Lanatoside C Induces G2/M Cell Cycle Arrest and Suppresses Cancer Cell Growth by Attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR Signaling Pathways



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Chemicals
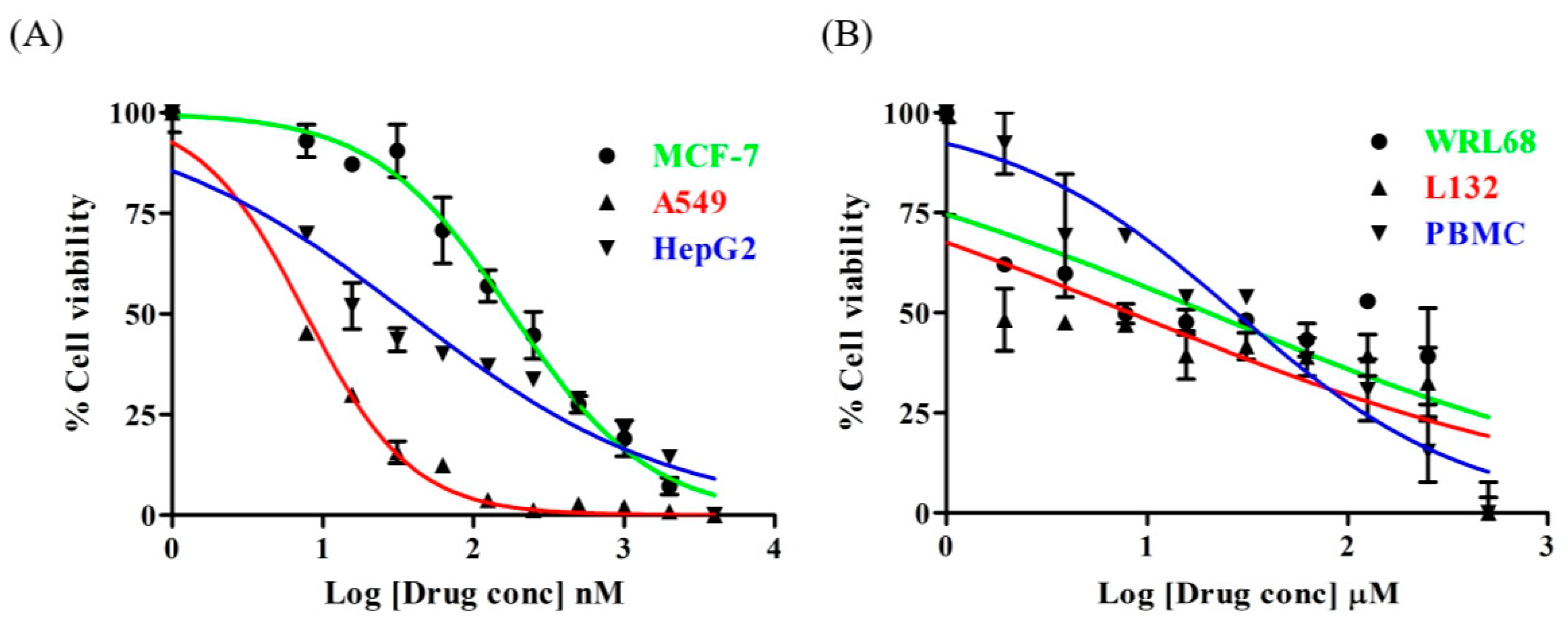
2.2. Cytotoxicity Assay
2.3. DNA Damage Assay
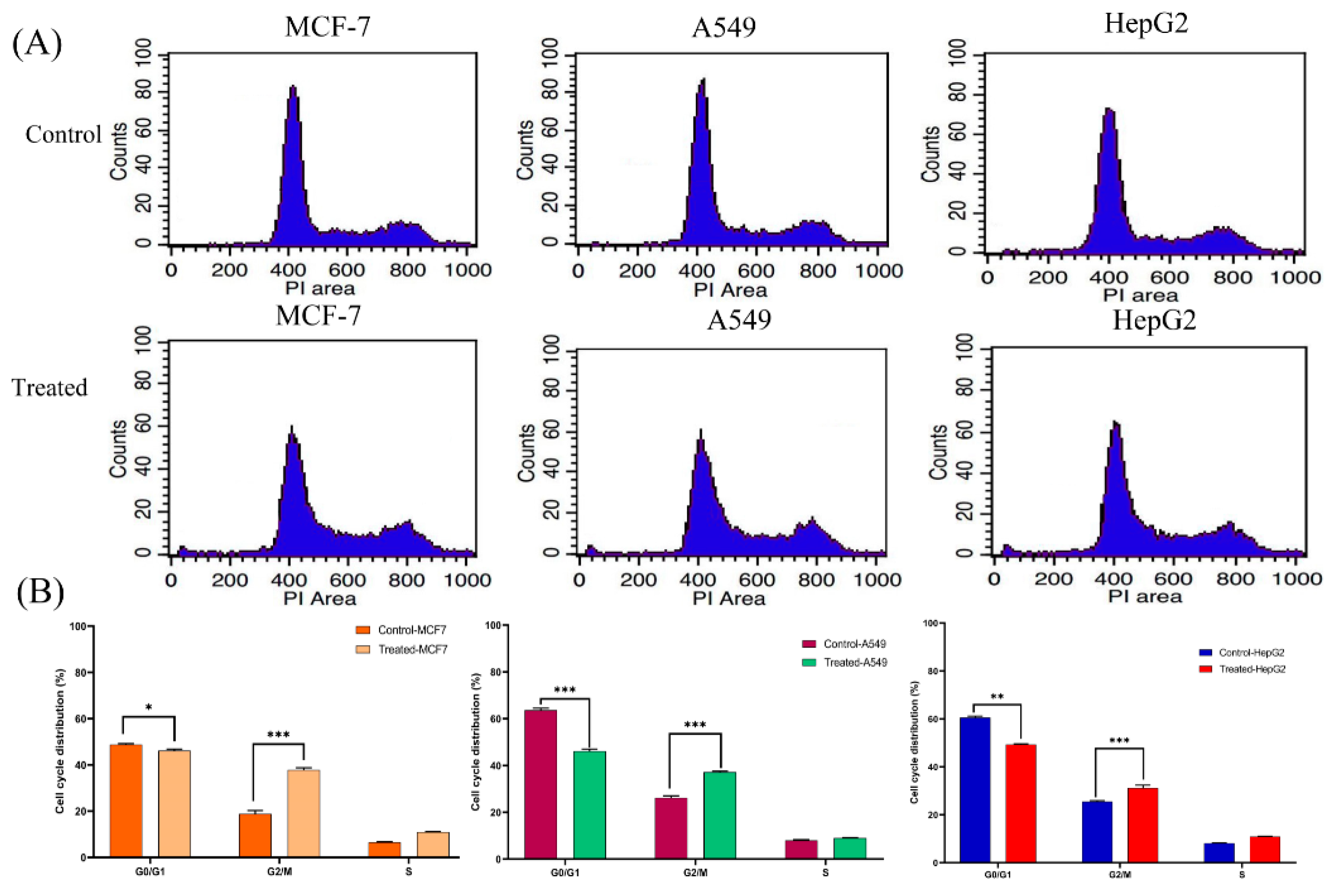
2.4. Cell Cycle Analysis By Flow Cytometry
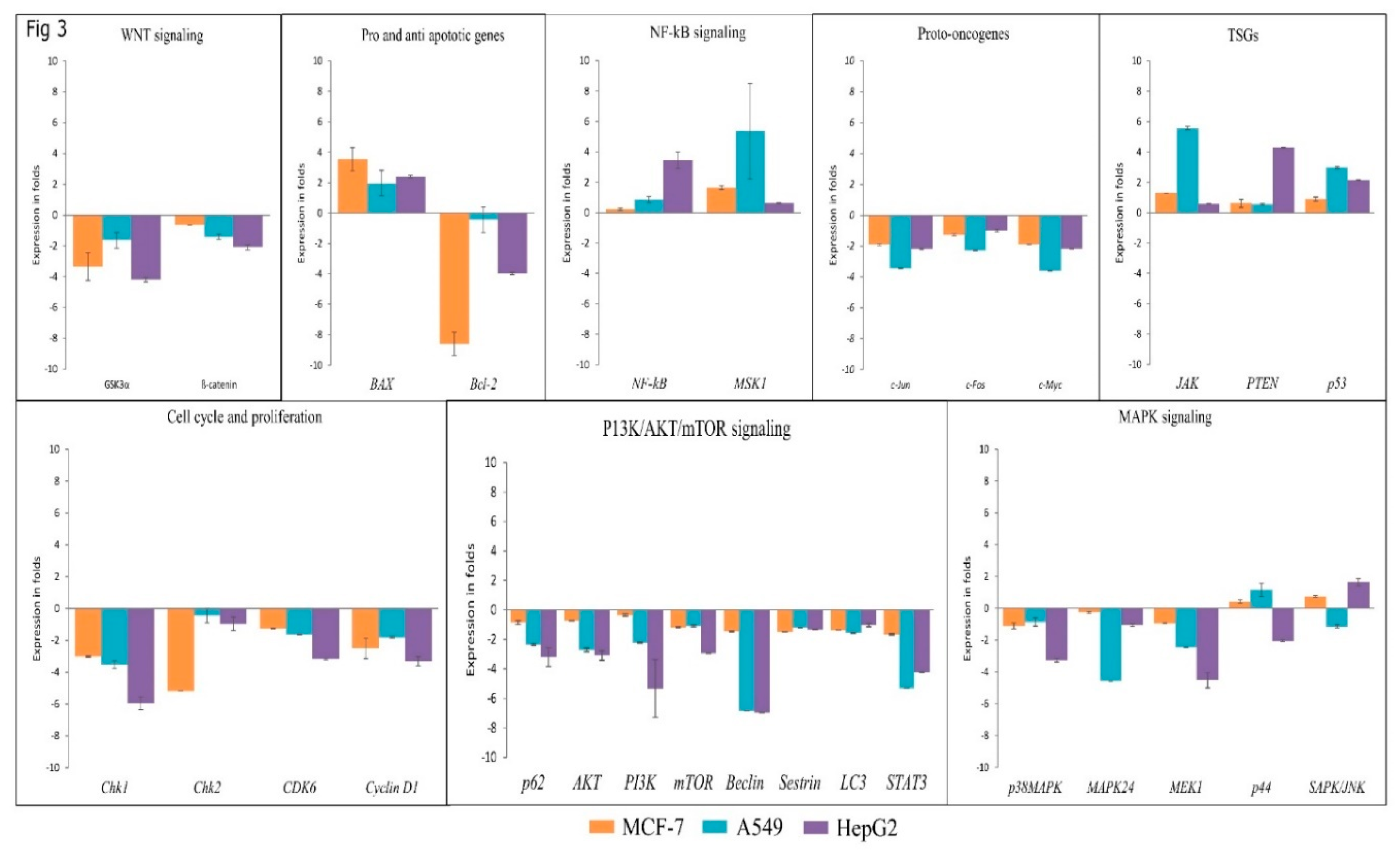
2.5. Real-Time PCR Analysis
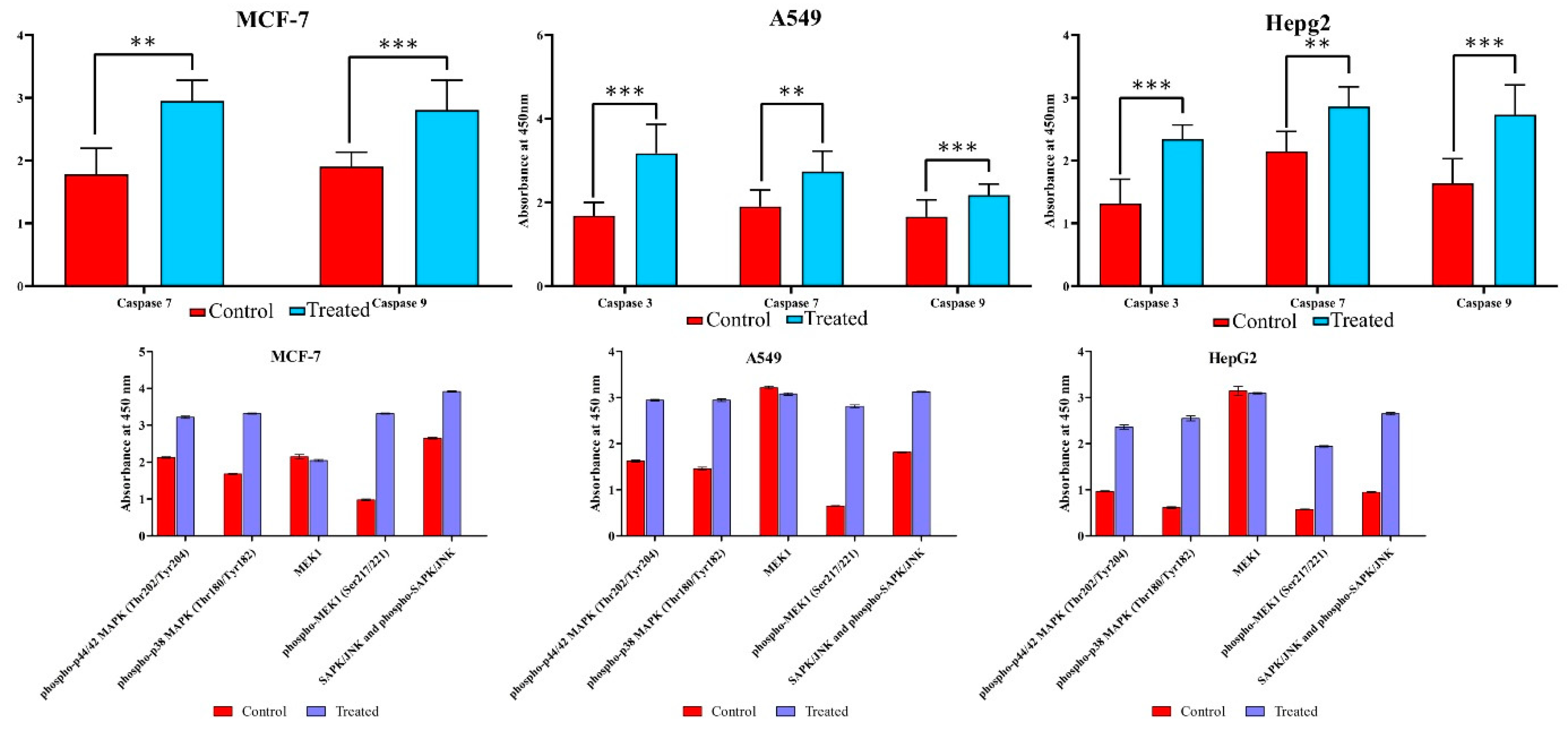
2.6. ELISA (Enzyme-Linked Immunosorbent Assay)
2.7. Immunoblotting Studies
2.8. Immunostaining Studies
2.9. In-silico Docking Analysis
2.10. Statistical Analysis
3. Results
3.1. Lanatoside C Exhibits Cytotoxic Effects Only on Cancer Cells
3.2. Lanatoside C Treatment Induces DNA Damage in Cancer Cell Lines
3.3. Lanatoside C Treatment Increases the Percentage of G2/GM and S Phase Cells in Cancer Cell Lines
3.4. Lanatoside C Inhibits Expression of G2/M Cell Cycle Regulator, MAPK, and PI3K/AKT Pathway Genes
3.5. Lanatoside C Down-Regulates BCL-2 and Up-Regulates BAX to Induce Apoptosis in Cancer Cell Lines
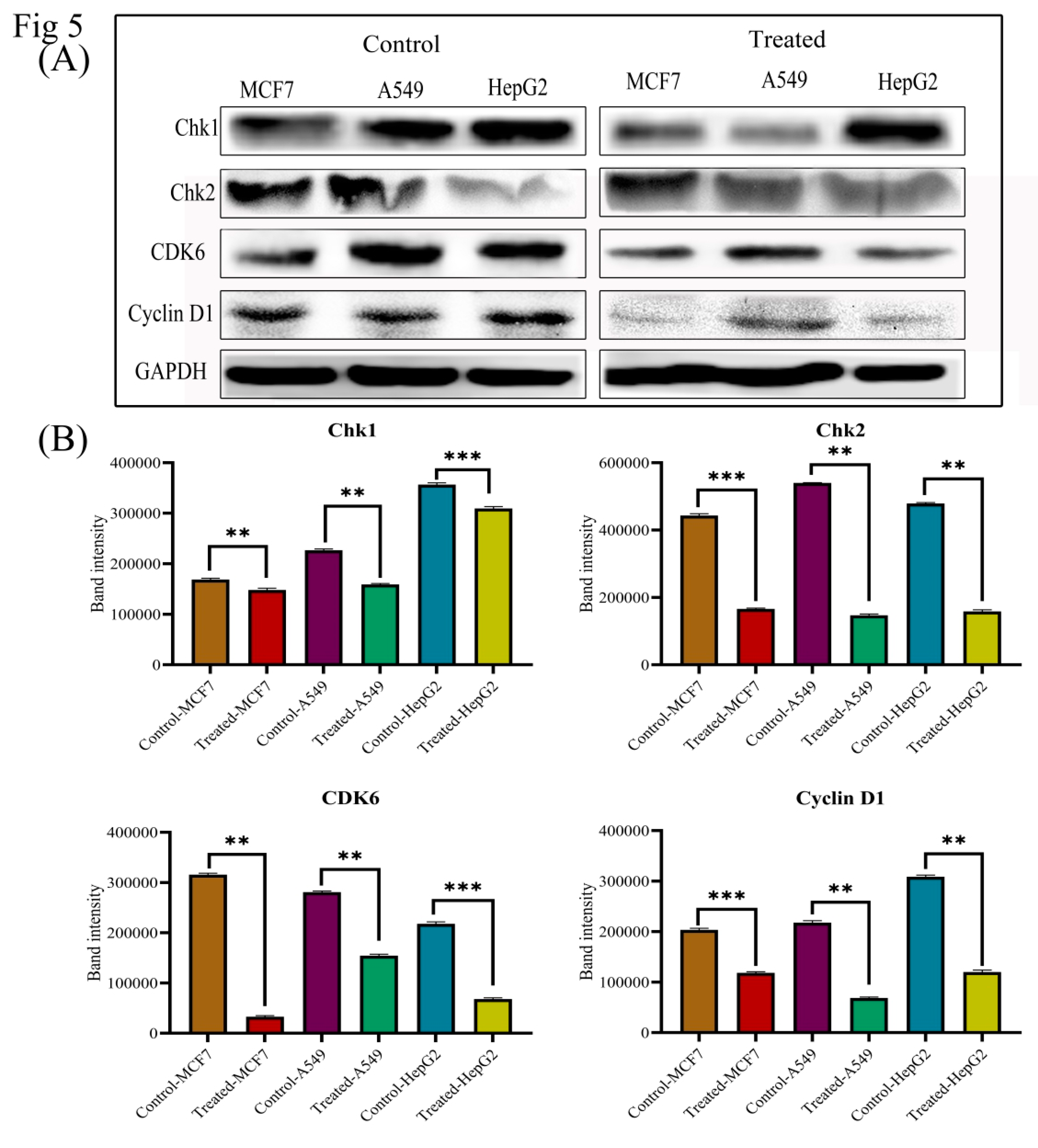
3.6. Lanatoside C Down-Regulates Cell Cycle Checkpoint Protein’s Expression to Exhibit Growth Arrest in Cancer Cell Lines
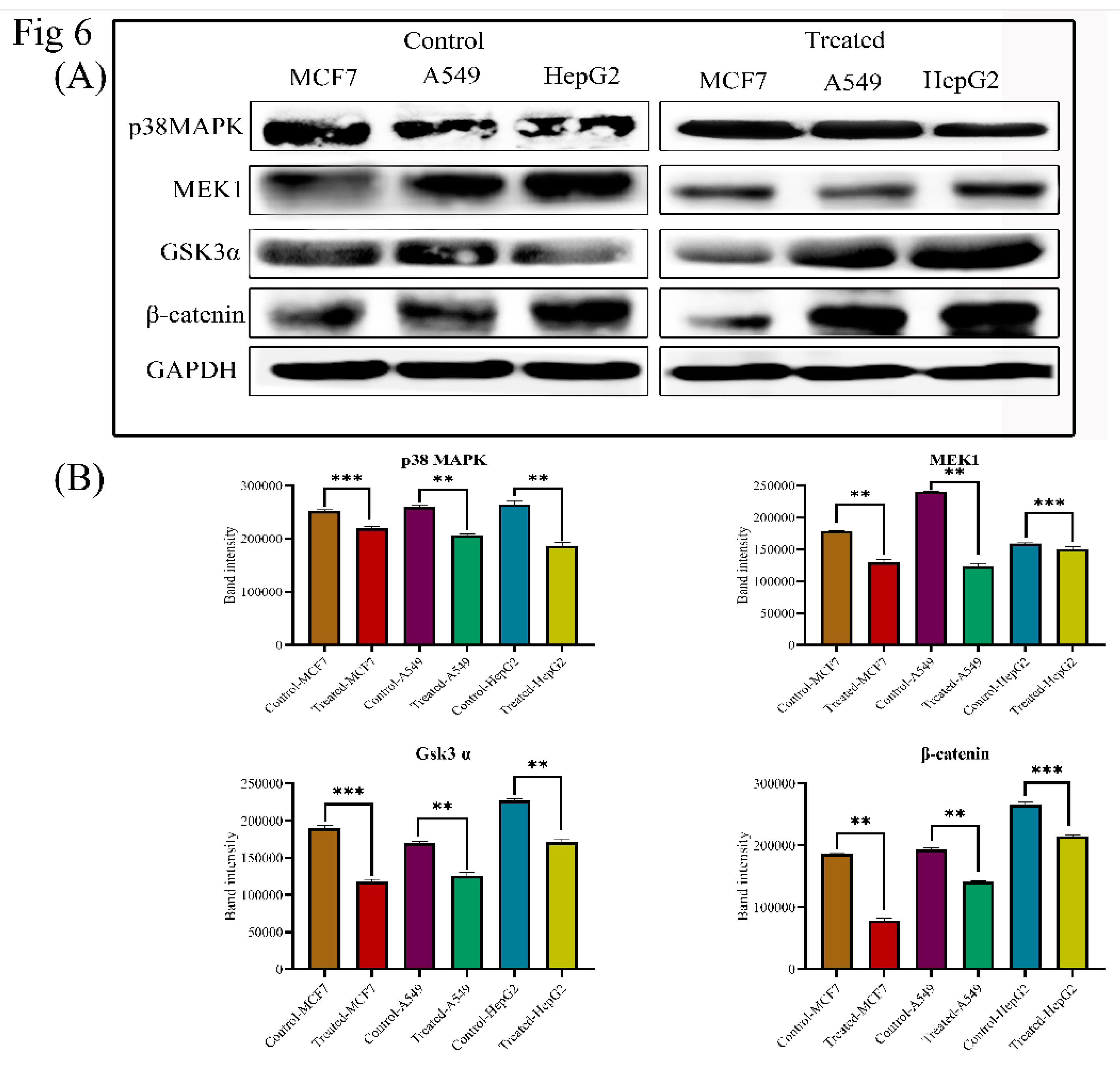
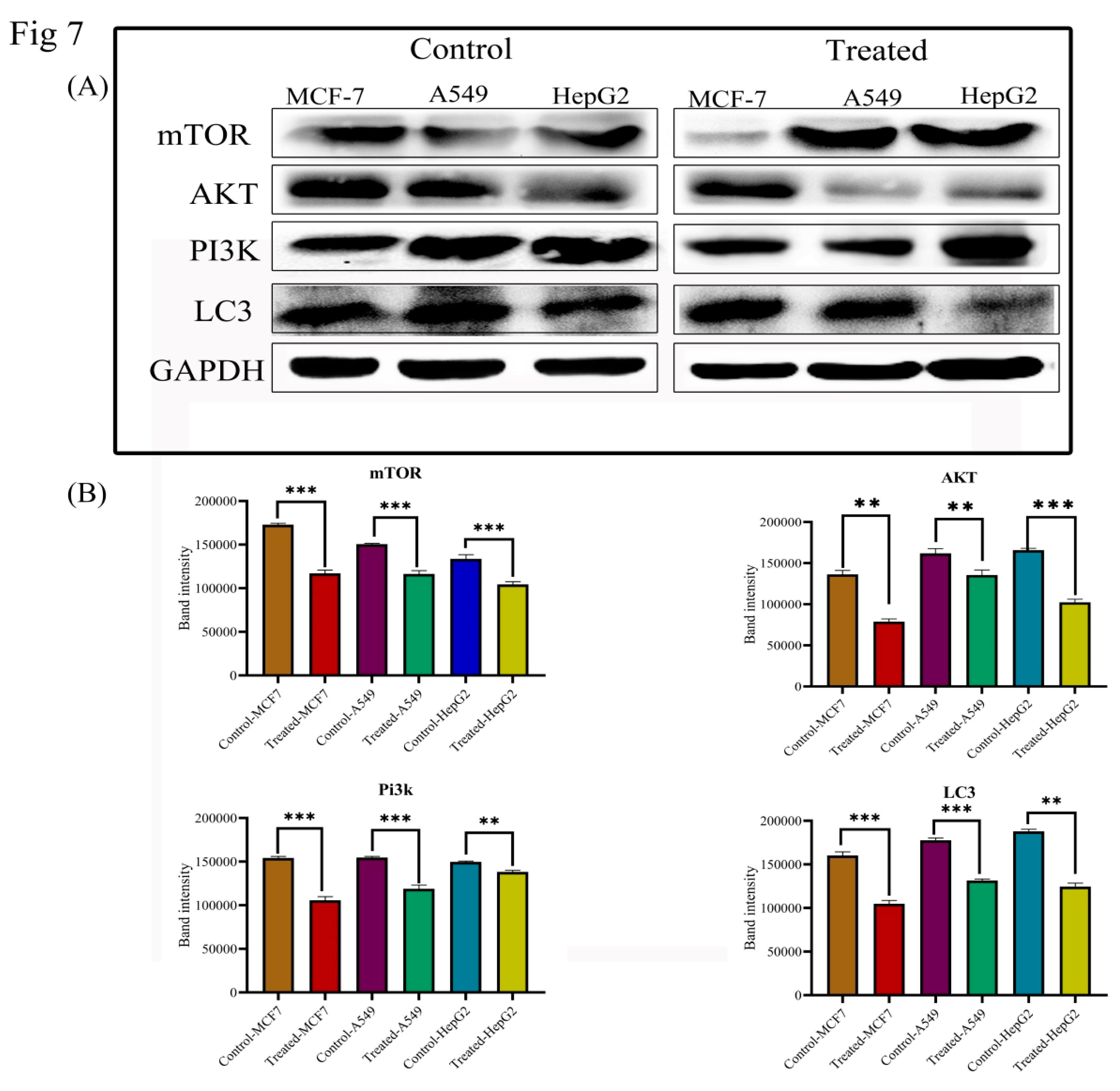
3.7. Lanatoside C Inhibits MAPK/Wnt, JAK-STAT, and PI3K/AKT/mTOR Pathways
3.8. Immunofluorescence Analysis Based Confirmation of Pathways Attenuated by Lanatoside C
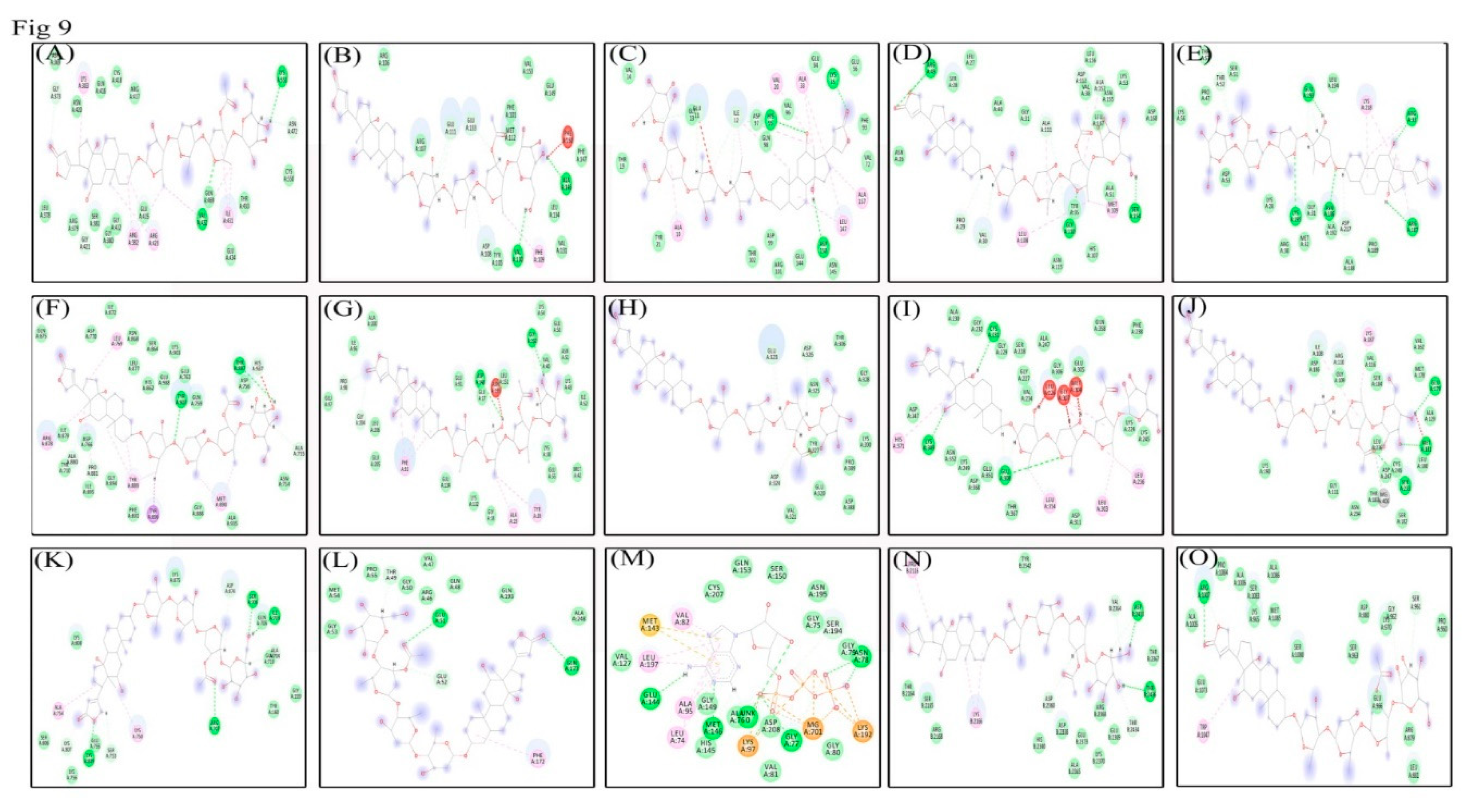
3.9. Molecular Docking Analysis Shows Lanatoside C can Potentially Inhibit Multiple Cancer Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics approval and consent to participate
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Peck-Radosavljevic, M. Drug Therapy for Advanced-Stage Liver Cancer. Liver Cancer 2014, 3, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Cerella, C.; Dicato, M.; Diederich, M. Assembling the puzzle of anti-cancer mechanisms triggered by cardiac glycosides. Mitochondrion 2013, 13, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.Y.; Chen, K.C.; Chen, H.; Seng, E.K.; Chu, J.J.H. Antiviral activity of lanatoside C against dengue virus infection. Antivir. Res. 2014, 111, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.G.; de Castro-Faria-Neto, H.C.; Da Silva, C.I.; Gonçalves-de-Albuquerque, C.F.; Silva, A.R.; De Amorim, L.M.; Freire, A.S.; Santelli, R.E.; Diniz, L.P.; Gomes, F.C.; et al. Na/K-ATPase as a target for anticancer drugs: Studies with perillyl alcohol. Mol. Cancer 2015, 14, 105. [Google Scholar] [CrossRef] [PubMed]
