Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
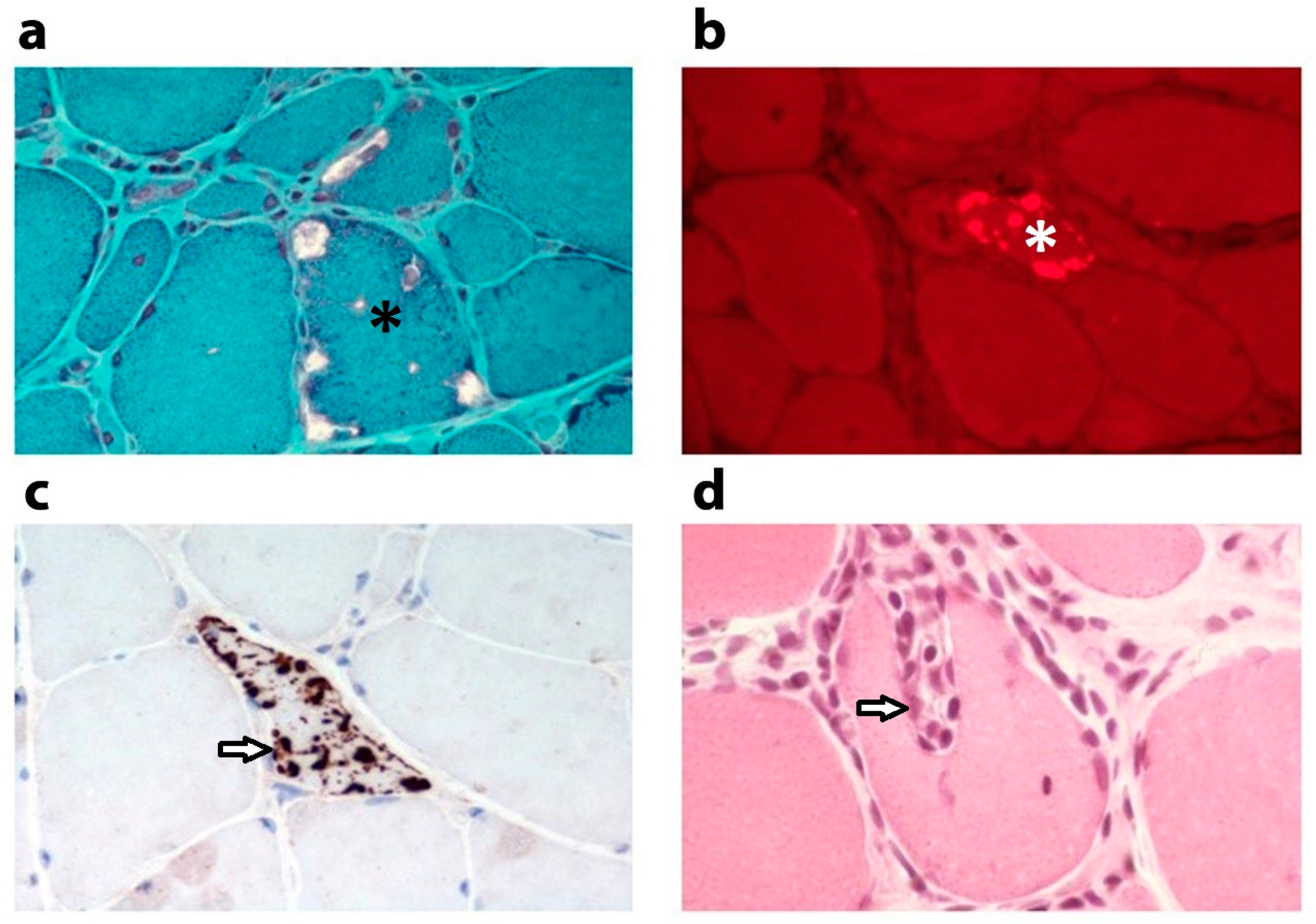
2. Altered Mitochondrial Appearance
3. Mitochondrial DNA Alterations
4. Mitochondrial Proliferation and Oxidative Phosphorylation Defects
5. Dysfunctional Mitophagy
6. Inflammation and Mitokines
7. Mitochondrial Defects in other Muscle Diseases
8. Consequence for Therapeutic Intervention
9. Conclusions
Funding
Conflicts of Interest
References
- Dimachkie, M.M.; Barohn, R.J. Inclusion body myositis. Semin. Neurol. 2012, 32, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; King Engel, W.; Nogalska, A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J. Neuropathol. Exp. Neurol. 2012, 71, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Ivanidze, J.; Hoffmann, R.; Lochmueller, H.; Engel, A.G.; Hohlfeld, R.; Dornmair, K. Inclusion body myositis: Laser microdissection reveals differential up-regulation of IFN-γ signaling cascade in attacked versus nonattacked myofibers. Am. J. Pathol. 2011, 179, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Breithaupt, M.; Schmidt, J. Update on treatment of inclusion body myositis. Curr. Rheumatol. Rep. 2013, 15, e329. [Google Scholar] [CrossRef] [PubMed]
- Pluk, H.; van Hoeve, B.J.; van Dooren, S.H.; Stammen-Vogelzangs, J.; van der Heijden, A.; Schelhaas, H.J.; Verbeek, M.M.; Badrising, U.A.; Arnardottir, S.; Gheorghe, K.; et al. Autoantibodies to cytosolic 5’-nucleotidase 1A in inclusion body myositis. Ann. Neurol. 2013, 73, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Rojana-Udomsart, A.; James, I.; Castle, A.; Needham, M.; Scott, A.; Day, T.; Kiers, L.; Corbett, A.; Sue, C.; Witt, C.; et al. High-resolution HLA-DRB1 genotyping in an Australian inclusion body myositis (s-IBM) cohort: An analysis of disease-associated alleles and diplotypes. J. Neuroimmunol. 2012, 250, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Oldfors, A.; Moslemi, A.-R.; Jonasson, L.; Ohlsson, M.; Kollberg, G.; Lindberg, C. Mitochondrial abnormalities in inclusion-body myositis. Neurology 2006, 66, S49–S55. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Nogalska, A.; D’Agostino, C.; Terracciano, C.; King Engel, W.; Askanas, V. Impaired autophagy in sporadic inclusion body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 2010, 177, 1377–1387. [Google Scholar] [CrossRef]
- Catalan-Garcia, M.; Garrabou, G.; Moren, C.; Guitart-Mampel, M.; Hernando, A.; Diaz-Ramos, A.; Gonzales-Casacuberta, I.; Juarez, D.-L.; Bano, M.; Enrich-Bengoa, J.; et al. Mitochondrial DNA disturbance and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin. Sci. 2016, 130, 1741–1751. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Tuppen, H.A.; Grady, J.P.; Vincent, A.; Blakely, E.L.; Reeve, A.K.; Taylor, R.W.; Picard, M.; Miller, J.; Turnbull, D.M. Complex mitochondrial DNA rearrangements in individual cells from patients with sporadic inclusion body myositis. Nucleic Acids Res 2016, 44, 5313–5329. [Google Scholar] [CrossRef] [PubMed]
- Moslemi, A.-R.; Lindberg, C.; Oldfors, A. Analysis of multiple mitochondrial DNA deletions in inclusion body myositis. Hum. Mutat. 1997, 10, 381–386. [Google Scholar] [CrossRef]
- Brady, S.; Wang, E.; Carver, J.; Hofer, M.; Diot, A.; Hilton, D.; Hilton-Jones, D.; Poulton, J.; Fratter, C. Low mitochondrial DNA copy number suggests abnormal mitophagy in inclusion body myositis. Neuromuscul. Disord. 2018, 28, S30. [Google Scholar] [CrossRef]
