Metformin: The Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer

 ,
,  ,
, 
Abstract
1. Background and Introduction
2. Biology of Metformin and Molecular Mechanism of Action
3. Cellular and Pre-Clinical Data
4. Clinical Data and Trials
5. Monotherapy vs. Combination Therapy
6. Challenges, Future Perspective, and Directions
6.1. Mixed Messages and Challenges
6.2. Metformin and Breast Cancer Biomarkers
6.3. Efficacy in Non-Diabetic Patients and Non-Diabetic Cancer Patients
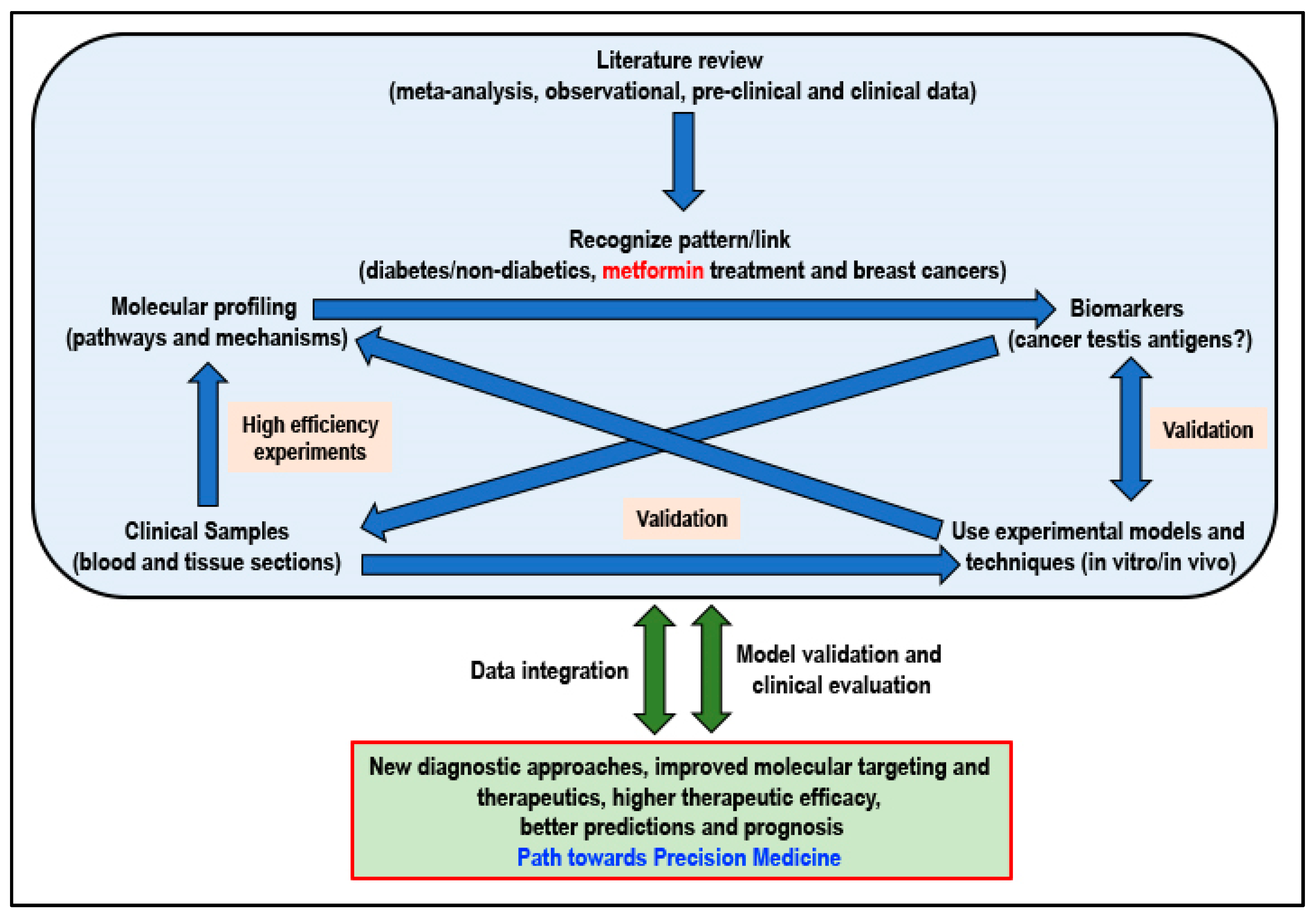
6.4. Future Directions—Metformin and a Proposed Path Towards Precision Medicine
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl CoA carboxylase |
| AKAP4 | A-kinase anchoring protein 4 |
| AMP | Adenosine monophosphate |
| AMPK | 5′ AMP activated protein kinase |
| ATP | Adenosine triphosphate |
| BAGE | B melanoma antigen |
| CTA | Cancer testis antigen |
| DDIT4/REDD1 | DNA Damage Inducible Transcript 4/Regulated in DNA Damage 1 |
| EMT | Epithelial mesenchymal transition |
| ER | Estrogen receptor |
| ERK | Extracellular signal-regulated kinase |
| ETC | Electron transport chain |
| HER2 | Human epidermal growth factor receptor 2 |
| IGF1 | Insulin-like growth factor 1 |
| IRS1 | Insulin receptor substrate 1 |
| MAGE | Melanoma antigen |
| MAPK | Mitogen activated protein kinase |
| MATE1/2 | Multidrug and toxin extrusion protein-1 and 2 |
| mTOR | mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa-B |
| OCT1/2/3 | Organic cation transporters 1, 2 and 3 |
| PAI-1 | Plasminogen activator inhibitor 1 |
| PI3K | Phosphoinositide 3-kinase |
| PMAT | Plasma membrane monoamine transporter |
| PR | Progesterone receptor |
| PTP | Permeability transition pore |
| SP17 | Sperm protein 17 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TNBC | Triple negative breast cancer |
| TNFα | Tumor necrosis factor alpha |
| TSC2 | Tuberous sclerosis 2 |
| UAP1 | UDP-N-acetylglucosamine pyrophosphorylase 1 |
| uPA | Urokinase-type plasminogen activator |
| UPR | Unfolded Protein Response |
| WHO | World Health Organization |
References
- World Health Organization. Available online: https://apps.who.int/iris/bitstream/handle/10665/204871/9789241565257_eng.pdf;jsessionid=CB10185391030DF727E80B7DC9747873?sequence=1 (accessed on 30 August 2019).
- European Society of Cardiology. Available online: https://www.escardio.org/Sub-specialty-communities/European-Association-of-Preventive-Cardiology-(EAPC)/News/global-statistics-on-diabetes (accessed on 30 August 2019).
- Leon, B.M.; Maddox, T.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 2015, 6, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Min, T.Z.; Stephens, M.W.; Kumar, P.; Chudleigh, R.A. Renal complications of diabetes. Br. Med. Bull. 2012, 104, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.M.; Varghese, E.; Varghese, S.; Busselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Said, G. Diabetic neuropathy—A review. Nat. Clin. Pr. Neurol. 2007, 3, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Blendea, M.C.; Thompson, M.J.; Malkani, S. Diabetes and Chronic Liver Disease: Etiology and Pitfalls in Monitoring. Clin. Diabetes 2010, 28, 139. [Google Scholar] [CrossRef]
- Boyle, P.; Boniol, M.; Koechlin, A.; Robertson, C.; Valentini, F.; Coppens, K.; Fairley, L.L.; Boniol, M.; Zheng, T.; Zhang, Y.; et al. Diabetes and breast cancer risk: A meta-analysis. Br. J. Cancer 2012, 107, 1608–1617. [Google Scholar] [CrossRef]
- Hardefeldt, P.J.; Edirimanne, S.; Eslick, G.D. Diabetes increases the risk of breast cancer: A meta-analysis. Endocr. Relat. Cancer 2012, 19, 793–803. [Google Scholar] [CrossRef]
- Larsson, S.C.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of breast cancer: A meta-analysis. Int. J. Cancer 2007, 121, 856–862. [Google Scholar] [CrossRef]
- Stattin, P.; Björ, O.; Ferrari, P.; Lukanova, A.; Lenner, P.; Lindahl, B.; Hallmans, G.; Kaaks, R. Prospective Study of Hyperglycemia and Cancer Risk. Diabetes Care 2007, 30, 561. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Day, C. Metformin: Its botanical background. Pract. Diabetes Int. 2004, 21, 115–117. [Google Scholar] [CrossRef]
- Leone, A.; Di Gennaro, E.; Bruzzese, F.; Avallone, A.; Budillon, A. New perspective for an old antidiabetic drug: Metformin as anticancer agent. Cancer Treat. Res. 2014, 159, 355–376. [Google Scholar] [CrossRef] [PubMed]
- Misbin, R.I. The Phantom of Lactic Acidosis due to Metformin in Patients With Diabetes. Diabetes Care 2004, 27, 1791. [Google Scholar] [CrossRef]
- Vecchio, S.; Protti, A. Metformin-induced lactic acidosis: No one left behind. Crit Care 2011, 15, 107. [Google Scholar] [CrossRef][Green Version]
- Fitzgerald, E.; Mathieu, S.; Ball, A. Metformin associated lactic acidosis. BMJ 2009, 339, b3660. [Google Scholar] [CrossRef]
- Marshall, S.M. 60 years of metformin use: A glance at the past and a look to the future. Diabetologia 2017, 60, 1561–1565. [Google Scholar] [CrossRef]
- Correia, S.; Carvalho, C.; Santos, M.S.; Seica, R.; Oliveira, C.R.; Moreira, P.I. Mechanisms of action of metformin in type 2 diabetes and associated complications: An overview. Mini Rev. Med. Chem. 2008, 8, 1343–1354. [Google Scholar] [CrossRef]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef]
- Iranshahy, M.; Rezaee, R.; Karimi, G. Hepatoprotective activity of metformin: A new mission for an old drug? Eur. J. Pharmacol. 2019, 850, 1–7. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Yanardag, R.; Ozsoy-Sacan, O.; Bolkent, S.; Orak, H.; Karabulut-Bulan, O. Protective effects of metformin treatment on the liver injury of streptozotocin-diabetic rats. Hum. Exp. Toxicol. 2005, 24, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Brackett, C.C. Clarifying metformin’s role and risks in liver dysfunction. J. Am. Pharm. Assoc. 2010, 50, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Corremans, R.; Vervaet, B.A.; D’Haese, P.C.; Neven, E.; Verhulst, A. Metformin: A Candidate Drug for Renal Diseases. Int. J. Mol. Sci. 2018, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Ma, J.; Liu, J.; Yu, H.; Chen, Y.; Wang, Q.; Xiang, L. Beneficial Effect of Metformin on Nerve Regeneration and Functional Recovery After Sciatic Nerve Crush Injury in Diabetic Rats. Neurochem. Res. 2016, 41, 1130–1137. [Google Scholar] [CrossRef]
- Mao-Ying, Q.-L.; Kavelaars, A.; Krukowski, K.; Huo, X.-J.; Zhou, W.; Price, T.J.; Cleeland, C.; Heijnen, C.J. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PLoS ONE 2014, 9, e100701. [Google Scholar] [CrossRef]
- Bahrambeigi, S.; Yousefi, B.; Rahimi, M.; Shafiei-Irannejad, V. Metformin; an old antidiabetic drug with new potentials in bone disorders. Biomed. Pharmacother. 2019, 109, 1593–1601. [Google Scholar] [CrossRef]
- Prattichizzo, F.; Giuliani, A.; Mensà, E.; Sabbatinelli, J.; De Nigris, V.; Rippo, M.R.; La Sala, L.; Procopio, A.D.; Olivieri, F.; Ceriello, A. Pleiotropic effects of metformin: Shaping the microbiome to manage type 2 diabetes and postpone ageing. Ageing Res. Rev. 2018, 48, 87–98. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Novelle, M.G.; Ali, A.; Dieguez, C.; Bernier, M.; de Cabo, R. Metformin: A Hopeful Promise in Aging Research. Cold Spring Harb. Perspect. Med. 2016, 6, a025932. [Google Scholar] [CrossRef] [PubMed]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [PubMed]
- DeCensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and Cancer Risk in Diabetic Patients: A Systematic Review and Meta-analysis. Cancer Prev. Res. 2010, 3, 1451. [Google Scholar] [CrossRef] [PubMed]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, S.; Chun, K.H.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kim, Y.S.; Woo, J.-T.; Nam, M.-S.; Baik, S.H.; et al. Metformin reduces the risk of cancer in patients with type 2 diabetes: An analysis based on the Korean National Diabetes Program Cohort. Medicine 2018, 97, e0036. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Liu, M.; Huang, Y.; Wu, K.; Huang, X.; Zhao, Y.; He, W.; Zhang, R. Metformin Use and Lung Cancer Risk in Diabetic Patients: A Systematic Review and Meta-Analysis. Dis. Markers 2019, 2019, 9. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef]
