Saturated Fatty Acid Activates T Cell Inflammation Through a Nicotinamide Nucleotide Transhydrogenase (NNT)-Dependent Mechanism

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Subjects Sample Collection
2.2. Isolation of PBMCs and T Cells
2.3. Immunoblotting
2.4. ROS Quantification
2.5. Mitochondrial Measures
2.6. Mitochondrial Membrane Potential
2.7. Measurement of Cellular Antioxidants
2.8. Measurement of Plasma Free Fatty Acid
2.9. Cytokine Analysis
2.10. Small Interference RNA
2.11. Statistical Analyses
3. Results
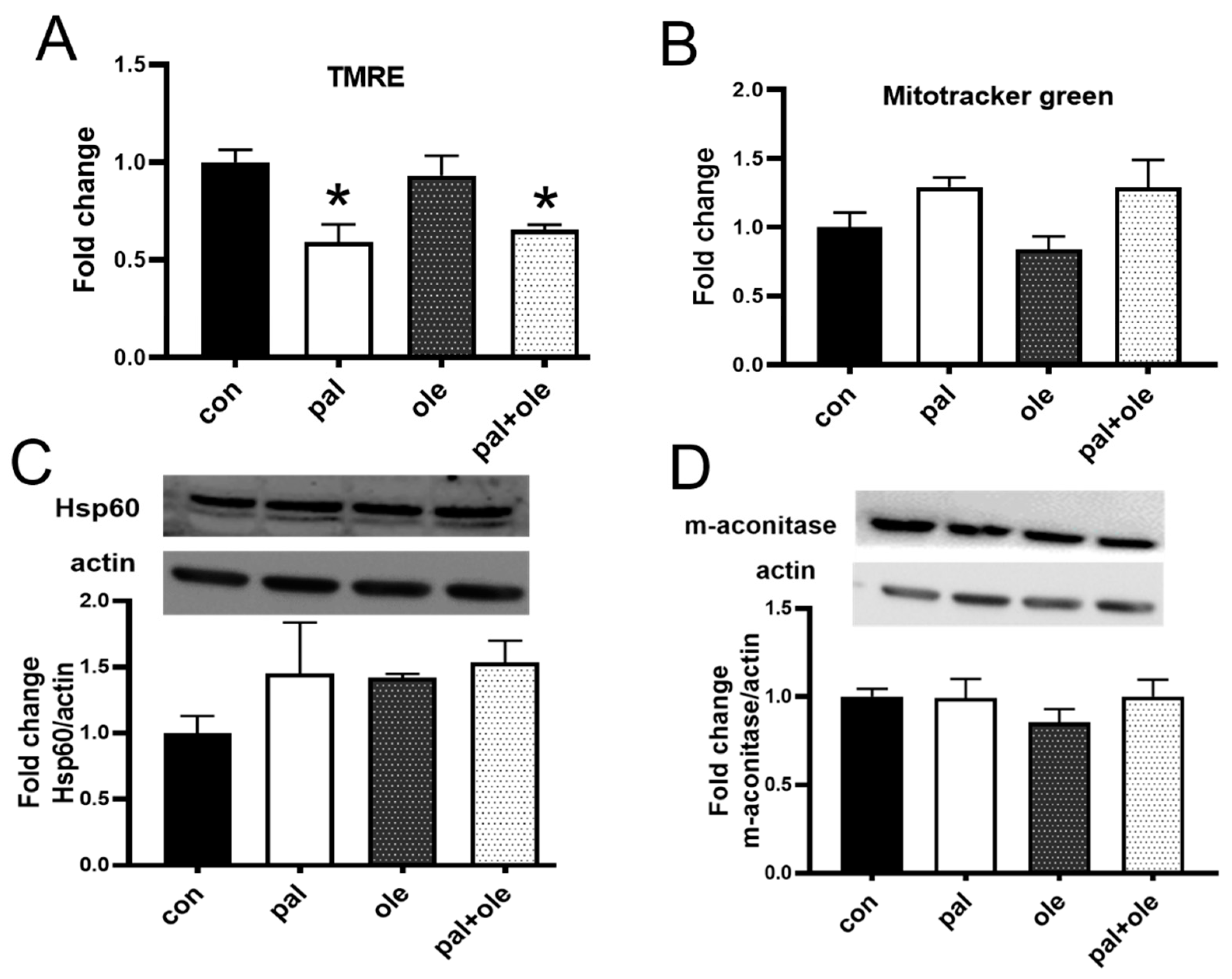
3.1. Palmitate Decreased PBMC Membrane Potential
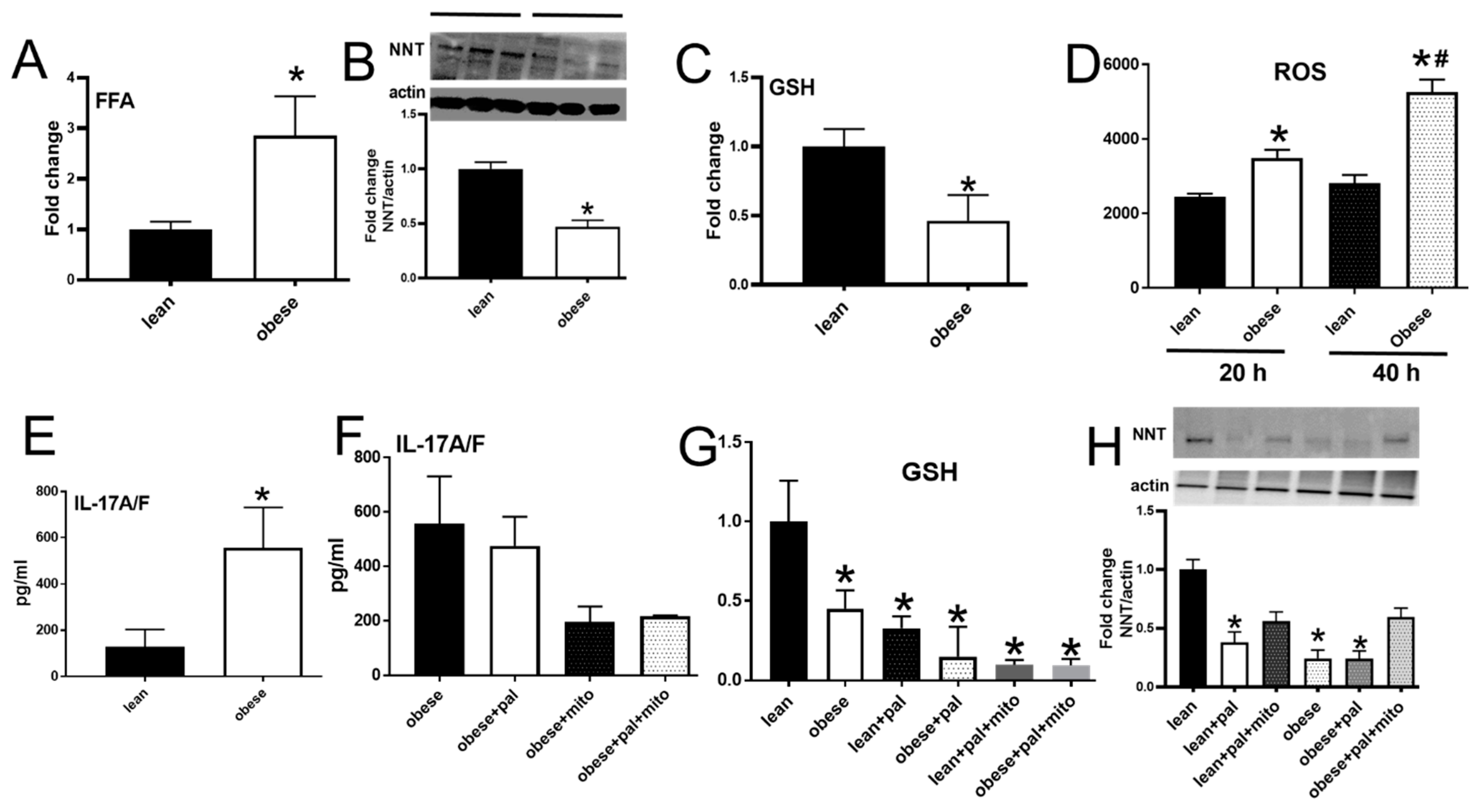
3.2. Palmitate Altered the Cellular Redox Balance
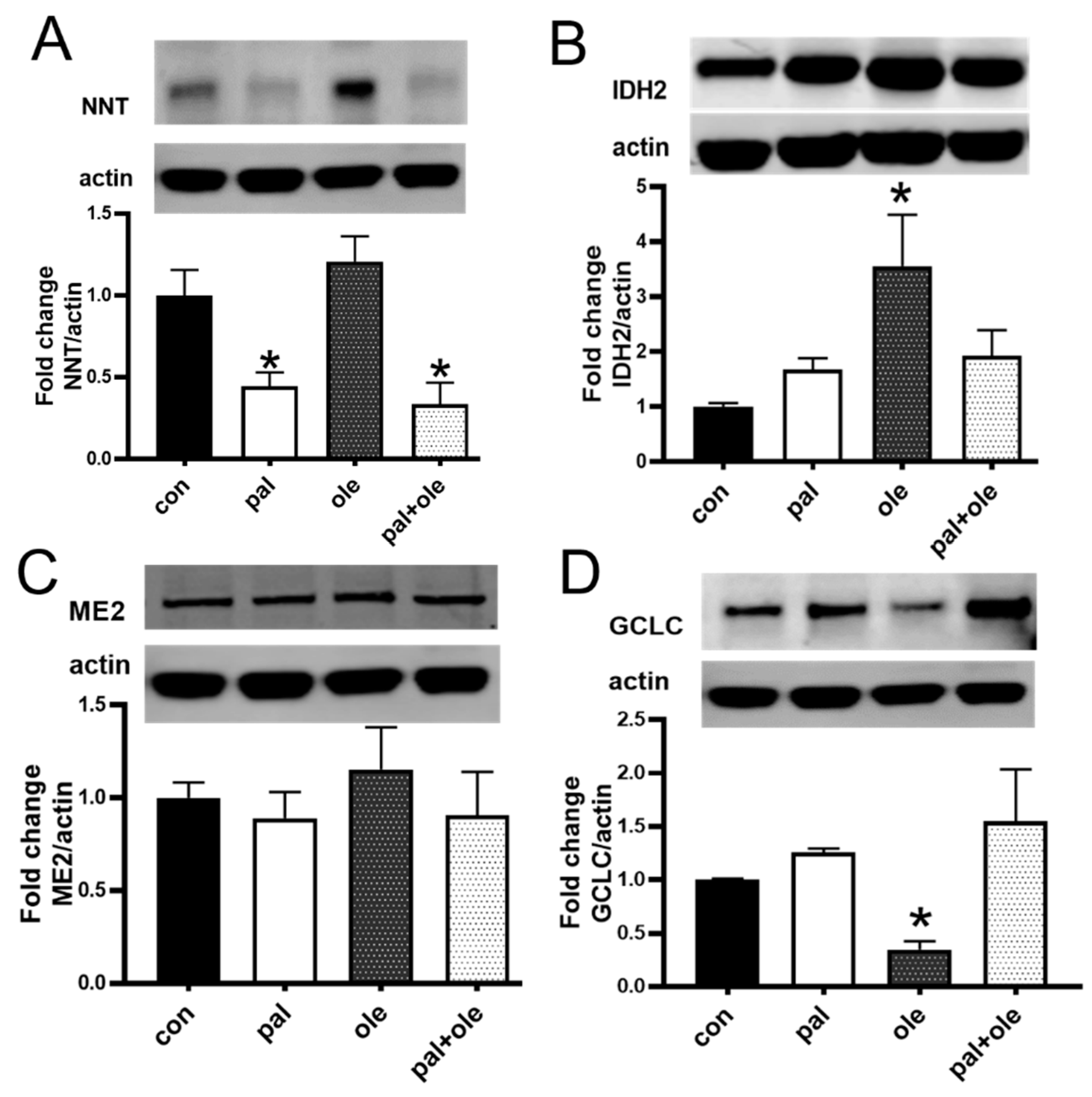
3.3. Palmitate Lowered the Expression of Nicotinamide Nucleotide Transhydrogenase
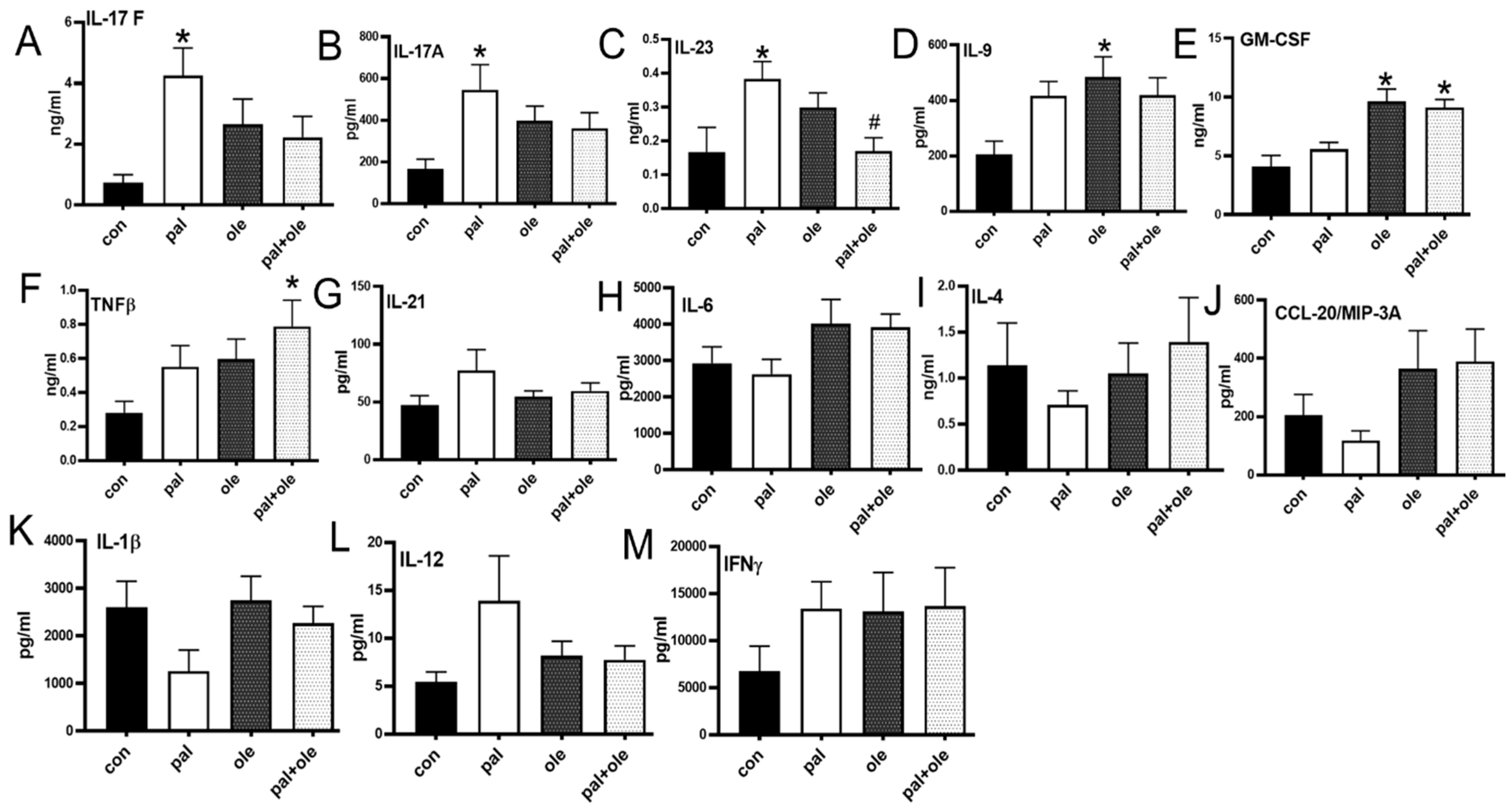
3.4. Palmitate Increased Proinflammatory Cytokine Production by PBMCs
3.5. NNT Protein Knockdown Recapitulated Palmitate-Induced Changes in PBMCs
3.6. PBMCs from Subjects with Obesity Mimicked Characteristics of Redox Imbalance Caused by Palmitate
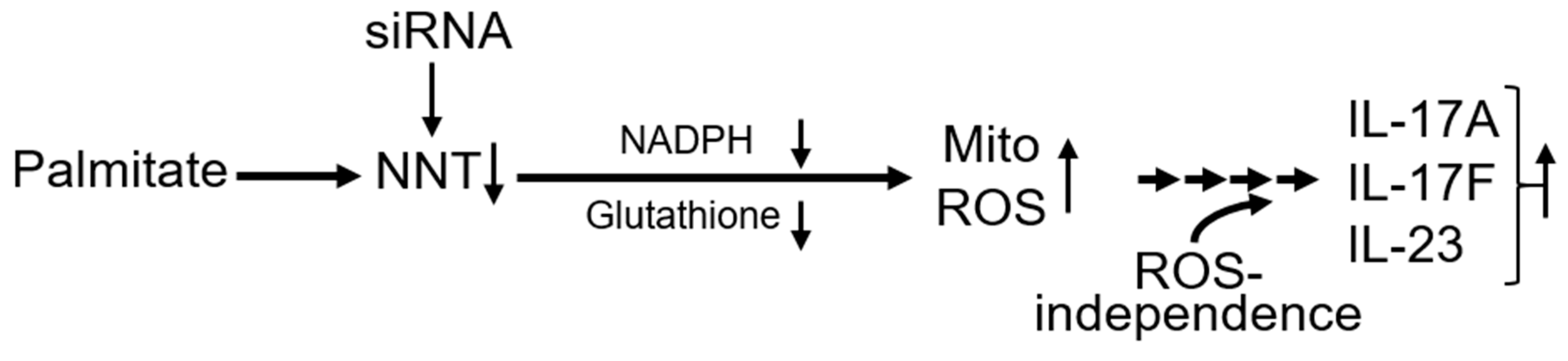
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salzano, S.; Checconi, P.; Hanschmann, E.M.; Lillig, C.H.; Dowler, L.D.; Chan, P.; Vaudry, D.; Mengozzi, M.; Coopo, L.; Sacre, S.; et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. USA 2014, 111, 12157–12162. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, H.R.; Gao, D.; Pararasa, C. Redox regulation in metabolic programming and inflammation. Redox. Biol 2017, 12, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Kienhofer, D.; Boeltz, S.; Hoffmann, M.H. Reactive oxygen homeostasis—The balance for preventing autoimmunity. Lupus 2016, 25, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J. Glutathione depletion is associated with augmenting a proinflammatory signal: Evidence for an antioxidant/pro-oxidant mechanism regulating cytokines in the alveolar epithelium. Cytokines Cell Mol. Ther. 2000, 6, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Ip, B.; Cilfone, N.; Belkina, A.C.; DeFuria, A.C.; Jagannathan-Bogdan, M.; Zhu, M.; Kuchibhatla, R.; McDonnell, M.E.; Xiao, Q.; Kepler, T.B.; et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFalpha production. Obesity (Silver. Spring) 2016, 24, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Rydstrom, J.; Hoek, J.B.; Ernster, L. On the oxidation of reduced nicotinamide dinucleotide phosphate by submitochondrial particles from beef heart. Biochim. Biophys. Acta 1973, 305, 694–698. [Google Scholar] [CrossRef]
- Busanello, E.N.B.; Figureueira, T.R.; Marques, A.C.; Navarro, C.D.C. Facilitation of Ca(2+) -induced opening of the mitochondrial permeability transition pore either by nicotinamide nucleotide transhydrogenase deficiency or statins treatment. Cell. Biol. Int. 2018, 42, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox. Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.Y.; Lin, Y.T.; Lin, G.; Wu, P.R.; Cheng, M.L. Nicotinamide nucleotide transhydrogenase (NNT) deficiency dysregulates mitochondrial retrograde signaling and impedes proliferation. Redox. Biol. 2017, 12, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Meimaridou, E.; Kowalczyk, J.; Guasti, L.; Hugues, C.R.; Wagner, F.; Frommolt, P.; Nürnberg, P.; Mann, N.P.; Banerjee, R.; Saka, H.N.; et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat. Genet. 2012, 44, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, J.A.; Figureueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.; Vercesi, A.E.; Castilho, R.F. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol Med. 2013, 63, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, V.M.; Meadows, N.A.; Bangert, M.; Lee, A.W.; Kadioglu, A.; Cox, R.D. Nicotinamide nucleotide transhydrogenase (NNT) acts as a novel modulator of macrophage inflammatory responses. FASEB J. 2012, 26, 3550–3562. [Google Scholar] [CrossRef] [PubMed]
- Abdelmagid, S.A.; Clarke, S.E.; Nielsen, D.E.; Badawi, A.; El-Sohemy, A.; Mutch, D.M.; Ma, D.W. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS ONE 2015, 10, e0116195. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Ruan, T.; Li, Y.; Ravindran, A.; Wan, X.; Nhan, J.K.; Walker, M.L.; Deeter, L.; Goodrich, R.; Johnson, E.; et al. Ceramide-Initiated Protein Phosphatase 2A Activation Contributes to Arterial Dysfunction In Vivo. Diabetes 2015, 64, 3914–3926. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Cho, J.M.; Park, S.K.; Ruan, T.; Li, Y.; Mueller, R.; Bean, T.; Reese, V.; Richardson, R.S.; Cai, J.; et al. Endothelial Cell Autophagy Maintains Shear Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Mueller, R.; Li, Y.; Ruan, T.; Kunz, D.; Goodrich, R.; Mills, T.; Deeter, L.; Sargsyan, A.; Anandh Babu, P.V.; et al. Impairment of autophagy in endothelial cells prevents shear-stress induced increases in nitric oxide bioavailability. Can. J. Physiol. Pharmacol. 2014, 92, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, A.; Lehmann, S.; Wirth, E.; Rondeau, J.M. The human IL-17A/F heterodimer: A two-faced cytokine with unique receptor recognition properties. Sci. Rep. 2017, 7, 8906. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Herrero, L.; Cipolleta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Budnick, I.; Singh, M.; Thiruppathi, M.; Altharshawi, K.; Elshabrawy, H.; Holterman, M.J.; Prabhakar, B.S. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. J. Interferon Cytokine Res. 2015, 35, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Thiruppathi, M.; Elshabrawy, H.A.; Alharshawi, K.; Kumar, P.; Prabhakar, B.S. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine 2015, 75, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Kaplan, M.H. A brief history of IL-9. J. Immunol. 2011, 186, 3283–3288. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab Clin. North. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Haymond, M.W.; Rizza, R.A.; Cryer, P.E.; Miles, J.M. Influence of body fat distribution on free fatty acid metabolism in obesity. J. Clin. Invest. 1989, 83, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Pongratz, R.L.; Kibbey, R.G.; Shulman, G.I.; Cline, G.W. Cytosolic and mitochondrial malic enzyme isoforms differentially control insulin secretion. J. Biol Chem. 2007, 282, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Mandella, R.D.; Sauer, L.A. The mitochondrial malic enzymes. I. Submitochondrial localization and purification and properties of the NAD(P)+-dependent enzyme from adrenal cortex. J. Biol Chem. 1975, 250, 5877–5884. [Google Scholar] [PubMed]
- Yin, F.; Sancheti, H.; Cadenas, E. Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim. Biophys. Acta 2012, 1817, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Naeemuddin, M.; Elchuri, S.; Yamaguchi, M.; Kozy, H.M.; Carlston, E.J.; Epstein, C.J. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum. Mol. Genet. 2006, 15, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Freeman, H.; Shimomura, K.; Cox, R.D.; Ashcroft, F.M. Nicotinamide nucleotide transhydrogenase: A link between insulin secretion, glucose metabolism and oxidative stress. Biochem. Soc. Trans. 2006, 34, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Freeman, H.; Shimomura, K.; Horner, E.; Cox, R.D.; Ashcroft, F.M. Nicotinamide nucleotide transhydrogenase: A key role in insulin secretion. Cell. Metab. 2006, 3, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.; Reifsnyder, P.C.; Malcolm, R.D.; Lucas, C.A.; MacGregor, G.R.; Zhang, W.; Leiter, E.H. Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity (Silver. Spring) 2010, 18, 1902–1905. [Google Scholar] [CrossRef] [PubMed]
