Wasabi Compound 6-(Methylsulfinyl) Hexyl Isothiocyanate Induces Cell Death with Coexisting Mitotic Arrest and Autophagy in Human Chronic Myelogenous Leukemia K562 Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Viability
2.4. Cell Cycle Analysis by Flow Cytometry
2.5. Detection of Phosphorylated Histone H3
2.6. Morphology by Light and Electronic Microscopy
2.7. Immunofluorescent Stain
2.8. Detection of Acidic Vesicular Organelles with Acridine Orange Staining
2.9. Western Blot Analysis
2.10. Immunoprecipitation Assay
2.11. RNA Interference
2.12. Assay for Differentiation Antigens
2.13. Statistical Analysis
3. Results
3.1. 6-(methylsulfinyl) hexyl isothiocyanate Inhibited K562 Cell Growth
3.2. 6-(methylsulfinyl) hexyl isothiocyanate Did Not Affect the Expression of Bcr-Abl Protein in K562 and K562-Resistant Cells
3.3. 6-(methylsulfinyl) hexyl isothiocyanate Induced Autophagy in Human K562 Cells
3.4. 6-(methylsulfinyl) hexyl isothiocyanate Affected Cell Cycle and Arrested in M Phase
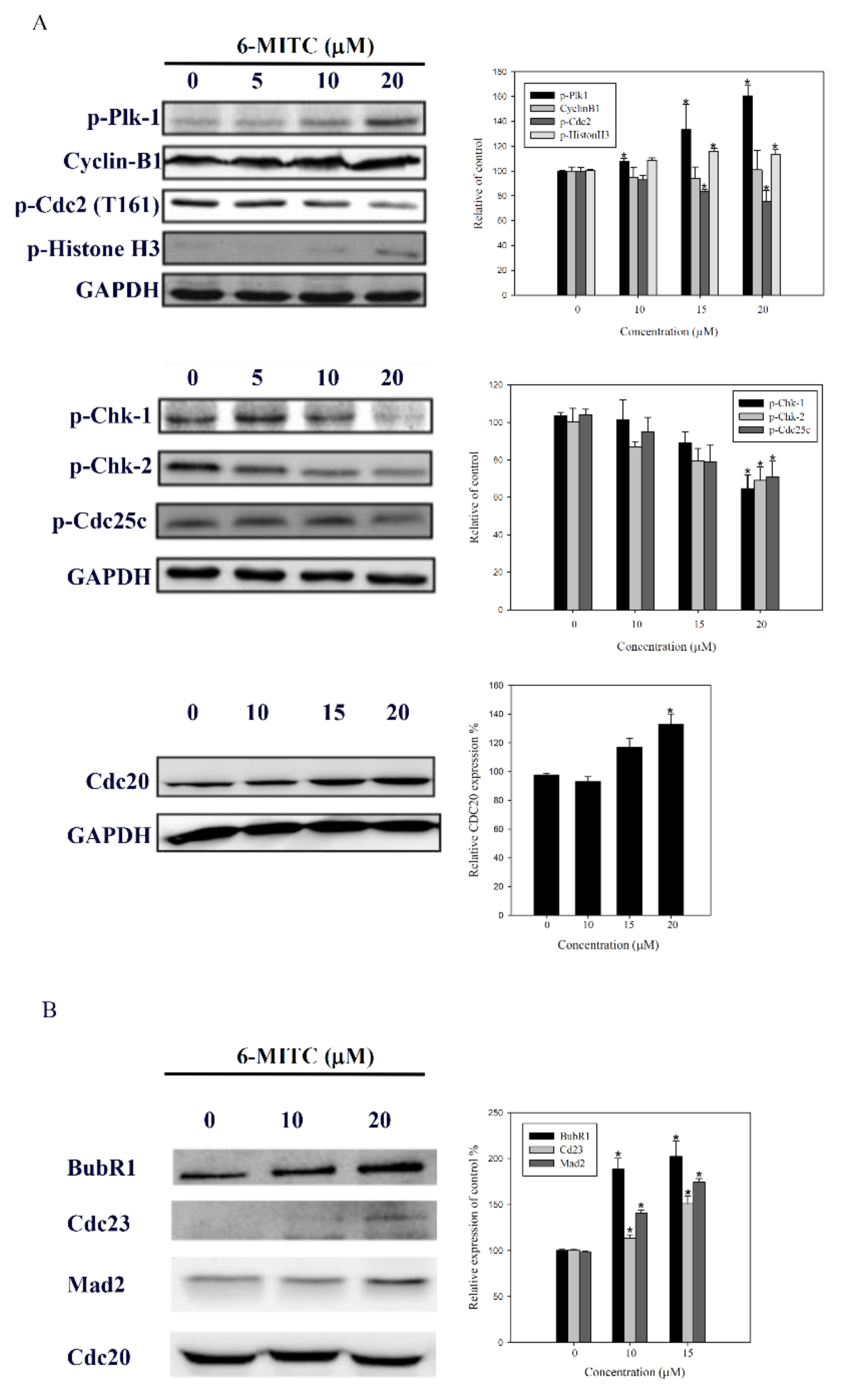
3.5. Molecular Mechanisms of 6-(methylsulfinyl) hexyl isothiocyanate on K562 Cells
3.6. Exclusion of Cell Differentiation by 6-(methylsulfinyl) hexyl isothiocyanate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef]
- Sell, S. Leukemia: Stem cells, maturation arrest, and differentiation therapy. Stem Cell Rev. 2005, 1, 197–205. [Google Scholar] [CrossRef]
- Stone, R.M. Optimizing treatment of chronic myeloid leukemia: A rational approach. Oncologist 2004, 9, 259–270. [Google Scholar] [CrossRef]
- Hernandez-Boluda, J.C.; Cervantes, F. Imatinib mesylate (Gleevec, Glivec): A new therapy for chronic myeloid leukemia and other malignancies. Drugs Today 2002, 38, 601–613. [Google Scholar] [CrossRef]
- Buchdunger, E.; Zimmermann, J.; Mett, H.; Meyer, T.; Muller, M.; Druker, B.J.; Lydon, N.B. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996, 56, 100–104. [Google Scholar]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Litzow, M.R. Imatinib resistance: Obstacles and opportunities. Arch. Pathol. Lab. Med. 2006, 130, 669–679. [Google Scholar] [CrossRef]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef]
- Hochhaus, A.; O’Brien, S.G.; Guilhot, F.; Druker, B.J.; Branford, S.; Foroni, L.; Goldman, J.M.; Muller, M.C.; Radich, J.P.; Rudoltz, M.; et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009, 23, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Cortes, J.E.; O’Brien, S.; Luthra, R.; Giles, F.; Verstovsek, S.; Faderl, S.; Thomas, D.; Garcia-Manero, G.; Rios, M.B.; et al. Long-term survival benefit and improved complete cytogenetic and molecular response rates with imatinib mesylate in Philadelphia chromosome-positive chronic-phase chronic myeloid leukemia after failure of interferon-alpha. Blood 2004, 104, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, T.; Riehm, B.; Berger, U.; Paschka, P.; Muller, M.C.; Kreil, S.; Merx, K.; Schwindel, U.; Schoch, C.; Hehlmann, R.; et al. Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center: A 4.5-year follow-up. Cancer 2005, 103, 1659–1669. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Kantarjian, H.; O’Brien, S.; Jabbour, E.; Borthakur, G.; Ravandi, F.; Verstovsek, S.; Shan, J.; Cortes, J. Outcome of patients with chronic myeloid leukemia with multiple ABL1 kinase domain mutations receiving tyrosine kinase inhibitor therapy. Haematologica 2011, 96, 918–921. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kepp, O.; Galluzzi, L.; Lipinski, M.; Yuan, J.; Kroemer, G. Cell death assays for drug discovery. Nat. Rev. Drug Discov. 2011, 10, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Kang, J.H.; Choi, S.; Jang, J.E.; Ramalingam, P.; Ko, Y.T.; Kim, S.Y.; Oh, S.H. Wasabia japonica is a potential functional food to prevent colitis via inhibiting the NF-kappaB signaling pathway. Food Funct. 2017, 8, 2865–2874. [Google Scholar] [CrossRef]
- Uto, T.; Fujii, M.; Hou, D.X. 6-(Methylsulfinyl)hexyl isothiocyanate suppresses inducible nitric oxide synthase expression through the inhibition of Janus kinase 2-mediated JNK pathway in lipopolysaccharide-activated murine macrophages. Biochem. Pharmacol. 2005, 70, 1211–1221. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Tarozzi, A.; Cantelli-Forti, G.; Hrelia, P. Neuroprotection by 6-(methylsulfinyl)hexyl isothiocyanate in a 6-hydroxydopamine mouse model of Parkinsons disease. Brain Res. 2014, 1589, 93–104. [Google Scholar] [CrossRef]
- Mizuno, K.; Kume, T.; Muto, C.; Takada-Takatori, Y.; Izumi, Y.; Sugimoto, H.; Akaike, A. Glutathione biosynthesis via activation of the nuclear factor E2-related factor 2 (Nrf2)—antioxidant-response element (ARE) pathway is essential for neuroprotective effects of sulforaphane and 6-(methylsulfinyl) hexyl isothiocyanate. J. Pharmacol. Sci. 2011, 115, 320–328. [Google Scholar] [CrossRef]
