated intracellular Ca2+ levels in HEK-293 cells; effect is blocked by SR141716. Induce neurite outgrowth in Neuro 2A cells. Several variants of RVD-hemopressin retain CB1R pharmacological activity. | [35,36,37] |
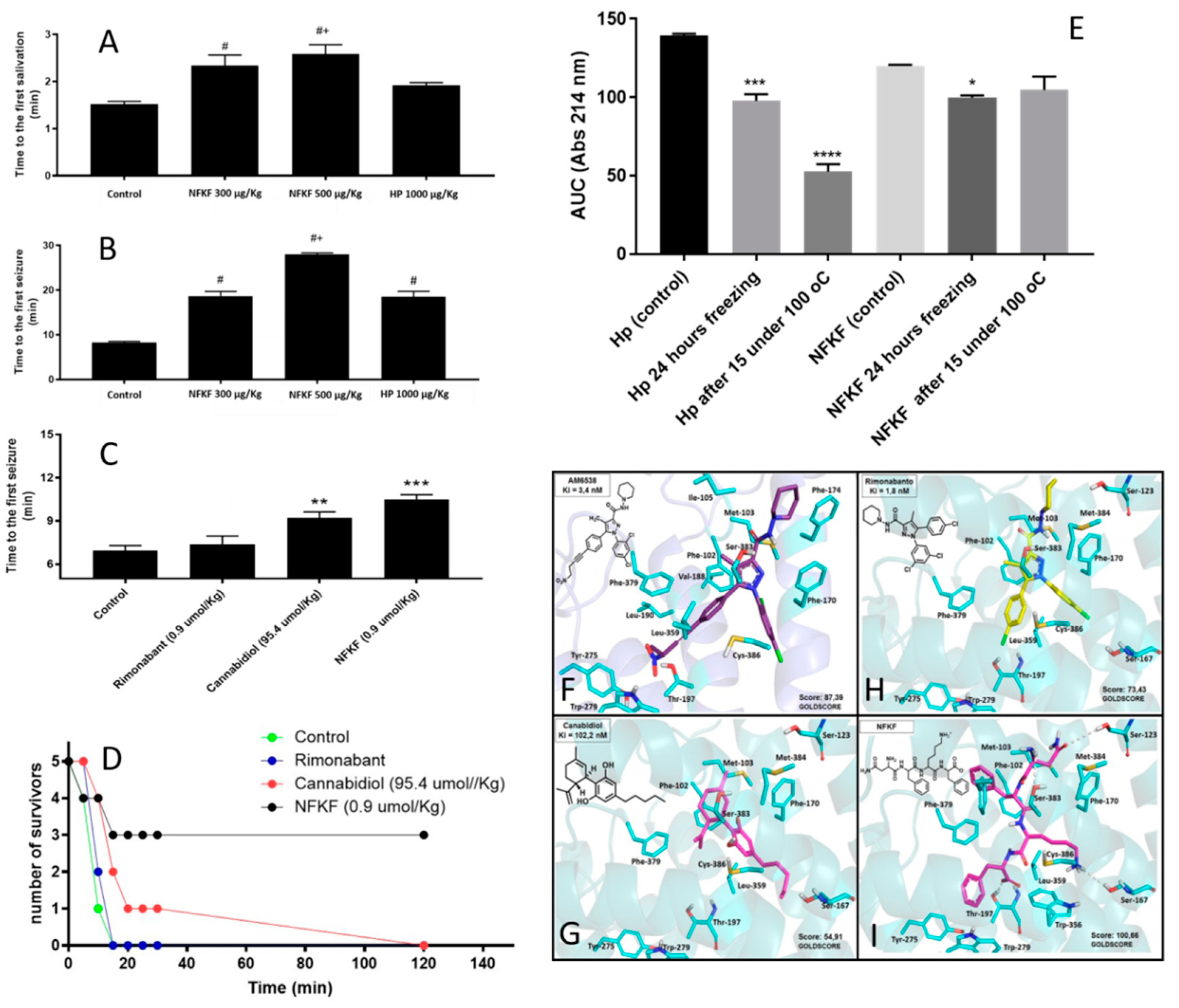
| NFKF | NFKF | Hemoglobin alpha-chain | Hemopressin (PVNFKFLSH; HP) and its smallest CB1R active fragment NFKF are both orally active, and delays symptoms and seizures of pilocarpine-induced seizures in mice. Orally administrated NFKF is 100 times more potent than cannabidiol in delaying the first seizure induced by pilocarpine in mice. Orally administrated NFKF is more efficient in protecting mice from death after pilocarpine-induced seizures. NFKF has the advantage of being more functionally stable than hemopressin after freezing and heating. | Molecular docking suggests that NFKF has a better Goldscore for binding to CB1R than AM6538, cannabidiol, and rimonabant. In vivo assays show that orally administrated NFKF is very efficient in preventing seizures and its symptoms in pilocarpine-induced mice model. NFKF administered orally is a potent cannabinoid for treating epilepsy seizures and has economic advantages over cannabidiol use. In vivo assays show that orally administrated NFKF-derived sequence NFKL has similar properties compared to NFKF, whereas NFK, FKL, NF, FK, KF, or KL shown no pharmacological activity in preventing or altering seizures and its symptoms in pilocarpine-induced mice model (data not shown). | Original data, presented herein. |
| Pep19 | DIIADDEPLT | None (synthetic non-natural peptide) | The original intracellular peptide is derived from peptidyl-prolyl cis–trans isomerase A (DITADDEPLT), and was rationally modified in specific amino acids to generate pep19 (DIIADDEPLT), which, compared to the natural intracellular peptide, shows a better inverse agonist activity binding to CB1R receptors, with a lack of undesired CNS effects. Changes in Pep19 amino acid sequence strongly affect its specificity and CB1R pharmacological properties. Pep19-induced uncoupling-protein 1 expression in both white adipose tissue