- Heiker, J.T.; Kern, M.; Kosacka, J.; Flehmig, G.; Stumvoll, M.; Shang, E.; Lohmann, T.; Dresler, M.; Kovacs, P.; Blüher, M.; et al. Nicotinamide nucleotide transhydrogenase mRNA expression is related to human obesity. Obesity (Silver. Spring) 2013, 21, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Heiker, J.T.; Gärtner, D.; Björnson, E.; Schön, M.R.; Flehmig, G.; Klöting, N.; Krohn, K.; Fasshauer, M.; Stumvoll, M.; et al. Extensive weight loss reveals distinct gene expression changes in human subcutaneous and visceral adipose tissue. Sci. Rep. 2015, 5, 14841. [Google Scholar] [CrossRef] [PubMed]
- Hafida, S.; Mirshahi, T.; Nikolajczyk, B.S. The impact of bariatric surgery on inflammation: Quenching the fire of obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 373–378. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lean | Obese | |
|---|---|---|
| Total N | 12 | 8 |
| Age, years [mean (range)] | 33 (28–45) | 39.6 (30–47) |
| A1c, % [mean (range)] | 5.1 (4.5–5.6) | 5.3 (5–5.5) |
| BMI, kg m−2 [mean (range)] * | 22.71 (21.1–24.4.) | 33.94 (30.4–40.8) |
| Females [N (%)] | 7 (58.33%) | 4 (50%) |
| Males [N (%)] | 5 (41.66%) | 4 (50%) |
| Protein | Catalog # | Company |
|---|---|---|
| NNT | ab110352 | Abcam |
| m-aconitase | ARP40390_P050 | Aviva biologicals |