- Li, D.; Yeung, S.C.; Hassan, M.M.; Konopleva, M.; Abbruzzese, J.L. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology 2009, 137, 482–488. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef]
- Bost, F.; Decoux-Poullot, A.G.; Tanti, J.F.; Clavel, S. Energy disruptors: Rising stars in anticancer therapy? Oncogenesis 2016, 5, e188. [Google Scholar] [CrossRef] [PubMed]
- Sośnicki, S.; Kapral, M.; Węglarz, L. Molecular targets of metformin antitumor action. Pharmacol. Rep. 2016, 68, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Daugan, M.; Dufaÿ Wojcicki, A.; d’Hayer, B.; Boudy, V. Metformin: An anti-diabetic drug to fight cancer. Pharmacol. Res. 2016, 113, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.M.; Hojlund, K.; Hother-Nielsen, O.; Stage, T.B.; Damkier, P.; Beck-Nielsen, H.; Brosen, K. Steady-state pharmacokinetics of metformin is independent of the OCT1 genotype in healthy volunteers. Eur J. Clin. Pharm. 2015, 71, 691–697. [Google Scholar] [CrossRef]
- Kinaan, M.; Ding, H.; Triggle, C.R. Metformin: An Old Drug for the Treatment of Diabetes but a New Drug for the Protection of the Endothelium. Med. Princ. Pract. 2015, 24, 401–415. [Google Scholar] [CrossRef]
- Liang, X.; Giacomini, K.M. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J. Pharm. Sci. 2017, 106, 2245–2250. [Google Scholar] [CrossRef]
- Choi, M.K.; Jin, Q.R.; Jin, H.E.; Shim, C.K.; Cho, D.Y.; Shin, J.G.; Song, I.S. Effects of tetraalkylammonium compounds with different affinities for organic cation transporters on the pharmacokinetics of metformin. Biopharm. Drug Dispos. 2007, 28, 501–510. [Google Scholar] [CrossRef]
- Kimura, N.; Masuda, S.; Tanihara, Y.; Ueo, H.; Okuda, M.; Katsura, T.; Inui, K. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab. Pharmacokinet. 2005, 20, 379–386. [Google Scholar] [CrossRef]
- Li, S.; Chen, Y.; Zhang, S.; More, S.S.; Huang, X.; Giacomini, K.M. Role of organic cation transporter 1, OCT1 in the pharmacokinetics and toxicity of cis-diammine(pyridine)chloroplatinum(II) and oxaliplatin in mice. Pharm. Res. 2011, 28, 610–625. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.C.; Liang, X.; Yee, S.W.; Geier, E.G.; Stocker, S.L.; Chen, L.; Giacomini, K.M. Targeted disruption of organic cation transporter 3 attenuates the pharmacologic response to metformin. Mol. Pharmacol. 2015, 88, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Teranishi, K.; Li, S.; Yee, S.W.; Hesselson, S.; Stryke, D.; Johns, S.J.; Ferrin, T.E.; Kwok, P.; Giacomini, K.M. Genetic variants in multidrug and toxic compound extrusion-1, hMATE1, alter transport function. Pharm. J. 2009, 9, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xia, L.; Wang, J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab. Dispos. 2007, 35, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Chien, H.C.; Yee, S.W.; Giacomini, M.M.; Chen, E.C.; Piao, M.; Hao, J.; Twelves, J.; Lepist, E.I.; Ray, A.S.; et al. Metformin Is a Substrate and Inhibitor of the Human Thiamine Transporter, THTR-2 (SLC19A3). Mol. Pharm. 2015, 12, 4301–4310. [Google Scholar] [CrossRef]
- Shu, Y.; Brown, C.; Castro, R.A.; Shi, R.J.; Lin, E.T.; Owen, R.P.; Sheardown, S.A.; Yue, L.; Burchard, E.G.; Brett, C.M.; et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharm. 2008, 83, 273–280. [Google Scholar] [CrossRef]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef]
- Cai, H.; Zhang, Y.; Han, T.K.; Everett, R.S.; Thakker, D.R. Cation-selective transporters are critical to the AMPK-mediated antiproliferative effects of metformin in human breast cancer cells. Int J. Cancer 2016, 138, 2281–2292. [Google Scholar] [CrossRef]
- Checkley, L.A.; Rudolph, M.C.; Wellberg, E.A.; Giles, E.D.; Wahdan-Alaswad, R.S.; Houck, J.A.; Edgerton, S.M.; Thor, A.D.; Schedin, P.; Anderson, S.M.; et al. Metformin Accumulation Correlates with Organic Cation Transporter 2 Protein Expression and Predicts Mammary Tumor Regression in Vivo. Cancer Prev. Res. 2017, 10, 198–207. [Google Scholar] [CrossRef]
- Chowdhury, S.; Yung, E.; Pintilie, M.; Muaddi, H.; Chaib, S.; Yeung, M.; Fusciello, M.; Sykes, J.; Pitcher, B.; Hagenkort, A.; et al. MATE2 Expression Is Associated with Cancer Cell Response to Metformin. PLoS ONE 2016, 11, e0165214. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Everett, R.S.; Thakker, D.R. Efficacious dose of metformin for breast cancer therapy is determined by cation transporter expression in tumours. Br. J. Pharmacol. 2019, 176, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, J.; Manzari, N.; Thompson, J.; Gudi, S.K.; Chhabra, M.; Naik, G.; Mousavi, S.M.; Varkaneh, H.K.; Clark, C.; Zhang, Y. The effect of metformin on biomarkers associated with breast cancer outcomes: A systematic review, meta-analysis, and dose-response of randomized clinical trials. Clin. Transl. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.M.; Ghosh, S.; Majeed, Y.; Arunachalam, G.; Emara, M.M.; Ding, H.; Triggle, C.R. Metformin represses glucose starvation induced autophagic response in microvascular endothelial cells and promotes cell death. Biochem. Pharmacol. 2017, 132, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Karnevi, E.; Said, K.; Andersson, R.; Rosendahl, A.H. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 2013, 13, 235. [Google Scholar] [CrossRef]
- Lee, J.; Hong, E.M.; Kim, J.H.; Jung, J.H.; Park, S.W.; Koh, D.H.; Choi, M.H.; Jang, H.J.; Kae, S.H. Metformin Induces Apoptosis and Inhibits Proliferation through the AMP-Activated Protein Kinase and Insulin-like Growth Factor 1 Receptor Pathways in the Bile Duct Cancer Cells. J. Cancer 2019, 10, 1734–1744. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin—mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143. [Google Scholar] [CrossRef]
- Rojas, L.B.; Gomes, M.B. Metformin: An old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 2013, 5, 6. [Google Scholar] [CrossRef]
- Triggle, C.R.; Ding, H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. 2017, 219, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, P.; Riondino, S.; Buonomo, O.; Palmirotta, R.; Guadagni, F.; Roselli, M. Type 2 Diabetes and Breast Cancer: The Interplay between Impaired Glucose Metabolism and Oxidant Stress. Oxid. Med. Cell Longev. 2015, 2015, 183928. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-κB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy—the barrier or the path. J. Mol. Cell Biol. 2018, 11, 293–305. [Google Scholar] [CrossRef]
- Lai, H.-H.; Li, J.-N.; Wang, M.-Y.; Huang, H.-Y.; Croce, C.M.; Sun, H.-L.; Lyu, Y.-J.; Kang, J.-W.; Chiu, C.-F.; Hung, M.-C.; et al. HIF-1α promotes autophagic proteolysis of Dicer and enhances tumor metastasis. J. Clin. Investig. 2018, 128, 625–643. [Google Scholar] [CrossRef]
- Villanueva, T. It’s nicer with DICER. Nat. Rev. Cancer 2010, 10, 531. [Google Scholar] [CrossRef]
- Martello, G.; Rosato, A.; Ferrari, F.; Manfrin, A.; Cordenonsi, M.; Dupont, S.; Enzo, E.; Guzzardo, V.; Rondina, M.; Spruce, T.; et al. A MicroRNA Targeting Dicer for Metastasis Control. Cell 2010, 141, 1195–1207. [Google Scholar] [CrossRef]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The Antidiabetic Drug Metformin Activates the AMP-Activated Protein Kinase Cascade via an Adenine Nucleotide-Independent Mechanism. Diabetes 2002, 51, 2420. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Meng, S.; Cao, J.; He, Q.; Xiong, L.; Chang, E.; Radovick, S.; Wondisford, F.E.; He, L. Metformin activates AMP-activated protein kinase by promoting formation of the αβγ heterotrimeric complex. J. Biol. Chem. 2015, 290, 3793–3802. [Google Scholar] [CrossRef]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, H.S.; Somasagara, R.R.; Bunting, K.D. Targeting MYC Dependence by Metabolic Inhibitors in Cancer. Genes 2017, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Reineke, L.C.; Knutsen, E.; Chen, M.; Pichler, M.; Ling, H.; Calin, G.A. Metformin blocks MYC protein synthesis in colorectal cancer via mTOR-4EBP-eIF4E and MNK1-eIF4G-eIF4E signaling. Mol. Oncol. 2018, 12, 1856–1870. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin Inhibits Cytokine-Induced Nuclear Factor κB Activation Via AMP-Activated Protein Kinase Activation in Vascular Endothelial Cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef]