and 3T3-L1 differentiated adipocytes activates pERK1/2 and AKT signaling pathways. Uncoupling-protein 1 expression induced by Pep19 in 3T3-L1 differentiated adipocytes is blocked by AM251, a CB1R receptor antagonist. | In vivo and in vitro inverse agonist of CB1R receptors; has the pharmacological advantage of not having undesired CNS cannabinoid activity; bind CB1R receptor as inverse agonist (EC50 = 0.49 nM); orally administrated in rats reduces adiposity index and body weight, and improves several metabolic parameters including reduction in the glucose, triacylglycerol, cholesterol, and blood pressure, without altering heart rate in obese rats. | [35,36]; Patent granted in USA (US9796760) and Europe (EP2878306). |
| FE2 | PGANAAAAKIQASFR | Neurogranin | Modulates AT1 and β1/2-adrenergic G-protein coupled receptors signal transduction in CHO and HEK293 cells. The mechanism of action likely involves competition to protein kinase C’s natural substrates, and binding to specific proteins or protein complex including dynamin 1, alpha-adaptin A2, alpha1- and beta2c-tubulin, vesicular fusion protein NSF, Rab GDP dissociation inhibitor, and several 14-3-3 isoforms. | Only if coupled to cell-penetrating peptide through a Cys–Cys bond that dissociate from the intracellular peptide upon internalization in HEK293 and CHO-S cells, this peptide at 80 µM concentration potentiates both angiotensin II and isoproterenol agonist action. The high concentration needed for pharmacological activity is probably due to the high degradation ratio of the free intracellular peptide, after it is released from cell-penetrating peptide into the cytosol. | [71] |
| FE3 | SSGAHGEEGSARIWKA | Cytochrome-c oxidase | Modulates AT1 and β1/2-adrenergic G-protein coupled receptors’ signal transduction in CHO and HEK293 cells. The mechanism of action is not related to competition to protein kinase C, whereas it binds to specific proteins or protein complex including dynamin 1, alpha1- and beta-tubulin, vesicular fusion protein NSF, amphyphisin 1, and alpha-adaptin C. It was observed to increase the interaction of both