- Buzkova, J.; Nikkanen, J.; Ahola, S.; Hakonen, A.H.; Sevastianova, K.; Hovinen, T.; Yki-Jarvinen, H.; Pietilainen, K.H.; Lonnqvist, T.; Velagapudi, V.; et al. Metabolomes of mitochondrial diseases and inclusion body myositis patients: Treatment targets and biomarkers. EMBO Mol. Med. 2018, e9091. [Google Scholar] [CrossRef] [PubMed]
- Samuels, D.C.; Schon, E.A.; Chinnery, P.F. Two direct repeats cause most human mtDNA deletions. Trends Genet. 2004, 20, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, U.; Roos, S.; Hedberg Oldfors, C.; Moslemi, A.-R.; Lindberg, C.; Oldfors, A. Mitochondrial pathology in inclusion body myositis. Neuromuscul. Disord. 2015, 25, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Rifai, Z.; Welle, S.; Kamp, C.; Thronton, C.A. Ragged red fibers in normal aging and inflammatory myopathy. Ann. Neurol. 1995, 37, 24–29. [Google Scholar] [CrossRef]
- Nishigaki, Y.; Tadesse, S.; Bonilla, E.; Shungu, D.; Hersh, S.; Keats, B.J.; Berlin, C.I.; Goldberg, M.F.; Vockley, J.; DiMauro, S.; et al. A novel mitochondrial tRNA(Leu(UUR)) mutation in a patient with features of MERRF and Kearns-Sayre syndrome. Neuromuscul. Disord. 2003, 13, 334–340. [Google Scholar] [CrossRef]
- Shabrokh, E.; Kavanaugh, J.; McMillan, R.; Pittman, J.; Hulver, M.; Frisard, M. Mitochondrial dysregulation in skeletal muscle from patients diagnosed with Alzheimer’s disease and sporadic inclusion body myositis. Open J. Mol. Integr. Physiol. 2014, 4, 11–19. [Google Scholar] [CrossRef]
- Dahlbom, K.; Lindberg, C.; Oldfors, A. Inclusion body myositis: Morphological clues to correct diagnosis. Neuromuscul. Disord. 2002, 12, 853–857. [Google Scholar] [CrossRef]
- Hamann, P.D.; Roux, B.T.; Heward, J.A.; Love, S.; McHugh, N.J.; Jones, S.W.; Lindsay, M.A. transcriptional profiling identifies differential expression of long non-coding RNAs in Jo-1 associated and inclusion body myositis. Sci. Rep. 2017, 7, e8024. [Google Scholar] [CrossRef] [PubMed]
- Georgantas, R.W.; Streicher, K.; Greenberg, S.A.; Greenlees, L.M.; Zhu, W.; Brohawn, P.Z.; Higgs, B.W.; Czapiga, M.; Morehouse, C.A.; Amato, A.; et al. Inhibition of myogenic microRNAs 1, 133, and 206 by inflammatory cytokines links inflammation and muscle degeneration in adult inflammatory myopathies. Arthritis Rheumatol. 2014, 66, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef]
- Nie, Y.; Sato, Y.; Wang, C.; Yue, F.; Kuang, S.; Gavin, T.P. Impaired exercise tolerance, mitochondrial biogenesis, and uscle fiber maintenance in miR-133a-deficient mice. FASEB J. 2016, 30, 3745–3758. [Google Scholar] [CrossRef]
- De Paepe, B.; Lefever, S.; Mestdagh, P. How long noncoding RNAs enforce their will on mitochondrial activity: Regulation of mitochondrial respiration, reactive oxygen species production, apoptosis, and metabolic reprogramming in cancer. Curr. Genet. 2018, 64, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, S.-B.; Wang, M.-R.; Yao, R.-W.; Wu, D.; Yang, L.; Chen, L.-L. Genome-wide screening of NEAT1 regulators reveals cross-regulation between paraspeckles and mitochondria. Nat. Cell Biol. 2018, 20, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; King Engel, W.; Nogalska, A. Sporadic inclsuion-body myositis: A degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochim. Biophys. Acta 2015, 1852, 633–643. [Google Scholar] [CrossRef]