- Sekino, N.; Kano, M.; Matsumoto, Y.; Sakata, H.; Akutsu, Y.; Hanari, N.; Murakami, K.; Toyozumi, T.; Takahashi, M.; Otsuka, R.; et al. Antitumor effects of metformin are a result of inhibiting nuclear factor kappa B nuclear translocation in esophageal squamous cell carcinoma. Cancer Sci. 2018, 109, 1066–1074. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Z.; Jin, P.; Yang, X.; Li, X.; Wei, X.; Wang, Y.; Long, S.; Zhang, T.; Chen, G.; et al. Metformin Suppresses Tumor Progression by Inactivating Stromal Fibroblasts in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 1291. [Google Scholar] [CrossRef]
- Li, P.; Zhao, M.; Parris, A.; Feng, X.; Yang, X. P53 is required for metformin-induced growth inhibition, senescence and apoptosis in breast cancer cells. Biochem. Biophys. Res. Commun. 2015, 464, 1267–1274. [Google Scholar] [CrossRef]
- Chen, L.; Ahmad, N.; Liu, X. Combining p53 stabilizers with metformin induces synergistic apoptosis through regulation of energy metabolism in castration-resistant prostate cancer. Cell Cycle 2016, 15, 840–849. [Google Scholar] [CrossRef][Green Version]
- Yi, Y.; Zhang, W.; Yi, J.; Xiao, Z.-X. Role of p53 Family Proteins in Metformin Anti-Cancer Activities. J. Cancer 2019, 10, 2434–2442. [Google Scholar] [CrossRef]
- Yi, G.; He, Z.; Zhou, X.; Xian, L.; Yuan, T.; Jia, X.; Hong, J.; He, L.; Liu, J. Low concentration of metformin induces a p53-dependent senescence in hepatoma cells via activation of the AMPK pathway. Int. J. Oncol. 2013, 43, 1503–1510. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Pulito, C.; Donzelli, S.; Muti, P.; Puzzo, L.; Strano, S.; Blandino, G. microRNAs and cancer metabolism reprogramming: The paradigm of metformin. Ann. Transl. Med. 2014, 2, 58. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat. Commun. 2012, 3, 865. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.O.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin Inhibits Mammalian Target of Rapamycin–Dependent Translation Initiation in Breast Cancer Cells. Cancer Res. 2007, 67, 10804. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.-P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649. [Google Scholar] [CrossRef]
- Gupta, S.; Roy, A.; Dwarakanath, B.S. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front. Oncol. 2017, 7, 68. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Sullivan, L.B.; Heiden, M.G. Understanding the complex-I-ty of metformin action: Limiting mitochondrial respiration to improve cancer therapy. BMC Biol. 2014, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Koido, M.; Haga, N.; Furuno, A.; Tsukahara, S.; Sakurai, J.; Tani, Y.; Sato, S.; Tomida, A. Mitochondrial deficiency impairs hypoxic induction of HIF-1 transcriptional activity and retards tumor growth. Oncotarget 2017, 8, 11841–11854. [Google Scholar] [CrossRef]
- Haga, N.; Saito, S.; Tsukumo, Y.; Sakurai, J.; Furuno, A.; Tsuruo, T.; Tomida, A. Mitochondria regulate the unfolded protein response leading to cancer cell survival under glucose deprivation conditions. Cancer Sci. 2010, 101, 1125–1132. [Google Scholar] [CrossRef]
- Ma, L.; Wei, J.; Wan, J.; Wang, W.; Wang, L.; Yuan, Y.; Yang, Z.; Liu, X.; Ming, L. Low glucose and metformin-induced apoptosis of human ovarian cancer cells is connected to ASK1 via mitochondrial and endoplasmic reticulum stress-associated pathways. J. Exp. Clin. Cancer Res. 2019, 38, 77. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Wolff, N.C.; Vega-Rubin-de-Celis, S.; Xie, X.-J.; Castrillon, D.H.; Kabbani, W.; Brugarolas, J. Cell-Type-Dependent Regulation of mTORC1 by REDD1 and the Tumor Suppressors TSC1/TSC2 and LKB1 in Response to Hypoxia. Mol. Cell. Biol. 2011, 31, 1870. [Google Scholar] [CrossRef]
- Katiyar, S.; Liu, E.; Knutzen, C.A.; Lang, E.S.; Lombardo, C.R.; Sankar, S.; Toth, J.I.; Petroski, M.D.; Ronai, Z.e.; Chiang, G.G. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep. 2009, 10, 866–872. [Google Scholar] [CrossRef]
- Tirado-Hurtado, I.; Fajardo, W.; Pinto, J.A. DNA Damage Inducible Transcript 4 Gene: The Switch of the Metabolism as Potential Target in Cancer. Front. Oncol. 2018, 8, 106. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.-F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, Independent of AMPK, Induces mTOR Inhibition and Cell-Cycle Arrest through REDD1. Cancer Res. 2011, 71, 4366. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Yi, Y.; Liu, Y.; Liu, X.; Keller, E.T.; Qian, C.-N.; Zhang, J.; Lu, Y. Metformin targets multiple signaling pathways in cancer. Chin. J. Cancer 2017, 36, 17. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, E. Rag GTPase in amino acid signaling. Amino Acids 2016, 48, 915–928. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Frank, A.R.; Jewell, J.L. Amino acid and small GTPase regulation of mTORC1. Cell. Logist. 2017, 7, e1378794. [Google Scholar] [CrossRef]
- Nicastro, R.; Sardu, A.; Panchaud, N.; De Virgilio, C. The Architecture of the Rag GTPase Signaling Network. Biomolecules 2017, 7, 48. [Google Scholar] [CrossRef]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Fu, K.; Jiang, C.; Huang, X.; Chan, W.C.; McKeithan, T. The Combination Of 2-DG and Metformin Inhibits The mTORC1 Pathway and Suppresses Aggressive B Cell Lymphoma Growth and Survival. Blood 2013, 122, 1665. [Google Scholar]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Saengboonmee, C.; Seubwai, W.; Cha’on, U.; Sawanyawisuth, K.; Wongkham, S.; Wongkham, C. Metformin Exerts Antiproliferative and Anti-metastatic Effects Against Cholangiocarcinoma Cells by Targeting STAT3 and NF-kB. Anticancer Res. 2017, 37, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Samuel, M.S.; Abotaleb, M.; Cheema, S.; Mamtani, R.; Büsselberg, D. The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers. Cancers 2018, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.O.; Niraula, S.; Chang, M.C.; Done, S.J.; Ennis, M.; McCready, D.R.; Leong, W.L.; Escallon, J.M.; Reedijk, M.; Goodwin, P.J.; et al. Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: A prospective window of opportunity neoadjuvant study. Breast Cancer Res. 2015, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Kuppusamy, G. Metformin in breast cancer: Preclinical and clinical evidence. Curr. Probl. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Oliveras-Ferraros, C.; Cufi, S.; Corominas-Faja, B.; Joven, J.; Martin-Castillo, B.; Vazquez-Martin, A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle 2012, 11, 2782–2792. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.; Fan, Z.; Edgerton, S.M.; Liu, B.; Deng, X.S.; Arnadottir, S.S.; Richer, J.K.; Anderson, S.M.; Thor, A.D. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle 2013, 12, 3759–3769. [Google Scholar] [CrossRef]
- Silvestri, A.; Palumbo, F.; Rasi, I.; Posca, D.; Pavlidou, T.; Paoluzi, S.; Castagnoli, L.; Cesareni, G. Metformin Induces Apoptosis and Downregulates Pyruvate Kinase M2 in Breast Cancer Cells Only When Grown in Nutrient-Poor Conditions. PLoS ONE 2015, 10, e0136250. [Google Scholar] [CrossRef]
- Zordoky, B.N.; Bark, D.; Soltys, C.L.; Sung, M.M.; Dyck, J.R. The anti-proliferative effect of metformin in triple-negative MDA-MB-231 breast cancer cells is highly dependent on glucose concentration: Implications for cancer therapy and prevention. Biochim. Biophys. Acta 2014, 1840, 1943–1957. [Google Scholar] [CrossRef]