calmodulin and 14-3-3 epsilon with mice brain proteins. | Only if coupled to cell-penetrating peptide through a Cys–Cys bond that dissociate from the intracellular peptide upon internalization in HEK293 and CHO-S cells, this peptide at 80 µM concentration potentiates both angiotensin II and isoproterenol agonist action. The high concentration needed for pharmacological activity is probably due to the high degradation ratio of the free intracellular peptide, after it is released from cell-penetrating peptide into the cytosol. | [71,72] |
| DBI | TVGDVNTDRPGLLDL | Acyl-CoA-binding protein | Intracellular peptide originally described as an agonist of benzodiazepine receptors and termed “diazepam-binding inhibitor” (DBI); facilitates the transport of glucose stimulated by insulin in 3T3-L1 adipocytes both in regular and insulin-resistant 3T3L1 differentiated adipocytes; binds to heat shock protein 8 only in epididymal adipose tissue extracts obtained from obese rats that were fed a Western diet. | DBI is a competitive inhibitor for the binding of [3H] diazepam to GABA receptors with a Ki of 4 µM concentration. DBI’s relative concentration was found to increase in the epididymal adipose tissue extracted from obese rats that were fed a Western diet, compared to non-obese rats that were fed a control diet. At concentrations of 0.1–1 nM, the peptide potentiated insulin-induced glucose uptake in 3T3-L1 differentiated adipocytes. DBI has no effects in glucose uptake in the absence of insulin or without being cell internalized through transient cell permeabilization with CHAPS 0.1%. | [11,73] |
| LDBI | GDVNTDRPGLLDL | Acyl-CoA-binding protein | LDBI is a shorter version of DBI lacking the two N-terminal amino acids. It was shown to facilitate glucose transport stimulated by insulin in 3T3-L1 adipocytes, both in regular and insulin-resistant 3T3L1 differentiated adipocytes. In addition to heat shock protein 8, LDBI specifically binds to additional proteins only in epididymal adipose tissue extracted from obese rats, including annexin A6, asporin, ATP synthase H+ transporting mitochondrial F1 