- Vaamonde-Garcia, C.; Riveiro-Naveira, R.R.; Valcarcel-Ares, M.N.; Hermida-Carballo, L.; Blanco, F.J.; Lopez-Armada, M.J. Mitochodnrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheumatol. 2012, 64, 2927–2936. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Miller, J.; Grady, J.P.; Rocha, M.C.; Taylor, R.W.; Turnbull, M.D. Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol. Appl. Neurobiol. 2015, 41, 288–303. [Google Scholar] [CrossRef]
- Mariappan, N.; Soorappan, R.N.; Haque, M.; Sriramula, S.; Francis, J. TNF-α-induced mitochondrial oxidative stress and cardiac dysfunction: Restoration by superoxide dismutase mimetic tempol. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2726–H2737. [Google Scholar] [CrossRef]
- Gidlund, E.-K.; von Walden, F.; Venojarvi, M.; Riserus, U.; Heinonen, O.J.; Norrbom, J.; Sundberg, C.J. Humanin skeletal muscle protein levels increase after resistance training in men with impaired glucose metabolism. Physiol. Rep. 2016, 4, e13063. [Google Scholar] [CrossRef]
- Suomalainen, A.; Elo, J.M.; Pietiläinen, K.H.; Hakonen, A.H.; Sevastianova, K.; Korpela, M.; Isohanni, P.; Marjavaara, S.K.; Tyni, T.; Kiuru-Enari, S.; et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: A diagnostic study. Lancet Neurol. 2011, 10, 806–818. [Google Scholar] [CrossRef]
- Xie, T.; Leung, P.S. Fibroblast growth factor 21: A regulator of metabolic disease and health span. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E292–E302. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, M.; Forsström, S.; Bottani, E.; Viscomi, C.; Baris, O.R.; Isoniemi, H.; Höckerstedt, K.; Österlund, P.; Hurme, M.; Jylhävä, J.; et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 2016, 87, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.K.; Ryu, D.; Kim, K.S.; Chang, J.Y.; Kim, Y.K.; Yi, H.S.; Kang, S.G.; Choi, M.J.; Lee, S.E.; Jung, S.B.; et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 2017, 216, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Kalko, S.G.; Jou, C.; Meznaric, M.; Rogac, M.; Jekovec-Vrhovsek, M.; Sciacco, M.; Moggio, M.; Fagiolari, G.; De Paepe, B.; De Meirleir, L.; et al. Transcriptomic profiling of thymidine kinase 2 deficient human skeletal muscle reveals activation of the p53 signaling pathway and identifies growth and differentiation 15 as a potential novel biomarker for mitochondrial myopathies. BMC Genom. 2014, 15, e91. [Google Scholar] [CrossRef] [PubMed]
- Sunitha, B.; Gayathri, N.; Kumar, M.; Keshava Prasad, T.S.; Nalini, A.; Padmanabhan, B.; Srinivas Bharath, M.M. Muscle biopsies from human muscle diseases with myopathic pathology reveal common alterations in mitochondrial function. J. Neurochem. 2016, 138, 174–191. [Google Scholar] [CrossRef]
- Temiz, P.; Weihl, C.C.; Pestronk, A. Inflammatory myopathies with mitochondrial pathology and protein aggregates. J. Neurol. Sci. 2009, 278, 25–29. [Google Scholar] [CrossRef]