- Fuke, Y.; Shinoda, S.; Nagata, I.; Sawaki, S.; Murata, M.; Ryoyama, K.; Koizumi, K.; Saiki, I.; Nomura, T. Preventive effect of oral administration of 6-(methylsulfinyl)hexyl isothiocyanate derived from wasabi (Wasabia japonica Matsum) against pulmonary metastasis of B16-BL6 mouse melanoma cells. Cancer Detect. Prev. 2006, 30, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Shinoda, S.; Yamori, T.; Sawaki, S.; Nagata, I.; Ryoyama, K.; Fuke, Y. Selective sensitivity to wasabi-derived 6-(methylsulfinyl)hexyl isothiocyanate of human breast cancer and melanoma cell lines studied in vitro. Cancer Detect. Prev. 2005, 29, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Tseng, W.S.; Lai, J.C.; Shieh, H.R.; Chi, C.W.; Chen, Y.J. Differential Pharmacological Activities of Oxygen Numbers on the Sulfoxide Moiety of Wasabi Compound 6-(Methylsulfinyl) Hexyl Isothiocyanate in Human Oral Cancer Cells. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Kuno, T.; Hirose, Y.; Yamada, Y.; Imaida, K.; Tatematsu, K.; Mori, Y.; Mori, H. Chemoprevention of 1,2-dimethylhydrazine-induced colonic preneoplastic lesions in Fischer rats by 6-methylsulfinylhexyl isothiocyanate, a wasabi derivative. Oncol. Lett. 2010, 1, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, M.; Cocchi, V.; Malaguti, M.; Barbalace, M.C.; Marchionni, S.; Hrelia, S.; Hrelia, P. 6-(Methylsulfonyl) hexyl isothiocyanate as potential chemopreventive agent: Molecular and cellular profile in leukaemia cell lines. Oncotarget 2017, 8, 111697–111714. [Google Scholar] [CrossRef]
- Chen, Y.J.; Huang, Y.C.; Tsai, T.H.; Liao, H.F. Effect of Wasabi Component 6-(Methylsulfinyl)hexyl Isothiocyanate and Derivatives on Human Pancreatic Cancer Cells. Evid. Based Complement. Altern. Med. ECAM 2014, 2014, 494739. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Wu, S.; Sakao, K.; Hou, D.X. Wasabi 6-(methylsulfinyl)hexyl isothiocyanate induces apoptosis in human colorectal cancer cells through p53-independent mitochondrial dysfunction pathway. Biofactors 2018. [Google Scholar] [CrossRef]
- Fuke, Y.; Hishinuma, M.; Namikawa, M.; Oishi, Y.; Matsuzaki, T. Wasabi-derived 6-(methylsulfinyl)hexyl isothiocyanate induces apoptosis in human breast cancer by possible involvement of the NF-kappaB pathways. Nutr. Cancer 2014, 66, 879–887. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Hsuan, S.W.; Chyau, C.C.; Hung, H.Y.; Chen, J.H.; Chou, F.P. The induction of apoptosis and autophagy by Wasabia japonica extract in colon cancer. Eur. J. Nutr. 2016, 55, 491–503. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Prescott, A.R.; Cooper, J.; Brachmann, S.M.; Wang, L.; Tang, X.; Backer, J.M.; Lucocq, J.M. Inhibition of autophagy in mitotic animal cells. Traffic 2002, 3, 878–893. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Kim, M.; Lipinski, M.; Li, J.; Kim, D.; Lu, T.; Shen, Y.; Rameh, L.; Yankner, B.; Tsai, L.H.; et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol. Cell 2010, 38, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.F.; Li, Z.H.; Du, W.W.; Xu, L.X.; Ren, J.L.; Li, X.L.; Fang, F.; Xie, Y.; Li, M.; Qian, G.H.; et al. Inhibiting PLK1 induces autophagy of acute myeloid leukemia cells via mammalian target of rapamycin pathway dephosphorylation. Oncol. Rep. 2017, 37, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Budina-Kolomets, A.; Balaburski, G.M.; Bondar, A.; Beeharry, N.; Yen, T.; Murphy, M.E. Comparison of the activity of three different HSP70 inhibitors on apoptosis, cell cycle arrest, autophagy inhibition, and HSP90 inhibition. Cancer Biol. Ther. 2014, 15, 194–199. [Google Scholar] [CrossRef]
- Maskey, D.; Yousefi, S.; Schmid, I.; Zlobec, I.; Perren, A.; Friis, R.; Simon, H.U. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat. Commun. 2013, 4, 2130. [Google Scholar] [CrossRef]
- Dilshara, M.G.; Jayasooriya, R.; Karunarathne, W.; Choi, Y.H.; Kim, G.Y. Camptothecin induces mitotic arrest through Mad2-Cdc20 complex by activating the JNK-mediated Sp1 pathway. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2019, 127, 143–155. [Google Scholar] [CrossRef]
- Liang, N.; Liu, X.; Zhang, S.; Sun, H. The role of Beclin 1 in IR-induced crosstalk between autophagy and G2/M cell cycle arrest. Cell. Signal. 2019, 62, 109353. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.-M.; Liao, H.-F.; Chi, C.-W.; Kou, Y.R.; Chen, Y.-J. Wasabi Compound 6-(Methylsulfinyl) Hexyl Isothiocyanate Induces Cell Death with Coexisting Mitotic Arrest and Autophagy in Human Chronic Myelogenous Leukemia K562 Cells. Biomolecules 2019, 9, 774. https://doi.org/10.3390/biom9120774
Wu K-M, Liao H-F, Chi C-W, Kou YR, Chen Y-J. Wasabi Compound 6-(Methylsulfinyl) Hexyl Isothiocyanate Induces Cell Death with Coexisting Mitotic Arrest and Autophagy in Human Chronic Myelogenous Leukemia K562 Cells. Biomolecules. 2019; 9(12):774. https://doi.org/10.3390/biom9120774
Chicago/Turabian StyleWu, Kun-Ming, Hui-Fen Liao, Chih-Wen Chi, Yu Ru Kou, and Yu-Jen Chen. 2019. "Wasabi Compound 6-(Methylsulfinyl) Hexyl Isothiocyanate Induces Cell Death with Coexisting Mitotic Arrest and Autophagy in Human Chronic Myelogenous Leukemia K562 Cells" Biomolecules 9, no. 12: 774. https://doi.org/10.3390/biom9120774
APA StyleWu, K.-M., Liao, H.-F., Chi, C.-W., Kou, Y. R., & Chen, Y.-J. (2019). Wasabi Compound 6-(Methylsulfinyl) Hexyl Isothiocyanate Induces Cell Death with Coexisting Mitotic Arrest and Autophagy in Human Chronic Myelogenous Leukemia K562 Cells. Biomolecules, 9(12), 774. https://doi.org/10.3390/biom9120774