complex beta polypeptide isoform CRA_a, complement component 4A, protein 1 (HMG-1), and Ig gamma-2A chain C region. | LDBI’s relative concentration was found to increase in the epididymal adipose tissue extracted from obese rats that were fed a Western diet compared to non-obese rats that were fed a control diet. At concentrations of 0.1–1 nM, the peptide potentiated insulin-induced glucose uptake in 3T3-L1 differentiated adipocytes. LDBI has no effects in glucose uptake in the absence of insulin or without being cell internalized through transient cell permeabilization with CHAPS 0.1%. | [73] |
| VFD-7 | VFDVELL | Peptidyl-prolyl cis–trans isomerase | In vitro, using surface plasmon resonance assay, it was found that the peptide inhibits the interaction of calmodulin and 14-3-3 with mice cytoplasmic brain proteins. It strongly inhibits the interaction of recombinant THOP1 with calmodulin at 1 and 10 µM concentrations; however, VFD7 is not able to disrupt this interaction after it is assembled. It stimulates the unconventional secretion of THOP1 at 10 µM concentration. It increases the concentration of Ca2+ in a dose-dependent manner starting at 10 µM concentration. | Intracellular VFD-7 quantification using MS with isotope labeling suggest that in HEK293 cells, its intracellular concentration is 16 ± 3 μM. Treatment of HEK293 cells with either 0.2 µM of epoxomicin or carfilzomib 1µM, for 1 h or 35 min, respectively, reduces more than 5 times the concentration of VFD7 in HEK293 cells, which may suggest its participation on clinical benefits obtained with proteasome inhibitors. | [29,72,74] |
| AGH | AGHLDDLPGALSAL | Hemoglobin alpha-chain | Identified in rat brain homogenates using the substrate-capture assay; inhibits peripheral hyperalgesia response through the activation of opioid receptors. | AGH (10 µ/paw) has peripheral antinociceptive effects on paw carrageenan-induced hyperalgesia in Wistar rats, which was antagonized by naloxone. However, AGH was neither observed to bind opioid receptors nor to have similar opioid analgesic central effects. | [75] |
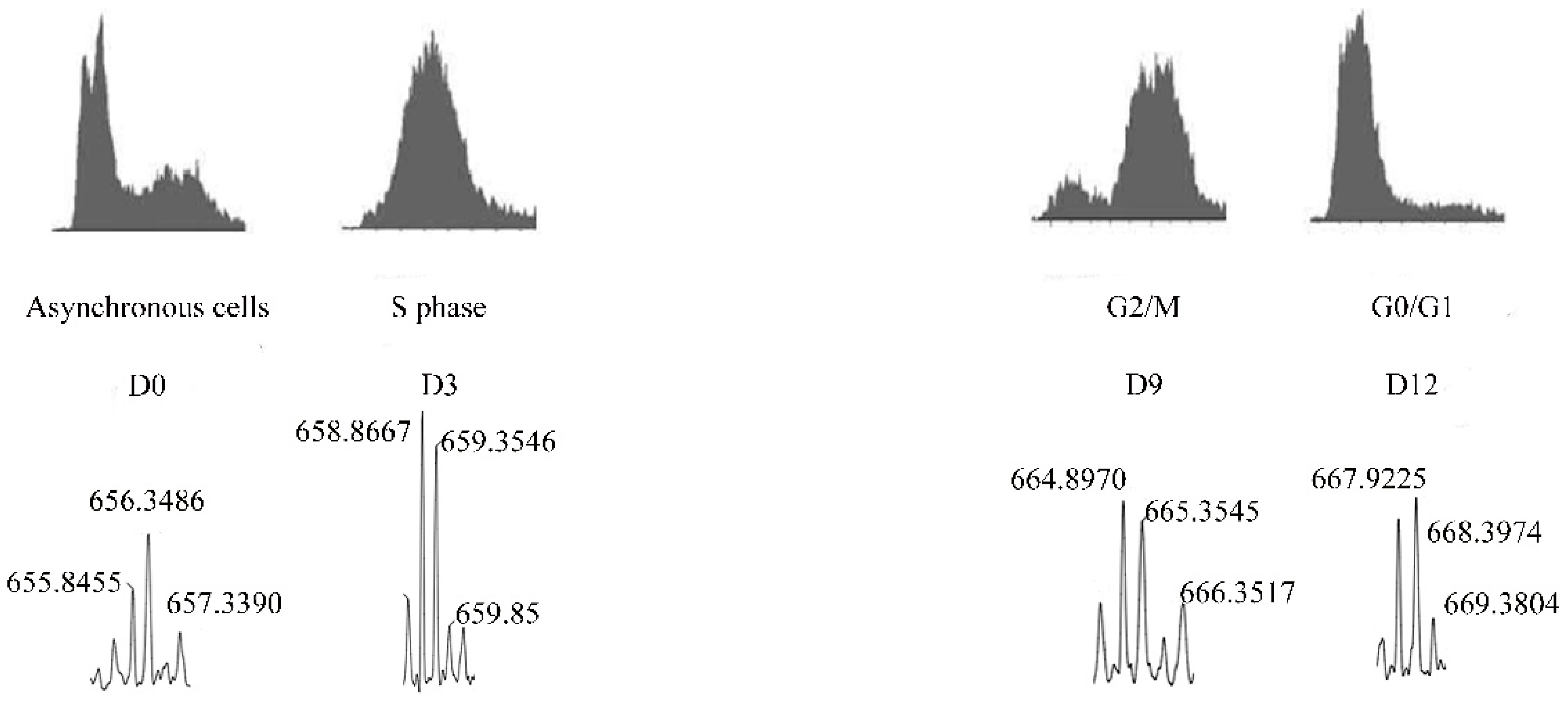