| Hsp60 | 12165S | Cell signaling technology |
| GCLC | ARP54577_P050 | Aviva biologicals |
| IDH2 | ab131263 | Abcam |
| B-Actin | 3700 | Cell signaling technology |
| Malic enzyme | 12399S | Cell signaling technology |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCambridge, G.; Agrawal, M.; Keady, A.; Kern, P.A.; Hasturk, H.; Nikolajczyk, B.S.; Bharath, L.P. Saturated Fatty Acid Activates T Cell Inflammation Through a Nicotinamide Nucleotide Transhydrogenase (NNT)-Dependent Mechanism. Biomolecules 2019, 9, 79. https://doi.org/10.3390/biom9020079
McCambridge G, Agrawal M, Keady A, Kern PA, Hasturk H, Nikolajczyk BS, Bharath LP. Saturated Fatty Acid Activates T Cell Inflammation Through a Nicotinamide Nucleotide Transhydrogenase (NNT)-Dependent Mechanism. Biomolecules. 2019; 9(2):79. https://doi.org/10.3390/biom9020079
Chicago/Turabian StyleMcCambridge, Grace, Madhur Agrawal, Alanna Keady, Philip A. Kern, Hatice Hasturk, Barbara S. Nikolajczyk, and Leena P. Bharath. 2019. "Saturated Fatty Acid Activates T Cell Inflammation Through a Nicotinamide Nucleotide Transhydrogenase (NNT)-Dependent Mechanism" Biomolecules 9, no. 2: 79. https://doi.org/10.3390/biom9020079
APA StyleMcCambridge, G., Agrawal, M., Keady, A., Kern, P. A., Hasturk, H., Nikolajczyk, B. S., & Bharath, L. P. (2019). Saturated Fatty Acid Activates T Cell Inflammation Through a Nicotinamide Nucleotide Transhydrogenase (NNT)-Dependent Mechanism. Biomolecules, 9(2), 79. https://doi.org/10.3390/biom9020079