- Queiroz, E.A.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.; Fortes, Z.B.; Khaper, N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fan, Z.; Edgerton, S.M.; Deng, X.S.; Alimova, I.N.; Lind, S.E.; Thor, A.D. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 2009, 8, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Wahdan-Alaswad, R.S.; Edgerton, S.M.; Salem, H.S.; Thor, A.D. Metformin Targets Glucose Metabolism in Triple Negative Breast Cancer. J. Oncol. Transl. Res. 2018, 4, 129. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-C.; Li, G.-Y.; Wang, B.; Han, S.-X.; Sun, X.; Jiang, Y.-N.; Shen, Y.-W.; Zhou, C.; Feng, J.; Lu, S.-Y.; et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-B downregulation. J. Exp. Clin. Cancer Res. 2019, 38, 235. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Gravel, S.-P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef]
- Xu, H.; Aldrich, M.C.; Chen, Q.; Liu, H.; Peterson, N.B.; Dai, Q.; Levy, M.; Shah, A.; Han, X.; Ruan, X.; et al. Validating drug repurposing signals using electronic health records: A case study of metformin associated with reduced cancer mortality. J. Am. Med. Inf. Assoc. 2015, 22, 179–191. [Google Scholar] [CrossRef]
- Hatoum, D.; McGowan, E.M. Recent Advances in the Use of Metformin: Can Treating Diabetes Prevent Breast Cancer? Biomed. Res. Int. 2015, 2015, 548436. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; McTiernan, A.; Wactawski-Wende, J.; Manson, J.E.; Aragaki, A.K.; Rohan, T.; Ipp, E.; Kaklamani, V.G.; Vitolins, M.; Wallace, R.; et al. Diabetes, metformin, and breast cancer in postmenopausal women. J. Clin. Oncol. 2012, 30, 2844–2852. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Meric-Bernstam, F. Metformin: A therapeutic opportunity in breast cancer. Clin. Cancer Res. 2010, 16, 1695–1700. [Google Scholar] [CrossRef]
- Campagnoli, C.; Berrino, F.; Venturelli, E.; Abba, C.; Biglia, N.; Brucato, T.; Cogliati, P.; Danese, S.; Donadio, M.; Zito, G.; et al. Metformin decreases circulating androgen and estrogen levels in nondiabetic women with breast cancer. Clin. Breast Cancer 2013, 13, 433–438. [Google Scholar] [CrossRef]
- Campagnoli, C.; Pasanisi, P.; Abba, C.; Ambroggio, S.; Biglia, N.; Brucato, T.; Colombero, R.; Danese, S.; Donadio, M.; Venturelli, E.; et al. Effect of different doses of metformin on serum testosterone and insulin in non-diabetic women with breast cancer: A randomized study. Clin. Breast Cancer 2012, 12, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Agbor-Tarh, D.; Bradbury, I.; Di Cosimo, S.; Azim, H.A., Jr.; Fumagalli, D.; Sarp, S.; Wolff, A.C.; Andersson, M.; Kroep, J.; et al. Impact of Diabetes, Insulin, and Metformin Use on the Outcome of Patients With Human Epidermal Growth Factor Receptor 2-Positive Primary Breast Cancer: Analysis From the ALTTO Phase III Randomized Trial. J. Clin. Oncol. 2017, 35, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.H.; Satkunam, M.; Pond, G.R.; Steinberg, G.R.; Blandino, G.; Schunemann, H.J.; Muti, P. Association of Metformin with Breast Cancer Incidence and Mortality in Patients with Type II Diabetes: A GRADE-Assessed Systematic Review and Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2018, 27, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Moulder, S.L.; Gonzalez-Angulo, A.M.; Ensor, J.; Murray, J.L.; Green, M.C.; Koenig, K.B.; Lee, M.H.; Hortobagyi, G.N.; Yeung, S.C. Phase I trial of exemestane in combination with metformin and rosiglitazone in nondiabetic obese postmenopausal women with hormone receptor-positive metastatic breast cancer. Cancer Chemother Pharm. 2013, 71, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Crew, K.D.; Refice, S.; Xiao, T.; Wang, A.; Feldman, S.M.; Taback, B.; Ahmad, A.; Cremers, S.; Hibshoosh, H.; et al. Presurgical trial of metformin in overweight and obese patients with newly diagnosed breast cancer. Cancer Investig. 2014, 32, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Zheng, T.; Hibshoosh, H.; Du, X.; Mundi, P.; Yang, J.; Refice, S.; Feldman, S.M.; Taback, B.; Connolly, E.; et al. Proteomic modulation in breast tumors after metformin exposure: Results from a “window of opportunity” trial. Clin. Transl. Oncol. 2017, 19, 180–188. [Google Scholar] [CrossRef]
- Lord, S.R.; Cheng, W.C.; Liu, D.; Gaude, E.; Haider, S.; Metcalf, T.; Patel, N.; Teoh, E.J.; Gleeson, F.; Bradley, K.; et al. Integrated Pharmacodynamic Analysis Identifies Two Metabolic Adaption Pathways to Metformin in Breast Cancer. Cell Metab. 2018, 28, 679–688.e4. [Google Scholar] [CrossRef]
- Kim, J.; Lim, W.; Kim, E.K.; Kim, M.K.; Paik, N.S.; Jeong, S.S.; Yoon, J.H.; Park, C.H.; Ahn, S.H.; Kim, L.S.; et al. Phase II randomized trial of neoadjuvant metformin plus letrozole versus placebo plus letrozole for estrogen receptor positive postmenopausal breast cancer (METEOR). BMC Cancer 2014, 14, 170. [Google Scholar] [CrossRef]
- Lega, I.C.; Fung, K.; Austin, P.C.; Lipscombe, L.L. Metformin and breast cancer stage at diagnosis: A population-based study. Curr. Oncol. 2017, 24, e85–e91. [Google Scholar] [CrossRef]
- Lega, I.C.; Austin, P.C.; Gruneir, A.; Goodwin, P.J.; Rochon, P.A.; Lipscombe, L.L. Association Between Metformin Therapy and Mortality After Breast Cancer. Diabetes Care 2013, 36, 3018. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Darko, K.O.; Tao, T.; Huang, Y.; Su, Q.; He, C.; Yin, T.; Liu, Z.; Yang, X. Combination of metformin with chemotherapeutic drugs via different molecular mechanisms. Cancer Treat. Rev. 2017, 54, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife 2013, 2, e00747. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Thaker, N.; De, A. Combined 2-deoxy glucose and metformin improves therapeutic efficacy of sodium-iodide symporter-mediated targeted radioiodine therapy in breast cancer cells. Breast Cancer 2015, 7, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Arbe, M.F.; Fondello, C.; Agnetti, L.; Álvarez, G.M.; Tellado, M.N.; Glikin, G.C.; Finocchiaro, L.M.E.; Villaverde, M.S. Inhibition of bioenergetic metabolism by the combination of metformin and 2-deoxyglucose highly decreases viability of feline mammary carcinoma cells. Res. Vet. Sci. 2017, 114, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Bizjak, M.; Malavašič, P.; Dolinar, K.; Pohar, J.; Pirkmajer, S.; Pavlin, M. Combined treatment with Metformin and 2-deoxy glucose induces detachment of viable MDA-MB-231 breast cancer cells in vitro. Sci. Rep. 2017, 7, 1761. [Google Scholar] [CrossRef]
- Wokoun, U.; Hellriegel, M.; Emons, G.; Grundker, C. Co-treatment of breast cancer cells with pharmacologic doses of 2-deoxy-D-glucose and metformin: Starving tumors. Oncol. Rep. 2017, 37, 2418–2424. [Google Scholar] [CrossRef]
- Xue, C.; Wang, C.; Sun, Y.; Meng, Q.; Liu, Z.; Huo, X.; Sun, P.; Sun, H.; Ma, X.; Ma, X.; et al. Targeting P-glycoprotein function, p53 and energy metabolism: Combination of metformin and 2-deoxyglucose reverses the multidrug resistance of MCF-7/Dox cells to doxorubicin. Oncotarget 2017, 8, 8622–8632. [Google Scholar] [CrossRef]
- Soo, J.S.-S.; Ng, C.-H.; Tan, S.H.; Malik, R.A.; Teh, Y.-C.; Tan, B.-S.; Ho, G.-F.; See, M.-H.; Taib, N.A.M.; Yip, C.-H.; et al. Metformin synergizes 5-fluorouracil, epirubicin, and cyclophosphamide (FEC) combination therapy through impairing intracellular ATP production and DNA repair in breast cancer stem cells. Apoptosis 2015, 20, 1373–1387. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Du, C.; Liu, Y.; Zhang, N.; Luo, F. Aspirin and metformin exhibit antitumor activity in murine breast cancer. Oncol. Rep. 2018, 39, 1414–1422. [Google Scholar] [CrossRef]
- Amaral, M.E.A.; Nery, L.R.; Leite, C.E.; de Azevedo Junior, W.F.; Campos, M.M. Pre-clinical effects of metformin and aspirin on the cell lines of different breast cancer subtypes. Investig. New Drugs 2018, 36, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, S.; Zarghami, N. Synergistic Growth Inhibitory Effects of Chrysin and Metformin Combination on Breast Cancer Cells through hTERT and Cyclin D1 Suppression. Asian Pac. J. Cancer Prev. 2018, 19, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Falah, R.R.; Talib, W.H.; Shbailat, S.J. Combination of metformin and curcumin targets breast cancer in mice by angiogenesis inhibition, immune system modulation and induction of p53 independent apoptosis. Ther. Adv. Med. Oncol. 2017, 9, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Farajzadeh, R.; Pilehvar-Soltanahmadi, Y.; Dadashpour, M.; Javidfar, S.; Lotfi-Attari, J.; Sadeghzadeh, H.; Shafiei-Irannejad, V.; Zarghami, N. Nano-encapsulated metformin-curcumin in PLGA/PEG inhibits synergistically growth and hTERT gene expression in human breast cancer cells. Artif. Cells Nanomed. Biotechnol. 2018, 46, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Menendez, J.A. Metformin inhibits RANKL and sensitizes cancer stem cells to denosumab. Cell Cycle 2017, 16, 1022–1028. [Google Scholar] [CrossRef]