- Langer, G.A. Relationship between myocardial contractility and the effects of digitalis on ionic exchange. Fed. Proc. 1977, 36, 2231–2234. [Google Scholar]
- Akera, T. The role of Na+, K+-ATPase in the inotropic action. Pharmacol. Rev. 1997, 29, 185–247. [Google Scholar]
- Prassas, I.; Diamandis, E.P. Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 2008, 7, 926. [Google Scholar] [CrossRef]
- Perne, A.; Muellner, M.K.; Steinrueck, M.; Craig-Mueller, N.; Mayerhofer, J.; Schwarzinger, I.; Sloane, M.; Uras, I.Z.; Hoermann, G.; Nijman, S.M.; et al. Cardiac glycosides induce cell death in human cells by inhibiting general protein synthesis. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Kaushik, V.; Yakisich, J.S.; Azad, N.; Kulkarni, Y.; Venkatadri, R.; Wright, C.; Rojanasakul, Y.; Iyer, A.K. Anti-tumor effects of cardiac glycosides on human lung cancer cells and lung tumorspheres. J. Cell. Physiol. 2017, 232, 2497–2507. [Google Scholar] [CrossRef]
- Badr, C.E.; Wurdinger, T.; Nilsson, J.; Niers, J.M.; Whalen, M.; Degterev, A.; Tannous, B.A. Lanatoside C sensitizes glioblastoma cells to tumor necrosis factor–related apoptosis-inducing ligand and induces an alternative cell death pathway. Neuro-Oncology 2011, 13, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; Kim, M.S.; Kim, W.; Um, J.H.; Shin, Y.J.; Song, J.Y.; Jeong, J.H. Lanatoside C suppressed colorectal cancer cell growth by inducing mitochondrial dysfunction and increased radiation sensitivity by impairing DNA damage repair. Oncotarget 2016, 7, 6074. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yu, K.; Wang, G.; Zhang, D.; Shi, C.; Ding, Y.; Hong, D.; Zhang, D.; He, H.; Sun, L.; et al. Lanatoside C inhibits cell proliferation and induces apoptosis through attenuating Wnt/β-catenin/c-Myc signaling pathway in human gastric cancer cell. Biochem. Pharmacol. 2018, 150, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.B.; Lee, R.C.; Chu, J.J.; Salmas, R.E.; Mavromoustakos, T.; Durdagi, S. Discovery of selective dengue virus inhibitors using combination of molecular fingerprint-based virtual screening protocols, structure-based pharmacophore model development, molecular dynamics simulations and in vitro studies. J. Mol. Graph. Model. 2018, 79, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Abu-Izneid, T.; Rauf, A.; Bawazeer, S.; Wadood, A.; Patel, S. Anti-Dengue, Cytotoxicity, Antifungal, and In Silico Study of the Newly Synthesized 3-O-Phospo--D-Glucopyranuronic Acid Compound. BioMed Res. Int. 2018, 2018. [Google Scholar] [CrossRef]