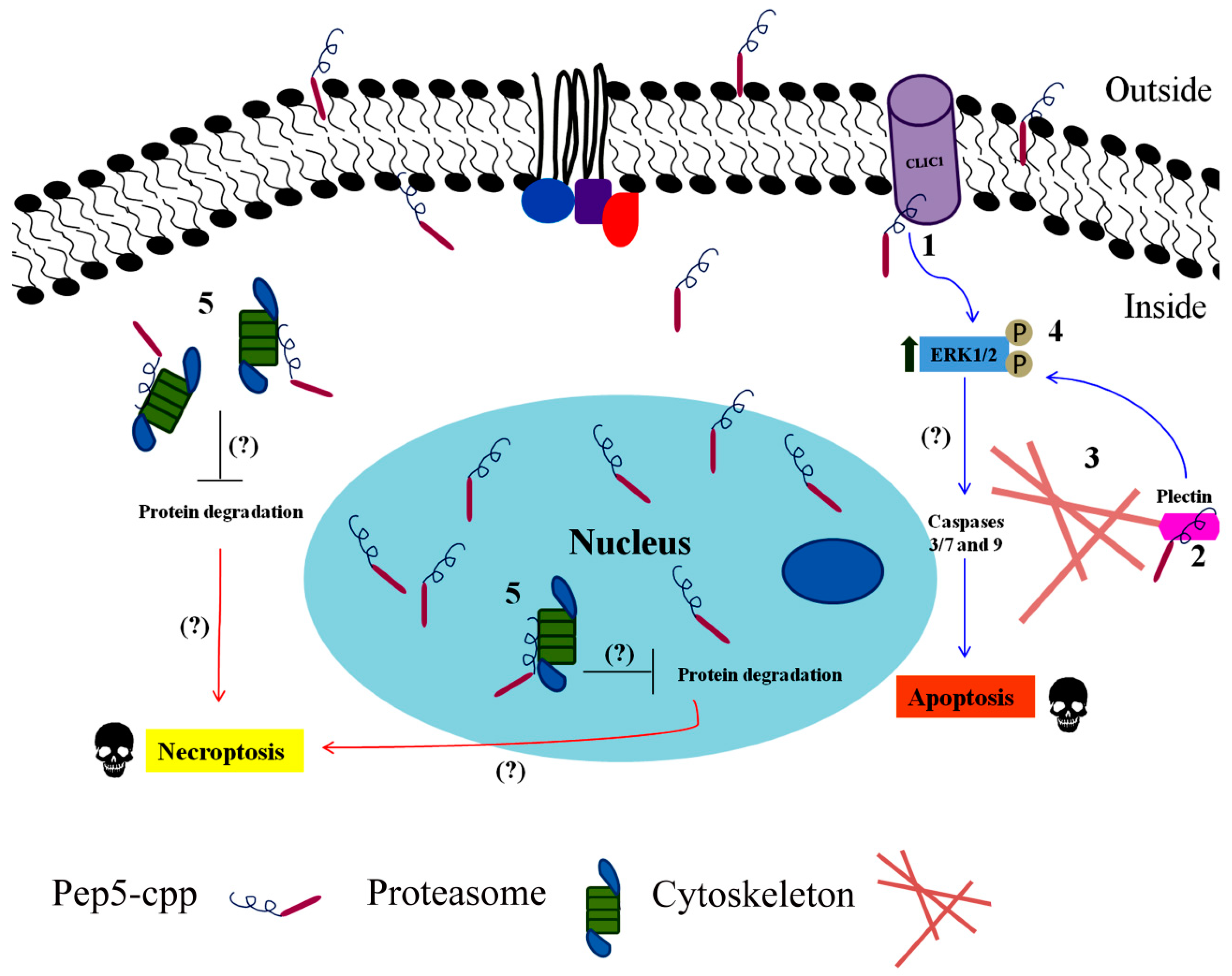
| Pep5 | WELVVLGKL | Cyclin D2 | Identified to increase in G1/S cell cycle of HeLa cells. Only if coupled to a cell-penetrating peptide (Pep5-cpp), the peptide induces cell death in several tumor cells, and in vivo reduces 50% of the size of C6 glioblastoma in rat brain. Pep5-cpp activates caspases 3/7 and 9, inhibits the phosphorylation of Akt2, activates p38α and -γ, and inhibits proteasome activity. N-terminal tryptophan removal as well as Leu to Ala substitutions totally abolishes the cell death activity by Pep5-cpp; the minimal pharmacological active sequence is WELVVL. Pep5-cpp also induces cell death in epimastigotes, trypomastigotes, and amastigotes forms of Trypanosoma cruzi parasites responsible for Chagas disease. At low doses, Pep5-cpp decreases the percentage of infected cells without any detectable toxic effects in mammalian host cells. The infective form of T. cruzi, i.e., trypomastigotes, pre-treated with Pep5-cpp was unable to infect LLC-MK2 cells. | Pep5-cpp (25 µM) cell death was significantly increased when the peptide was added at G1/S or S phases of the cell cycle compared to the effects of the peptide on asynchronous cells. Pep5-cpp treatment caused a major disruption of the stress F-actin fibers’ integrity after only 4 h of treatment at 25 µM. ERK1/2 phosphorylation is increased following pep5-cpp treatment in both asynchronous or synchronized cells; however, if added to cells synchronized in S phase, pep5-cpp induces a significant increase in ERK1/2 phosphorylation that remains high for more than 4 h. In mammalian cells, pep5 binds to different proteins depending of the cell cycle phases; however, at least two proteins, plectin and cytosolic cloride channel (CLIC1), were targeted by pep5 in either asynchronous or synchronized MDA-MB-231 cells. In Tripanossoma cruzi, a different set of specific proteins were identified to bind pep5, including calmodulin-ubiquitin-associated protein, GTPase activating protein, and a putative protein kinase. | [76,77,78] |