- Haugrud, A.B.; Zhuang, Y.; Coppock, J.D.; Miskimins, W.K. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 539–550. [Google Scholar] [CrossRef]
- Hong, S.-E.; Jin, H.-O.; Kim, H.-A.; Seong, M.-K.; Kim, E.-K.; Ye, S.-K.; Choe, T.-B.; Lee, J.K.; Kim, J.-I.; Park, I.-C.; et al. Targeting HIF-1α is a prerequisite for cell sensitivity to dichloroacetate (DCA) and metformin. Biochem. Biophys. Res. Commun. 2016, 469, 164–170. [Google Scholar] [CrossRef]
- Cooper, A.C.; Fleming, I.N.; Phyu, S.M.; Smith, T.A.D. Changes in [18F]Fluoro-2-deoxy-d-glucose incorporation induced by doxorubicin and anti-HER antibodies by breast cancer cells modulated by co-treatment with metformin and its effects on intracellular signalling. J. Cancer Res. Clin. Oncol. 2015, 141, 1523–1532. [Google Scholar] [CrossRef]
- Lu, Z.; Long, Y.; Cun, X.; Wang, X.; Li, J.; Mei, L.; Yang, Y.; Li, M.; Zhang, Z.; He, Q. A size-shrinkable nanoparticle-based combined anti-tumor and anti-inflammatory strategy for enhanced cancer therapy. Nanoscale 2018, 10, 9957–9970. [Google Scholar] [CrossRef]
- Shafiei-Irannejad, V.; Samadi, N.; Salehi, R.; Yousefi, B.; Rahimi, M.; Akbarzadeh, A.; Zarghami, N. Reversion of Multidrug Resistance by Co-Encapsulation of Doxorubicin and Metformin in Poly(lactide-co-glycolide)-d-α-tocopheryl Polyethylene Glycol 1000 Succinate Nanoparticles. Pharm. Res. 2018, 35, 119. [Google Scholar] [CrossRef]
- Shafiei-Irannejad, V.; Samadi, N.; Yousefi, B.; Salehi, R.; Velaei, K.; Zarghami, N. Metformin enhances doxorubicin sensitivity via inhibition of doxorubicin efflux in P-gp-overexpressing MCF-7 cells. Chem. Biol. Drug Des. 2018, 91, 269–276. [Google Scholar] [CrossRef]
- El-Ashmawy, N.E.; Khedr, N.F.; El-Bahrawy, H.A.; Abo Mansour, H.E. Metformin augments doxorubicin cytotoxicity in mammary carcinoma through activation of adenosine monophosphate protein kinase pathway. Tumor Biol. 2017, 39, 1010428317692235. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Zhi, P.; You, J.; Gao, J.-Q. Metformin synergistically suppress tumor growth with doxorubicin and reverse drug resistance by inhibiting the expression and function of P-glycoprotein in MCF7/ADR cells and xenograft models. Oncotarget 2018, 9, 2158–2174. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.-K.I.; Du, X.; Rayannavar, V.; Hopkins, B.; Shaw, J.; Bessler, E.; Thomas, T.; Pires, M.M.; Keniry, M.; Parsons, R.E.; et al. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget 2014, 5, 10503–10517. [Google Scholar] [CrossRef] [PubMed]
- Ariaans, G.; Jalving, M.; Vries, E.G.E.d.; Jong, S.d. Anti-tumor effects of everolimus and metformin are complementary and glucose-dependent in breast cancer cells. BMC Cancer 2017, 17, 232. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, J.; Li, L.; Fan, C.; Sun, Y. Combined Use of Metformin and Everolimus Is Synergistic in the Treatment of Breast Cancer Cells. Oncol. Res. 2014, 22, 193–201. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhu, W.; Yang, B.; Chai, R.; Liu, T.; Li, F.; Ren, G.; Ji, S.; Liu, S.; Li, G. The co-treatment of metformin with flavone synergistically induces apoptosis through inhibition of PI3K/AKT pathway in breast cancer cells. Oncol. Lett. 2018, 15, 5952–5958. [Google Scholar] [CrossRef]
- Bojková, B.; Kajo, K.; Kisková, T.; Kubatka, P.; Žúbor, P.; Solár, P.; Péč, M.; Adamkov, M. Metformin and melatonin inhibit DMBA-induced mammary tumorigenesis in rats fed a high-fat diet. Anti-Cancer Drugs 2018, 29, 128–135. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, S.; Zong, Q.; Yin, Z. Co-delivery of Metformin and Paclitaxel Via Folate-Modified pH-Sensitive Micelles for Enhanced Anti-tumor Efficacy. AAPS PharmSciTech 2018, 19, 2395–2406. [Google Scholar] [CrossRef]
- Chatran, M.; Pilehvar-Soltanahmadi, Y.; Dadashpour, M.; Faramarzi, L.; Rasouli, S.; Jafari-Gharabaghlou, D.; Asbaghi, N.; Zarghami, N. Synergistic anti-proliferative effects of metformin and silibinin combination on T47D breast cancer cells via hTERT and cyclin D1 inhibition. Drug Res. 2018, 68, 710–716. [Google Scholar] [CrossRef]
- Yeo, S.K.; Paul, R.; Haas, M.; Wang, C.; Guan, J.-L. Improved efficacy of mitochondrial disrupting agents upon inhibition of autophagy in a mouse model of BRCA1-deficient breast cancer. Autophagy 2018, 14, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guo, Y.; Chen, S.; Zhong, C.; Xue, Y.; Zhang, Y.; Lai, X.; Wei, Y.; Yu, S.; Zhang, J.; et al. Metformin enhances tamoxifen-mediated tumor growth inhibition in ER-positive breast carcinoma. BMC Cancer 2014, 14, 172. [Google Scholar] [CrossRef] [PubMed]
- Banala, V.T.; Sharma, S.; Barnwal, P.; Urandur, S.; Shukla, R.P.; Ahmad, N.; Mittapelly, N.; Pandey, G.; Dwivedi, M.; Kalleti, N.; et al. Synchronized Ratiometric Codelivery of Metformin and Topotecan through Engineered Nanocarrier Facilitates In Vivo Synergistic Precision Levels at Tumor Site. Adv. Healthc. Mater. 2018, 7, e1800300. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.S.; Li, H.X.; Li, C.Y.; Zhang, S.Y.; Chen, J.; Wang, Q.L.; Gao, J.M.; Liang, J.Q.; Gao, M.T.; Wu, Y.J. Synergistic antitumor activity of vitamin D3 combined with metformin in human breast carcinoma MDA-MB-231 cells involves m-TOR related signaling pathways. Pharmazie 2015, 70, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Gao, C.; Guo, M.; Law, B.Y.K.; Xu, Y. Effects of metformin treatment on radiotherapy efficacy in patients with cancer and diabetes: A systematic review and meta-analysis. Cancer Manag. Res. 2018, 10, 4881–4890. [Google Scholar] [CrossRef]
- Ferro, A.; Goyal, S.; Kim, S.; Wu, H.; Taunk, N.K.; Schiff, D.; Pirlamarla, A.; Haffty, B.G. Evaluation of Diabetic Patients with Breast Cancer Treated with Metformin during Adjuvant Radiotherapy. Int. J. Breast Cancer 2013, 2013, 7. [Google Scholar] [CrossRef]
- Keywan, M.; Dheyauldeen, S.; Ahmed, E.M.; Masoud, N.; Bagher, F. Metformin as a Radiation Modifier; Implications to Normal Tissue Protection and Tumor Sensitization. Curr. Clin. Pharmacol. 2019, 14, 41–53. [Google Scholar] [CrossRef]
- Brown, S.L.; Kolozsvary, A.; Isrow, D.M.; Al Feghali, K.; Lapanowski, K.; Jenrow, K.A.; Kim, J.H. A Novel Mechanism of High Dose Radiation Sensitization by Metformin. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Suissa, S.; Azoulay, L. Metformin and Cancer: Mounting Evidence Against an Association. Diabetes Care 2014, 37, 1786. [Google Scholar] [CrossRef]
- Mamtani, R.; Pfanzelter, N.; Haynes, K.; Finkelman, B.S.; Wang, X.; Keefe, S.M.; Haas, N.B.; Vaughn, D.J.; Lewis, J.D. Incidence of Bladder Cancer in Patients With Type 2 Diabetes Treated With Metformin or Sulfonylureas. Diabetes Care 2014, 37, 1910. [Google Scholar] [CrossRef][Green Version]
- Samuel, S.M.; Satheesh, N.J.; Ghosh, S.; Büsselberg, D.; Majeed, Y.; Ding, H.; Triggle, C.R. Treatment with a Combination of Metformin and 2-Deoxyglucose Upregulates Thrombospondin-1 in Microvascular Endothelial Cells: Implications in Anti-Angiogenic Cancer Therapy. Cancers 2019, 11, 1737. [Google Scholar] [CrossRef]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.M.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS ONE 2013, 8, e71583. [Google Scholar] [CrossRef] [PubMed]
- Suissa, S.; Azoulay, L. Metformin and the Risk of Cancer. Diabetes Care 2012, 35, 2665. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Metformin and cancer: Doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 2010, 9, 1057–1064. [Google Scholar] [CrossRef]