- Ammeux, N.; Housden, B.E.; Georgiadis, A.; Hu, Y.; Perrimon, N. Mapping signaling pathway cross-talk in Drosophila cells. Proc. Natl. Acad. Sci. USA 2016, 113, 9940–9945. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Zhang, J.; Tian, X.J.; Chen, Y.J.; Wang, W.; Watkins, S.; Xing, J. Pathway crosstalk enables cells to interpret TGF-β duration. npj Syst. Biol. Appl. 2018, 4, 18. [Google Scholar] [CrossRef]
- Timmermans-Sprang, E.P.; Gracanin, A.; Mol, J.A. High basal Wnt signaling is further induced by PI3K/mTor inhibition but sensitive to cSRC inhibition in mammary carcinoma cell lines with HER2/3 overexpression. BMC Cancer 2015, 15, 545. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Pavlovic, D. The role of cardiotonic steroids in the pathogenesis of cardiomyopathy in chronic kidney disease. Nephron Clin. Pract. 2014, 128, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Cerella, C.; Simões, C.M.; Diederich, M. Anticancer and immunogenic properties of cardiac glycosides. Molecules 2017, 22, 1932. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, E.; Akhtar, S.; Khan, M.K.; Lohani, M.; Arif, J.M.; Siddiqui, M.H. Molecular docking analysis of aplysin analogs targeting survivin protein. Bioinformation 2017, 13, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, M.I.; Steinbrecher, T.; Schmid, R. Fascaplysin as a specific inhibitor for CDK4: Insights from molecular modelling. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Sarvagalla, S.; Singh, V.K.; Ke, Y.Y.; Shiao, H.Y.; Lin, W.H.; Hsieh, H.P.; Hsu, J.T.; Coumar, M.S. Identification of ligand efficient, fragment-like hits from an HTS library: Structure-based virtual screening and docking investigations of 2H-and 3H-pyrazolo tautomers for Aurora kinase A selectivity. J. Comput.-Aided Mol. Des. 2015, 29, 89–100. [Google Scholar] [CrossRef]
- Hashemzaei, M.; Delarami Far, A.; Yari, A.; Heravi, R.E.; Tabrizian, K.; Taghdisi, S.M.; Sadegh, S.E.; Tsarouhas, K.; Kouretas, D.; Tzanakakis, G.; et al. Anticancer and apoptosis-inducing effects of quercetin in vitro and in vivo. Oncol. Rep. 2017, 38, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.F.; Weng, C.J.; Sethi, G.; Hu, D.N. Natural bioactives and phytochemicals serve in cancer treatment and prevention. Evid.-Based Complement. Alternat Med. 2013, 2013. [Google Scholar] [CrossRef]