- Matsubara, S.; Bokuda, K.; Asao, Y.; Morishima, R.; Sugaya, K.; Miyamoto, K.; Koide, R.; Komori, T.; Suzuki, S.; Nishino, I. Mitophagy in three cases of immune-mediated necrotizing myopathy associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies: Ultrastructural and immunohistochemical studies. Neuromuscul. Disord. 2018, 28, 283–288. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Allenbach, Y.; Grelet, E.; Ueberschlag, V.; Echaniz-Lagun, A.; Lannes, B.; Alsaleh, G.; Charles, A.L.; Singh, F.; et al. IFN-β-induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis. Acta Neuropathol. 2017, 134, 655–666. [Google Scholar] [CrossRef]
- Woo, M.; Chung, S.J.; Nonaka, I. Perifascicular atrophic fibers in childhood dermatomyositis with particular reference to mitochondrial changes. J. Neurol. Sci. 1988, 88, 133–143. [Google Scholar] [CrossRef]
- Gambelli, S.; Dotti, M.T.; Malandrini, A.; Mondelli, M.; Stromillo, M.L.; Gaudiano, C.; Federico, A. Mitochondrial alterations in muscle biopsies of patients on statin therapy. J. Submicrosc. Cytol. Pathol. 2004, 36, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Stringer, H.A.J.; Sohi, G.K.; Maguire, J.A.; Cote, H.C.F. Decreased skeletal muscle mitochondrial DNA in patients with statin-induced myopathy. J. Neurol. Sci. 2013, 325, 142–147. [Google Scholar] [CrossRef]
- Paiva, H.; Thelen, K.M.; Van Coster, R.; Smet, J.; De Paepe, B.; Mattila, K.M.; Lakkso, J.; Lehtimaki, T.; von Bergmann, K.; Lutjohann, D.; et al. High dose statins and skeletal muscle metabolism in humans: A randomized, controlled trial. Clin. Pharmacol. Ther. 2005, 78, 60–68. [Google Scholar] [CrossRef]
- Amato, A.A.; Badrising, U.; Benveniste, O.; Needham, M.; Chinoy, H.; Wu, M.; Koumaras, B.; de Vera, A.; Papanicolaou, D.A.; Hanna, M.G. RESILIENT: A randomized, double-blind, placebo-controlled study of bimagrumab in patients with sporadic inclusion body myositis. Neurology 2017, 88, 1–111. [Google Scholar]
- Johnson, L.G.; Collier, K.E.; Edwards, D.J.; Philippe, D.L.; Eastwood, P.R.; Walters, S.E.; Thickbroom, G.W.; Mastaglia, F.L. Improvement in aerobic capacity after an exercise program in sporadic inclusion body myositis. Clin. Neuromusc. Dis. 2009, 10, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Lira, V.A.; Greene, N.P. Exercise training-induced regulation of mitochondrial quality. Exerc. Sport Sci. Rev. 2012, 40, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Pohnert, S.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.; Williams, R.; Yan, Z. Exercise stimulates PGC-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M. Treatment of mitochondrial disorders: Antioxidants and beyond. J. Child Neurol. 2014, 29, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Raj Joshi, P.; Vetterke, M.; Hauburger, A.; Tacik, P.; Stoltenburg, G.; Hanisch, F. Functional relevance of mitochondrial abnormalities in sporadic inclusion body myositis. J. Clin. Neurosci. 2014, 21, 1959–1963. [Google Scholar] [CrossRef]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Paepe, B. Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras. Biomolecules 2019, 9, 15. https://doi.org/10.3390/biom9010015
De Paepe B. Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras. Biomolecules. 2019; 9(1):15. https://doi.org/10.3390/biom9010015
Chicago/Turabian StyleDe Paepe, Boel. 2019. "Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras" Biomolecules 9, no. 1: 15. https://doi.org/10.3390/biom9010015
APA StyleDe Paepe, B. (2019). Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras. Biomolecules, 9(1), 15. https://doi.org/10.3390/biom9010015