- Aljofan, M.; Riethmacher, D. Anticancer activity of metformin: A systematic review of the literature. Future Sci. OA 2019, 5, FSO410. [Google Scholar] [CrossRef]
- Scherbakov, A.M.; Sorokin, D.V.; Tatarskiy, V.V., Jr.; Prokhorov, N.S.; Semina, S.E.; Berstein, L.M.; Krasil’nikov, M.A. The phenomenon of acquired resistance to metformin in breast cancer cells: The interaction of growth pathways and estrogen receptor signaling. IUBMB Life 2016, 68, 281–292. [Google Scholar] [CrossRef]
- National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/diagnosis-staging/diagnosis/tumor-markers-fact-sheet (accessed on 15 January 2018).
- National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/causes-prevention/genetics/brca-fact-sheet (accessed on 15 January 2018).
- Li, Y.; Li, J.; Wang, Y.; Zhang, Y.; Chu, J.; Sun, C.; Fu, Z.; Huang, Y.; Zhang, H.; Yuan, H.; et al. Roles of cancer/testis antigens (CTAs) in breast cancer. Cancer Lett. 2017, 399, 64–73. [Google Scholar] [CrossRef]
- Fratta, E.; Coral, S.; Covre, A.; Parisi, G.; Colizzi, F.; Danielli, R.; Nicolay, H.J.; Sigalotti, L.; Maio, M. The biology of cancer testis antigens: Putative function, regulation and therapeutic potential. Mol. Oncol. 2011, 5, 164–182. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Andersen, M.H.; Ditzel, H.J. Oncogenic cancer/testis antigens: Prime candidates for immunotherapy. Oncotarget 2015, 6, 15772–15787. [Google Scholar] [CrossRef]
- Ademuyiwa, F.O.; Bshara, W.; Attwood, K.; Morrison, C.; Edge, S.B.; Karpf, A.R.; James, S.A.; Ambrosone, C.B.; O’Connor, T.L.; Levine, E.G.; et al. NY-ESO-1 cancer testis antigen demonstrates high immunogenicity in triple negative breast cancer. PLoS ONE 2012, 7, e38783. [Google Scholar] [CrossRef]
- Badovinac Crnjevic, T.; Spagnoli, G.; Juretic, A.; Jakic-Razumovic, J.; Podolski, P.; Saric, N. High expression of MAGE-A10 cancer-testis antigen in triple-negative breast cancer. Med. Oncol. 2012, 29, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Balafoutas, D.; zur Hausen, A.; Mayer, S.; Hirschfeld, M.; Jaeger, M.; Denschlag, D.; Gitsch, G.; Jungbluth, A.; Stickeler, E. Cancer testis antigens and NY-BR-1 expression in primary breast cancer: Prognostic and therapeutic implications. BMC Cancer 2013, 13, 271. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Viale, G.; Ghioni, M.; Jungbluth, A.A.; Bagnardi, V.; Spagnoli, G.C.; Neville, A.M.; Nole, F.; Rotmensz, N.; Goldhirsch, A. Cancer-testis antigen expression in triple-negative breast cancer. Ann. Oncol. 2011, 22, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, L.; Pedretti, E.; Figueroa, J.A.; Chiaramonte, R.; Colombo, M.; Chapman, C.; Grizzi, F.; Patrinicola, F.; Kast, W.M.; Nguyen, D.D.; et al. Cancer testis antigen Sperm Protein 17 as a new target for triple negative breast cancer immunotherapy. Oncotarget 2017, 8, 74378–74390. [Google Scholar] [CrossRef] [PubMed]
- Rastgoosalami, M.; Memar, B.; Aledavood, S.A.; Fanipakdel, A. Evaluation of MAGE-1 Cancer-Testis Antigen Expression in Invasive Breast Cancer and its Correlation with Prognostic Factors. Iran. J. Cancer Prev. 2016, 9, e4404. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Jagadish, N.; Gupta, A.; Bhatnagar, A.; Suri, A. A novel cancer testis antigen, A-kinase anchor protein 4 (AKAP4) is a potential biomarker for breast cancer. PLoS ONE 2013, 8, e57095. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, M.; Bolton, L.M.; Johnson, P.; Elliott, T.; Murray, N. Breast cancer is a promising target for vaccination using cancer-testis antigens known to elicit immune responses. Breast Cancer Res. 2007, 9, R46. [Google Scholar] [CrossRef]
- Soffer, D.; Shi, J.; Chung, J.; Schottinger, J.E.; Wallner, L.P.; Chlebowski, R.T.; Lentz, S.E.; Haque, R. Metformin and breast and gynecological cancer risk among women with diabetes. BMJ Open Diabetes Res. Care 2015, 3, e000049. [Google Scholar] [CrossRef]
- Camacho, L.; Dasgupta, A.; Jiralerspong, S. Metformin in breast cancer - an evolving mystery. Breast Cancer Res. 2015, 17, 88. [Google Scholar] [CrossRef]
- Podo, F.; Buydens, L.M.C.; Degani, H.; Hilhorst, R.; Klipp, E.; Gribbestad, I.S.; Van Huffel, S.; van Laarhoven, H.W.; Luts, J.; Monleon, D.; et al. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 2010, 4, 209–229. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Serial No:. | Type of Cancer | Total Number of Registered Trials | Completed | Active, not Recruiting | Active, Recruiting | Terminated | Withdrawn | Unknown Status # |
|---|---|---|---|---|---|---|---|---|
| 1 | Breast Cancers | 44 | 14 | 4 | 14 | 5 | 1 | 6 |
| 2 | Prostate Cancers | 27 | 6 | 5 | 9 | 2 | 4 | 1 |
| 3 | Colorectal Cancers | 20 | 4 | 3 | 7 | 6 | 0 | 0 |
| 4 | Lung Cancers | 19 | 4 | 6 | 3 | 5 | 0 | 1 |
| 5 | Oral Cancers | 5 | 0 | 2 | 3 | 0 | 0 | 0 |
| 6 | Head & Neck Cancers | 9 | 1 | 3 | 3 | 2 | 0 | 0 |
| Serial No: | Name/ID | Trial Phase | Intervention Using Metformin | Objectives | Type of Cancer | Clinicaltrials.Gov ID (NCT Number)/Status Completion Year or Estimated Primary Completion Year | Publications/References |
|---|---|---|---|---|---|---|---|
| 1 | Clinical and Biologic Effects of Metformin in Early Stage Breast Cancer | Phase II | Metformin | To determine if taking metformin prior to surgery can reduce cell proliferation rates in tumor tissue | Breast Cancer | NCT00897884/Completed July 2011 | [136] |
| 2 | Effect of Metformin on Breast Cancer Metabolism | Phase II | Metformin | Measure metformin induced effects in phosphorylation of S6K, 4E-BP-1 and AMPK via immunohistochemical analysis | Breast Cancer | NCT01266486/Completed May 2014 | [160] |
| 3 | Metformin in Breast Cancer, Visualized with Positron Emission Tomography | Phase I | Radiation: 11C-metformin | Metformin uptake in breast cancer | Breast Cancer | NCT02882581/Completed Oct 2017 | No results posted |
| 4 | A Trial of Standard Chemotherapy with Metformin (vs Placebo) in Women with Metastatic Breast Cancer | Phase II | Metformin + standard chemotherapy (containing anthracyclines, platinum, taxanes or capecitabine) vs. placebo + standard chemotherapy | Progression free survival | Metastatic Breast Cancer | NCT01310231/Completed Mar 2018 | [162,163] |
| 5 | Study of Erlotinib and Metformin in Triple Negative Breast Cancer | Phase I | Erlotinib + Metformin | The maximum tolerated dose of metformin in combination with a fixed dose of 150 mg erlotinib daily | Breast Cancer | NCT01650506/Completed June 2016 | No results posted |
| 6 | Neoadjuvant Letrozole Plus Metformin vs Letrozole Plus Placebo for ER-positive Postmenopausal Breast Cancer | Phase II | Letrozole + Metformin vs. Letrozole + Placebo | Clinical response rate | Hormone Receptor Positive Malignant Neoplasm of Breast | NCT01589367/Completed Aug 2018 | [161] |
| 7 | Metformin Hydrochloride vs. Placebo in Overweight or Obese Patients at Elevated Risk for Breast Cancer | Phase I (Early) | Metformin vs. Placebo | Changes in the phosphorylation of proteins after metformin exposure | Breast Cancer and Obesity | NCT01793948/Completed Jan 2018 | No results posted |
| 8 | Efficacy and Safety of Adjuvant Metformin for Operable Breast Cancer Patients | Phase II | Metformin (500 mg/1000 mg) vs. Placebo | Weight loss | Breast Cancer | NCT00909506/Completed Dec 2011 | No results posted |
| 9 | Myocet + Cyclophosphamide + Metformin vs. Myocet + Cyclophosphamide in 1st Line Treatment of HER2 Neg. Metastatic Breast Cancer Patients | Phase II | Metformin + Myocet + Cyclophosphamide vs. Myocet + Cyclophosphamide | Progression-free survival | Human Epidermal Growth Factor 2 Negative Carcinoma of Breast | NCT01885013/Completed May 2015 | No results posted |
| 10 | The Impact of Obesity and Obesity Treatments on Breast Cancer | Phase I | Exemestane vs. Exemestane + Avandamet (Metformin + Rosiglitazone) | Dose-limiting toxicity | Breast Cancer | NCT00933309/Completed Aug 2012 | [157] |
| 11 | Metformin Pre-surgical Pilot Study | Phase II | Metformin | Effects of metformin on AMPK/mTOR signaling pathway | Breast Cancer | NCT00930579/Completed Oct 2011 | [158,159] |