- Hsieh, Y.S.; Yang, S.F.; Sethi, G.; Hu, D.N. Natural bioactives in cancer treatment and prevention. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Bishayee, A.; Sethi, G. Bioactive natural products in cancer prevention and therapy: Progress and promise. Semin. Cancer Biol. 2016, 40, 1–3. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential Role of Natural Compounds as Anti-Angiogenic Agents in Cancer. Curr. Vasc. Pharmacol. 2017, 15, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Sethi, G.; Ahn, K.S. Ophiopogonin D modulates multiple oncogenic signaling pathways, leading to suppression of proliferation and chemosensitization of human lung cancer cells. Phytomedicine 2018, 40, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kar, S.; Lai, X.; Cai, W.; Arfuso, F.; Sethi, G.; Lobie, P.E.; Goh, B.C.; Lim, L.H.; Hartman, M.; et al. Triple negative breast cancer in Asia: An insider’s view. Cancer Treat. Rev. 2018, 62, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.W.; Chen, T.H.; Huang, H.L.; Chang, Y.W.; HuangFu, W.C.; Lee, Y.C.; Teng, C.M.; Pan, S.L. Lanatoside C, a cardiac glycoside, acts through protein kinase Cδ to cause apoptosis of human hepatocellular carcinoma cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Stenkvist, B. Cardenolides and cancer. Anti-Cancer Drug 2001, 12, 635–636. [Google Scholar] [CrossRef]
- Newman, R.A.; Yang, P.; Pawlus, A.D.; Block, K.I. Cardiac glycosides as novel cancer therapeutic agents. Mol. Interv. 2008, 8, 36. [Google Scholar] [CrossRef]
- Durmaz, I.; Guven, E.B.; Ersahin, T.; Ozturk, M.; Calis, I.; Cetin-Atalay, R. Liver cancer cells are sensitive to Lanatoside C induced cell death independent of their PTEN status. Phytomedicine 2016, 23, 42–51. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Silver, D.P.; Livingston, D.M. Mechanisms of BRCA1 tumor suppression. Cancer Discov. 2012, 2, 679–684. [Google Scholar] [CrossRef]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, H.; Liu, W.; Wu, J.; Yue, X.; Wang, J.; Quan, L.; Liu, H.; Guo, L.; Wang, Z.; et al. Ganoderic acid A exerts antitumor activity against MDA-MB-231 human breast cancer cells by inhibiting the Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway. Oncol. Lett. 2018, 16, 6515–6521. [Google Scholar] [CrossRef] [PubMed]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.; Xiang, M.; Frank, D. STAT3 Activity and Function in Cancer: Modulation by STAT5 and miR-146b. Cancers 2014, 6, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Müller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Chen, X.Q.; Huang, Y.; Chen, N.; Zeng, H. The multikinase inhibitor sorafenib induces caspase-dependent apoptosis in PC-3 prostate cancer cells. Asian J. Androl. 2010, 12, 527–534. [Google Scholar] [CrossRef]