| EL28 | VGSELIQKY | Human 19S ATPase regulatory subunit 4 | Peptide identified after its relative concentration increased in HeLa cells following treatment with gamma interferon. Intracellular peptide activator of immune proteasome and proliferation of CD8+. | In vitro, EL28 (50 µM) increased the chymotrypsin, trypsin, and caspase-like proteasome activities. In vivo only when linked to a cell-penetrating peptide, EL28 (100µM) potentiated the ability of interferon-gamma to stimulate the expression of the immunoproteasome β5i subunit, and increase the proliferation of CD8+ T-cells. The EL28-cell-penetrating peptide improved and positively modulated the secondary IgG anti-bovine serum albumin immune responsiveness elicited in high antibody-responder mice. | [79] |
| PepH | SEGTKAVTKYTSSK | Histone H2B | In Neuro2A cells, PepH bound to a cell-penetrating peptide (PepH-cpp, 50 μM) showed a protective effect against cell death. PepH-cpp (10–50 μM) significantly prevented Neuro2A cells death induced by lipopolysaccharide. | Decreased in the anterior temporal lobe of brains of patients with schizophrenia when compared with healthy individuals (postmortem). | [30] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Araujo, C.B.; Heimann, A.S.; Remer, R.A.; Russo, L.C.; Colquhoun, A.; Forti, F.L.; Ferro, E.S. Intracellular Peptides in Cell Biology and Pharmacology. Biomolecules 2019, 9, 150. https://doi.org/10.3390/biom9040150
de Araujo CB, Heimann AS, Remer RA, Russo LC, Colquhoun A, Forti FL, Ferro ES. Intracellular Peptides in Cell Biology and Pharmacology. Biomolecules. 2019; 9(4):150. https://doi.org/10.3390/biom9040150
Chicago/Turabian Stylede Araujo, Christiane B., Andrea S. Heimann, Ricardo A. Remer, Lilian C. Russo, Alison Colquhoun, Fábio L. Forti, and Emer S. Ferro. 2019. "Intracellular Peptides in Cell Biology and Pharmacology" Biomolecules 9, no. 4: 150. https://doi.org/10.3390/biom9040150
APA Stylede Araujo, C. B., Heimann, A. S., Remer, R. A., Russo, L. C., Colquhoun, A., Forti, F. L., & Ferro, E. S. (2019). Intracellular Peptides in Cell Biology and Pharmacology. Biomolecules, 9(4), 150. https://doi.org/10.3390/biom9040150