| 12 | I-SPY 2 TRIAL: Neoadjuvant and Personalized Adaptive Novel Agents to Treat Breast Cancer | Phase II | AMG 479 (Ganitumab) + Metformin | Comparing the efficacy of novel drugs in combination with standard chemotherapy with the efficacy of standard therapy alone and identification of improved treatment regimens for subjects on the basis of molecular characteristics (biomarker signatures) pertaining to their disease | Breast Neoplasms/Cancer/Tumors | NCT01042379/Active-Recruiting Dec 2020 | No results posted |
| 13 | Randomized Trial of Neo-adjuvant Chemotherapy with or without Metformin for HER2 Positive Operable Breast Cancer | Phase II | Chemotherapy (Taxotere, Carboplatin, Herceptin + Pertuzumab) vs. Chemotherapy + Metformin | Pathologic complete response | HER2-positive Breast Cancer | NCT03238495/Active-Recruiting Sep 2019 | No results posted |
| 14 | Pre-Surgical Trial of the Combination of Metformin and Atorvastatin in Newly Diagnosed Operable Breast Cancer | Phase I (Early) | Metformin + Atorvastatin (pre-treatment, prior to breast surgery) | Change in the tissue levels of the proliferation marker Ki-67. Tumor proliferation as measured by the natural expression of Ki.67 staining of breast cancer cells | Breast Cancer, Breast Tumors, Cancer of Breast | NCT01980823/Active-Recruiting Dec 2021 | No results posted |
| 15 | Metformin Hydrochloride in Preventing Breast Cancer in Patients with Atypical Hyperplasia or In Situ Breast Cancer | Phase III | Metformin vs. Placebo | Test for the presence or absence of cytological atypia in unilateral or bilateral RPFNA aspirates after 12 and 24 months | Atypical Ductal Breast Hyperplasia, BRCA1 Mutation Carrier, BRCA2 Mutation Carrier, Ductal Breast Carcinoma in Situ, Lobular Breast Carcinoma in Situ | NCT01905046/Active-Recruiting Jun 2022 | No results posted |
| 16 | NeoMET Study in Neoadjuvant Treatment of Breast Cancer | Phase II | Metformin + chemotherapy (docetaxel + epirubicin + cyclophosphamide) vs. chemotherapy | Pathologic complete response rate | Breast Cancer | NCT01929811/Active-Recruiting Sep 2021 | No results posted |
| 17 | Neoadjuvant FDC with Melatonin or Metformin for Locally Advanced Breast Cancer (MBC1) | Phase II | Metformin + chemotherapy (fluoruracil + doxorubicin + cyclophosphamide) vs. Melatonin + chemotherapy vs. Chemotherapy | Response rate and pathomorphological response | Breast Cancer | NCT02506777/Active-Recruiting Aug 2020 | No results posted |
| 18 | Neoadjuvant Toremifene with Melatonin or Metformin for Locally Advanced Breast Cancer (MBC1 | Phase II | Metformin + toremifene vs. Melatonin + toremifene vs. Toremifene | Response rate and pathomorphological response | Breast Cancer | NCT02506790/Active-Recruiting Aug 2020 | No results posted |
| 19 | Metformin Hydrochloride and Doxycycline in Treating Patients with Localized Breast or Uterine Cancer | Phase II | Metformin + doxycycline vs. Doxycycline | Change in the percent of stromal cells expressing Caveolin-1 (Cav1) at an intensity of 1+ or greater as assessed by immunohistochemistry | Breast Carcinoma | NCT02874430/Active-Recruiting Feb 2021 | No results posted |
| 20 | Evaluation of the effect of Metformin on Metastatic Breast Cancer as Adjuvant Treatment | Phase I | Metformin | Disease progression through tumor size | Metastatic Breast Cancer | NCT04143282/Active-Recruiting Dec 2019 | No results posted |
| Conventional Chemotherapeutic Drug or Treatment Modality | Effect and Possible Mechanism of Action | Cells/in vivo Model Used | Ref. |
|---|---|---|---|
| 2-Deoxyglucose (2DG) | Improved the efficacy of sodium-iodide symporter-mediated targeted radioiodine therapy breast cancer cells. | MCF7, MDA-MB-231 | [166] |
| Inhibited decreased bioenergetic metabolism and decreased viability in feline mammary carcinoma cells. | AlRB (HER2+++ve), AlRATN (HER2-ve) | [167] | |
| Induced AMPK dependent detachment and decrease in proliferation of viable breast cancer cells in vitro. | MCF7, MDA-MB-231 | [168] | |
| Significantly reduced cell viability and increased PARP cleavage associated apoptosis. | MDA-MB-231, HCC1806 | [169] | |
| Reversed multidrug resistance, increased doxorubicin (DOX) accumulation, resumed p53 function via inhibition of MDM2 and MDM4 leading to G2/M cell cycle arrest and apoptosis, inhibited glucose uptake, production of lactate, fatty acid and ATP and downregulated the Akt/mTOR pathway. | MCF7/DOX resistant cells | [170] | |
| 5-Fluorouracil, Epirubicin and Cyclophosphamide (FEC) | Metformin synergizes FEC combination therapy via AMPK dependent mechanism in non-stem/parental breast cancer cells, while in cancer stem cells (CSCs) the synergistic effect of the combination treatment was found to be independent of AMPK. In CSCs, while metformin accelerated glucose consumption and lactate production, the production of intracellular ATP was significantly diminished leading to energy stress and impairment of the ability of CSCs to repair the FEC induced DNA damage. | MCF7, MDA-MB-231, MDA-MB-468, HCC1937, SKBR3, T47D, MCF10A, MRC-5 (human embryonic lung fibroblasts), breast CSCs | [171] |
| Aspirin | Aspirin and metformin in combination synergistically activated apoptotic cancer cell death in vitro and reduced tumor growth in vivo facilitated by enhancing the secretion of TGFβ1. Reducing the estrogen levels in circulation or its inhibition maximized the anti-tumor activity of the combinatorial drug. | 4T1, BALB/c mice inoculated with 4T1 cells | [172] |
| Metformin treatment alone altered morphology decreased viability and migration of ER+ve MCF7 cells. The combination of aspirin and metformin synergistically altered morphology decreased viability and migration in TNBC MDA-MB-231 cells. HER2+ve SK-BR-3 cells showed a partial response to monotherapy (aspirin or metformin) and combinatorial therapy (aspirin and metformin). | MCF7, SK-BR-3, MDA-MB-231, | [173] | |
| Chrysin | Synergistic growth inhibitory effects due to suppression of hTERT and cyclin D1 gene expression | T47D | [174] |
| Curcumin | Inhibition of tumor proliferation and growth associated with reduced VEGF expression and angiogenesis, induction of p53 independent apoptosis, and activation of Th2 related immune response with no toxicity. | EMT6/P cells, BALB/c mice inoculated with EMT6/P cells | [175] |
| Combination of PEGylated PLGA nanoparticle co-encapsulated metformin and curcumin exhibited dosage dependent toxicity and synergistic antiproliferative effect causing significant cell cycle/growth arrest in the cancer cells. The hTERT gene expression was significantly inhibited in cells treated with the nano-formulation of metformin-curcumin when compared to delivery of either metformin or curcumin alone. | T47D | [176] | |
| Denosumab | BRCA1 haplo-insufficiency driven RANKL gene overexpression was hampered by metformin treatment and disrupted the RANKL mediated auto-regulatory feedback in CSCs thereby sensitizing the CSC to denosumab and synergistically reducing the cancer initiating cell population and their capacity for self-renewal. | Breast CSCs, MDA-MB-436 | [177] |
| Dichloroacetic Acid (DCA) | DCA and metformin when used in combination synergistically induced caspase dependent apoptosis in cancer cells. Metformin-associated oxidative stress-induced damage was amplified by DCA treatment associated pyruvate dehydrogenase kinase 1 inhibition thereby reducing metformin mediated lactate production. | MCF7, T47D, MCF10A | [178] |
| The combinatorial therapeutic strategy using DCA and metformin inhibited key glycolytic enzymes—hexokinase 2, lactate dehydrogenase A, and enolase 1. An activation of HIF1α abolished the effect of the combination therapy and reversed the inhibition on the expression of the glycolytic enzymes and reduced cell death. | MCF7, H1299, HDF, MCF10A | [179] | |
| Doxorubicin (DOX) + 2-Deoxy-2-(F)-Fluoro-d-Glucose (2FDG) | Metformin treatment increased pAMPK levels while the levels of pAkt and pERK decreased. 2FDG incorporation and phosphorylation increased upon metformin treatment. | MDA-MB-453, MDA-MB-468, SK-BR-3, BT474 | [180] |