- Chung, T.W.; Lin, S.C.; Su, J.H.; Chen, Y.K.; Lin, C.C.; Chan, H.L. Sinularin induces DNA damage, G2/M phase arrest, and apoptosis in human hepatocellular carcinoma cells. BMC Complement. Altern. Med. 2017, 17, 62. [Google Scholar] [CrossRef]
- Gao, Q.L.; Ye, F.; Xing, H.; Xie, D.X.; Lu, Y.P.; Zhou, J.F.; Ma, D. Down-regulation of Chk1/Chk2 gene expression increases apoptosis in irradiated HeLa cells and its mechanism. Chin. J. Oncol. 2009, 31, 178–182. [Google Scholar]
- Biliran, H.; Wang, Y.; Banerjee, S.; Xu, H.; Heng, H.; Thakur, A.; Bollig, A.; Sarkar, F.H.; Liao, J.D. Overexpression of Cyclin D1 Promotes Tumor Cell Growth and Confers Resistance to Cisplatin-Mediated Apoptosis in an Elastase-myc Transgene–Expressing Pancreatic Tumor Cell Line. Clin. Cancer Res. 2005, 11, 6075–6086. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Esmaeili, M.A.; Farimani, M.M.; Kiaei, M. Anticancer effect of calycopterin via PI3K/Akt and MAPK signaling pathways, ROS-mediated pathway and mitochondrial dysfunction in hepatoblastoma cancer (HepG2) cells. Mol. Cell Biochem. 2014, 397, 17–31. [Google Scholar] [CrossRef]
- Woo, C.C.; Hsu, A.; Kumar, A.P.; Sethi, G.; Tan, K.H. Thymoquinone inhibits tumor growth and induces apoptosis in a breast cancer xenograft mouse model: The role of p38 MAPK and ROS. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, C.; Bae, H.; Lee, J.H.; Baek, S.H.; Nam, D.; Chung, W.S.; Shim, B.S.; Lee, S.G.; Kim, S.H.; et al. 6-Shogaol exerts anti-proliferative and pro-apoptotic effects through the modulation of STAT3 and MAPKs signaling pathways. Mol. Carcinog. 2015, 54, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Xie, Y.; Tao, L.; Tang, D. Both ERK1 and ERK2 kinases promote G2/M arrest in etoposide-treated MCF7 cells by facilitating ATM activation. Cell Signal. 2010, 22, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, Y.; Liu, G.; Liu, H.; Zhu, F.; Ji, H.; Li, B. C-Phycocyanin exerts anti-cancer effects via the MAPK signaling pathway in MDA-MB-231 cells. Cancer Cell Int. 2018, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Ong, M.S.; Cai, W.; Yuan, Y.; Leong, H.C.; Tan, T.Z.; Mohammad, A.; You, M.L.; Arfuso, F.; Goh, B.C.; Warrier, S.; et al. ‘Lnc’-ing Wnt in female reproductive cancers: Therapeutic potential of long non-coding RNAs in Wnt signalling. Br. J. Pharmacol. 2017, 174, 4684–4700. [Google Scholar] [CrossRef]
- Watson, A.L.; Rahrmann, E.P.; Moriarity, B.S.; Choi, K.; Conboy, C.B.; Greeley, A.D.; Halfond, A.L.; Anderson, L.K.; Wahl, B.R.; Keng, V.W.; et al. Canonical Wnt/β-catenin signaling drives human Schwann cell transformation, progression, and tumor maintenance. Cancer Discov. 2013, 3, 674–689. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.; Wu, Y.; Shi, K.; Bing, L.; Hao, J. Wnt/β-catenin signaling pathway upregulates c-Myc expression to promote cell proliferation of P19 teratocarcinoma cells. Anat. Rec. 2012, 295, 2104–2113. [Google Scholar] [CrossRef]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014, 5, 1437. [Google Scholar] [CrossRef]
- Baek, S.H.; Ko, J.H.; Lee, J.H.; Kim, C.; Lee, H.; Nam, D.; Lee, J.; Lee, S.G.; Yang, W.M.; Um, J.Y.; et al. Ginkgolic acid inhibits invasion and migration and TGF-β-induced EMT of lung cancer cells through PI3K/Akt/mTOR inactivation. J. Cell Physiol. 2017, 232, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Manu, K.A.; Chen, L.; Li, F.; Rajendran, P.; Subramaniam, A.; Lam, P.; Kumar, A.P.; Sethi, G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis 2011, 16, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Park, K.R.; Nam, D.; Yun, H.M.; Lee, S.G.; Jang, H.J.; Sethi, G.; Cho, S.K.; Ahn, K.S. β-Caryophyllene oxide inhibits growth and induces apoptosis through the suppression of PI3K/AKT/mTOR/S6K1 pathways and ROS-mediated MAPKs activation. Cancer Lett. 2011, 312, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.U.; Sangai, T.; Akcakanat, A.; Chen, H.; Wei, C.; Meric-Bernstam, F. Vertical inhibition of the PI3K/Akt/mTOR pathway is synergistic in breast cancer. Oncogenesis 2017, 6, 385. [Google Scholar] [CrossRef] [PubMed]
- Phang, C.W.; Karsani, S.A.; Sethi, G.; Abd Malek, S.N. Flavokawain C Inhibits Cell Cycle and Promotes Apoptosis, Associated with Endoplasmic Reticulum Stress and Regulation of MAPKs and Akt Signaling Pathways in HCT 116 Human Colon Carcinoma Cells. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Zhang, W.J.; Fan, Q.X.; Wang, L.X. Licochalcone A inhibits PI3K/Akt/mTOR signaling pathway activation and promotes autophagy in breast cancer cells. Oncology Lett. 2018, 15, 1869–1873. [Google Scholar] [CrossRef]
- Hossan, M.S.; Chan, Z.Y.; Collins, H.M.; Shipton, F.N.; Butler, M.S.; Rahmatullah, M.; Lee, J.B.; Gershkovich, P.; Kagan, L.; Khoo, T.J.; et al. Cardiac glycoside cerberin exerts anticancer activity through PI3K/AKT/mTOR signal transduction inhibition. Cancer Lett. 2019, 453, 57–73. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Total Length of Comet | Length of Head | Length of Tail | Head DNA (%) | Tail DNA (%) | Tail Movement | Overall Tail Movement (OTM) |