| DOX | Nanoparticle co-encapsulated metformin and DOX achieved good tumor penetration, inhibited NF-κB activity, and decreased TNFα and IL6 expressions leading to the significant decrease in cancer cell proliferation. The nano-formulation of metformin and DOX showed a therapeutic effect in the treatment of lipopolysaccharide (LPS)-induced pulmonary metastasis model of murine 4T1 cells. | 4T1, BALB/c mice inoculated with 4T1 cells | [181] |
| Nanoparticle co-encapsulated metformin and DOX (dual drug loaded) treatment showed increased toxicity and apoptotic cell death in DOX resistant MCF7 cells. The enhanced efficiency and cytotoxicity were attributed the to the intracellular accumulation of the drugs via enhanced cellular uptake and reduction in drug efflux leading to significant energy stress (reduced cellular ATP) and inhibition of multidrug resistance (MDR) mediating P-glycoprotein (P-gp). | MCF7, MCF7/DOX resistant cells | [182] | |
| Metformin and DOX dual drug-loaded nanoparticles effectively reduced P-gp expression and activity, increased energy stress as evidenced by reduced intracellular ATP levels, and sensitized the cells to DOX induced apoptotic cell death. | MCF7, MCF7/DOX resistant cells | [183] | |
| Metformin treatment associated AMPK dependent anti-tumor effect was observed in addition to the inhibition of NF-κB and cyclin D1 gene expression. Combinatorial treatment was found to be more effective in decreasing tumor volume and improve overall rate of survival in the animals. Higher rates of apoptosis were observed in histopathological samples derived from animals to which the combination treatment was administered. Metformin treatment-associated reduction in the P-gp expression and elimination of Ki-67 positive cancer cells were observed in MCF7/ADR tumor xenografts. | The Ehrlich ascites carcinoma cells (derived from mouse breast adenocarcinoma cells) were implanted and allowed to multiply in the peritoneal cavity of Swiss albino mice. Solid Ehrlich carcinoma were derived by implanting EAC cells subcutaneously in Swiss albino mice. | [184] | |
| The combinatorial treatment synergistically reversed DOX resistance both in vitro and in vivo. Metformin inhibited tumor growth. The cytotoxic effects of metformin were enhanced by increasing the levels of ROS while the levels of ATP levels depleted. | MCF7/ADR cells-DOX resistant cells, subcutaneously implanted MCF7/ADR cells in nu/nu mice | [185] | |
| Erlotinib | The combination treatment synergistically induced apoptotic cell death and reduced the phosphorylation of EGFR, Akt, S6, and 4EBP1, prevented colony formation and inhibited mammosphere outgrowth. | MDA-MB-468, MDA-MB-157, MDA-MB-435S, MDA-MB-436, MDA-MB-231, MX-1, MCF7, BT20, L56Br-C1, CAOV-3, HCC1143, HCC1806, HCC1937, HCC1987, HCC70, HCC38, BT549, mice implanted with MDA-MB-468 cells into the mammary fat pad | [186] |
| Everolimus | Metformin-induced additive effects were observed when used as a co-treatment with everolimus and inhibited cell proliferation and colony formation ability. The additive effect of the combinatorial treatment was also related to the inhibition of mitochondrial respiration and mTOR growth signaling. | MCF7, MDA-MB-231, T47D | [187] |
| Inhibition of cell proliferation and tumor growth was observed both in cultures and mouse xenograft models treated with a combination of everolimus and metformin. Significant decrease in the levels of phosphorylated S6 ribosomal protein and 4E-BP1 was observed upon combination treatment. | HCC1428, MDA-MB-468, BT549, BALB/c mice inoculated with HCC1428 cells | [188] | |
| Flavone | Significant inhibition of cell viability, increased apoptosis, decrease in the expression of murine double minute X (MDMX), and activation of p53 via the PI3K/Akt pathway was observed in the combination drutreated cells. Apoptosis was mediated by decrease in Bcl2 and increase in the levels of Bax and caspase 3. | MCF10A, MCF7, MDA-MB-231 | [189] |
| Melatonin | DMBA induced tumor incidence, tumor growth, and volume were reduced by the combination treatment. The apoptotic stimulation observed in the cancer cells was attributed to the activation of caspase 3. | 7, 12-dimethylbenz[a]anthracene (DMBA) induced in vivo rat model of breast cancer | [190] |
| Paclitaxel (PTX) | Co-delivery of PTX and metformin using a folate-modified amphiphilic and biodegradable biomaterial synergistically decreased cell proliferation and induced apoptosis through the toll-like receptor (TLR) signaling via the modulation of the TLR-MyD88-ERK pathway (responsible for tumor growth, progression, metastasis, and drug resistance). | 4T1 cells, BALB/c mice inoculated with 4T1 cells | [191] |
| Silibinin | Synergistic effect on growth inhibition of cancer cells was observed when silibinin and metformin were used in combination. Downregulation of hTERT and cyclin D1 was observed with the combinatorial therapeutic approach. | T47D | [192] |
| Spautin-1 | Deletion of the essential autophagy gene, Rb1cc1, suppressed tumorigenesis in BRCA1-deficient mice, while tumor growth and distribution of histological subtypes were not affected by loss of Rb1cc1. Co-treatment using spautin-1 (autophagy inhibitor) and metformin (mitochondrial complex-1 inhibitor) efficiently reduced the oxidative respiratory capacity, colony forming ability, and tumor growth. | Tumor cells derived from BRCA1-deficient tumors, Rb1cc1+/+ brca1F/F trp53F/F K14-Cre mice, Rb1cc1F/+ brca1F/F trp53F/F K14-Cre mice and Rb1cc1F/F brca1F/F trp53F/F K14-Cre mice, athymic nude-Foxn1nu mice transplanted with tumor cells derived from BRCA1-deficient tumors | [193] |
| Tamoxifen | The dosage of tamoxifen required for growth inhibition of cells was much lower when combined with metformin than when used as a monotherapy. The combination treatment inhibited cellular proliferation, DNA replication activity, colony formation, and activated apoptotic cell death in ER+ve breast cancer cells. The involvement of the Bax/Bcl2 apoptotic pathway and the AMPK/mTOR/p70S6 growth pathways were implicated in the beneficial effects of this combinatorial therapy approach. | MCF7, ZR-75-1 | [194] |
| Topotecan | Metformin and topotecan dual drug carrier nanoparticles were found to be synergistically cytotoxic for the breast cancer cells, effectively promoting cell death via mitochondrial membrane depolarization and cell cycle arrest. | MDA-MB-231, 4T1 | [195] |
| Vitamin D3 | The combination treatment of metformin and vitamin D3 in synergistically inhibited cell proliferation and activated apoptosis in breast cancer cells. Mechanisms involving activation of AMPK, upregulation of Bax, cleavage of caspase 3, and inhibition of pBcl2, c-Myc, pIGF-IR, pmTOR, pP70S6K, and pS6 were implicated in anti-cancer activity of the combinatorial treatment. | MDA-MB-231 | [196] |
| Radiation | A higher tumor response to radiation was observed in diabetic breast cancer patients who received metformin and partly yielded survival benefits. | Meta-analysis/Clinical | [197] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samuel, S.M.; Varghese, E.; Kubatka, P.; Triggle, C.R.; Büsselberg, D. Metformin: The Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer. Biomolecules 2019, 9, 846. https://doi.org/10.3390/biom9120846
Samuel SM, Varghese E, Kubatka P, Triggle CR, Büsselberg D. Metformin: The Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer. Biomolecules. 2019; 9(12):846. https://doi.org/10.3390/biom9120846
Chicago/Turabian StyleSamuel, Samson Mathews, Elizabeth Varghese, Peter Kubatka, Chris R. Triggle, and Dietrich Büsselberg. 2019. "Metformin: The Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer" Biomolecules 9, no. 12: 846. https://doi.org/10.3390/biom9120846
APA StyleSamuel, S. M., Varghese, E., Kubatka, P., Triggle, C. R., & Büsselberg, D. (2019). Metformin: The Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer. Biomolecules, 9(12), 846. https://doi.org/10.3390/biom9120846