|---|---|---|---|---|---|---|---|
| MCF7-control | 62 ± 5 | 45 ± 3 | 14 ± 6 | 81 ± 6 | 18 ± 5 | 6. ± 2 | 7 ± 1 |
| MCF7-Treated | 252 ± 13 | 89 ± 6 | 162 ± 16 | 19 ± 5 | 80 ± 13 | 427 ± 36 | 261 ± 22 |
| A549-control | 112 ± 9 | 94 ± 6 | 18 ± 4 | 92 ± 8 | 9 ± 4 | 3 ± 1 | 6 ± 3 |
| A549-Treated | 321 ± 14 | 69 ± 5 | 251 ± 13 | 18 ± 2 | 81 ± 13 | 416 ± 42 | 221 ± 23 |
| HepG2-control | 132 ± 8 | 86 ± 7 | 46 ± 6 | 82 ± 12 | 17 ± 3 | 9 ± 4 | 64 ± 1 |
| HepG2-Treated | 362 ± 9 | 98 ± 6 | 264 ± 19 | 11 ± 2 | 88 ± 12 | 361 ± 41 | 112 ± 18 |
| S.no | PDB ID | Libdock Score | No. of H Bonds | Interacting Residues |
|---|---|---|---|---|
| 1. | 1BG1 | 170.564 | 5 | H bonds: GLY373, GLY421, GLN469, ASN472, LYS551. Interacting residues: ASP369, LEU378, ARG379, GLY380, SER381, ARG382, LYS383, GLU415, GLN416, ARG417, CYS418, ASN420, GLY422, ARG423, ILE431, VAL432, THR433, ASN472, CYS550. |
| 2. | 1OVE | 140.632 | 8 | H bonds: PRO29, VAL30, VAL38, ARG49, GLY110, ALA111, SER154, ALA157. Interacting residues: ASN26, LEU27, SER28, GLY31, ALA40, ALA51, LYS53, HIS107, LEU108, MET109, ASP112, ASN115, ASN155, LEU156, LEU167, ASP168. |
| 3. | 1VKX | 124.57 | 7 | H bonds: ARG33, THR52, ASN186, ARG187, GLU193, LYS195, ASP217. Interacting residues: LYS28, ARG30, GLY31, MET32, PRO47, SER51, ASP53, LYS56, THR57, ALA188, PRO189, ALA192, LEU194, LYS218. |
| 4. | 1WOK | 180.235 | 7 | H bonds: ALA755, ASP756, ALA880, PRO881, THR887, TYR907, HIS937. Interacting residues: TYR710, ASN754, GLN759, GLU763, ASP766, LEU769, ASP770, HIS862, SER864, ASN868, ILE872, GLN875, LEU877, ARG878, ILE879, GLY888, TYR889, MET890, PHE891, GLY894, ILE895, TYR896, LYS903, ALA935, GLU988. |
| 5. | 2CBZ | 102 | 4 | H bonds: GLN713, GLN714, TRP716, GLN718. Interacting residues: TYR710, PRO712, PHE728, SER689. |
| 6. | 2E9P | 161.897 | 5 | H bonds: GLU17, PRO98, ASP148, GLY150, GLY204. Interacting residues: GLY18, ALA19, TYR20, LYS38, MET42, LYS43, GLU50, ASN51, ILE52, LYS54, GLU55, GLU91, PHE93, ILE96, GLU97, PRO98, LYS132, GLU134, ASN135, LEU151, ALA200, GLU205, LEU206. |
| 7. | 2JDO | 51.2917 | 3 | H bonds: GLU323, ASP324, ASP326. Interacting residues: THR306, GLU320, VAL321, ASN325, TYR327, GLY328, ASP388, PRO389, LYS390. |
| 8. | CDK4 or CYCLIN D1 | 172.401 | 6 | H bonds: GLU11, ILE12, GLY13, LYS35, HIS95, ASP158. Interacting residues: ALA10, VAL14, THR19, VAL20, TYR21, ALA33, LYS35, GLU56, VAL72, PHE93, GLU94, VAL96, ASP97, GLN98, ASP99, ARG101, THR102, GLU144, ASN145, LEU147. |
| 9 | 2O21 | 116.154 | 5 | H bonds: ASP108, GLU111, VAL130, GLU133, ALA146. Interacting residues: PHE101, TYR105, ARG106, ARG107, PHE109, MET112, VAL131, LEU134, PHE147, GLU149, PHE150, VAL153. |
| 10 | 3ALN | 102 | 5 | H bonds: ILE108, ARG110, GLU179, MET181, SER233. Interacting residues: GLY109, GLY111, VAL116, ALA129, VAL162, MET178, LEU180, SER182, THR183, SER184, ASP186, LYS187, LYS190, ASN234, LEU236, CYS246, ASP247. |
| 11 | 3EYG | 87.0358 | 3 | H bonds: SER961, GLY962, ARG1007. Interacting residues: ARG879, ASP880, LEU881, PRO960, SER963, LYS965, GLU966, LYS970, ALA1005, ALA1006, TRP1047, GLU1073, SER1080, SER1083, PRO1084, MET1085, ALA1086. |
| 12 | 3L54 | 105 | 7 | H bonds: ILE703, ALA704, SER706, ARG707, SER753, LYS809, LUY807. Interacting residues: GLY159, TYR160, GLN705, GLN710, LYS750, ALA754, GLU755, LYS756, LYS808, ASP874, LYS875. |
| 13 | 3NUP | 97.4948 | 4 | H bonds: THR49, GLU51, GLU52, GLN173. Interacting residues: ARG46, VAL47, GLN48, GLY50, GLY53, MET54, PRO55, PHE172, GLN193, ALA248. |
| 14 | 3VVH | 137.537 | 8 | H bonds: ALA76, GLY77, ASN78, LYS97, GLU144, MET146, SER194. Interacting residues: LEU74, GLY75, GLY79, GLY80, VAL81, VAL82, ALA95, VAL127, MET143, HIS145, GLY149, SER150, GLN153, LYS192, ASN195, LEU197, CYS207, ASP208. |
| 15 | 4JSP | 83.6223 | 5 | H bonds: ASP2360, VAL2364, ASP2433, THR2434, THR2436. Interacting residues: PRO2116, THR2164, SER2165, LYS2166, ARG2168, ASP2338, HIS2340, ALA2365, THR2367, ARG2368, GLU2369, LYS2370, GLU2373, TYR2542. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, D.; Kumavath, R.; Ghosh, P.; Barh, D. Lanatoside C Induces G2/M Cell Cycle Arrest and Suppresses Cancer Cell Growth by Attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR Signaling Pathways. Biomolecules 2019, 9, 792. https://doi.org/10.3390/biom9120792
Reddy D, Kumavath R, Ghosh P, Barh D. Lanatoside C Induces G2/M Cell Cycle Arrest and Suppresses Cancer Cell Growth by Attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR Signaling Pathways. Biomolecules. 2019; 9(12):792. https://doi.org/10.3390/biom9120792
Chicago/Turabian StyleReddy, Dhanasekhar, Ranjith Kumavath, Preetam Ghosh, and Debmalya Barh. 2019. "Lanatoside C Induces G2/M Cell Cycle Arrest and Suppresses Cancer Cell Growth by Attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR Signaling Pathways" Biomolecules 9, no. 12: 792. https://doi.org/10.3390/biom9120792
APA StyleReddy, D., Kumavath, R., Ghosh, P., & Barh, D. (2019). Lanatoside C Induces G2/M Cell Cycle Arrest and Suppresses Cancer Cell Growth by Attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR Signaling Pathways. Biomolecules, 9(12), 792. https://doi.org/10.3390/biom9120792