Dynamical Rearrangement of Human Epidermal Growth Factor Receptor 2 upon Antibody Binding: Effects on the Dimerization

 ,
,  , ,
, ,  ,
,  , ,
, ,  ,
,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Model Construction
2.2. Molecular Dynamics Simulations
2.3. Solvent-Accessible Surface Area Analysis
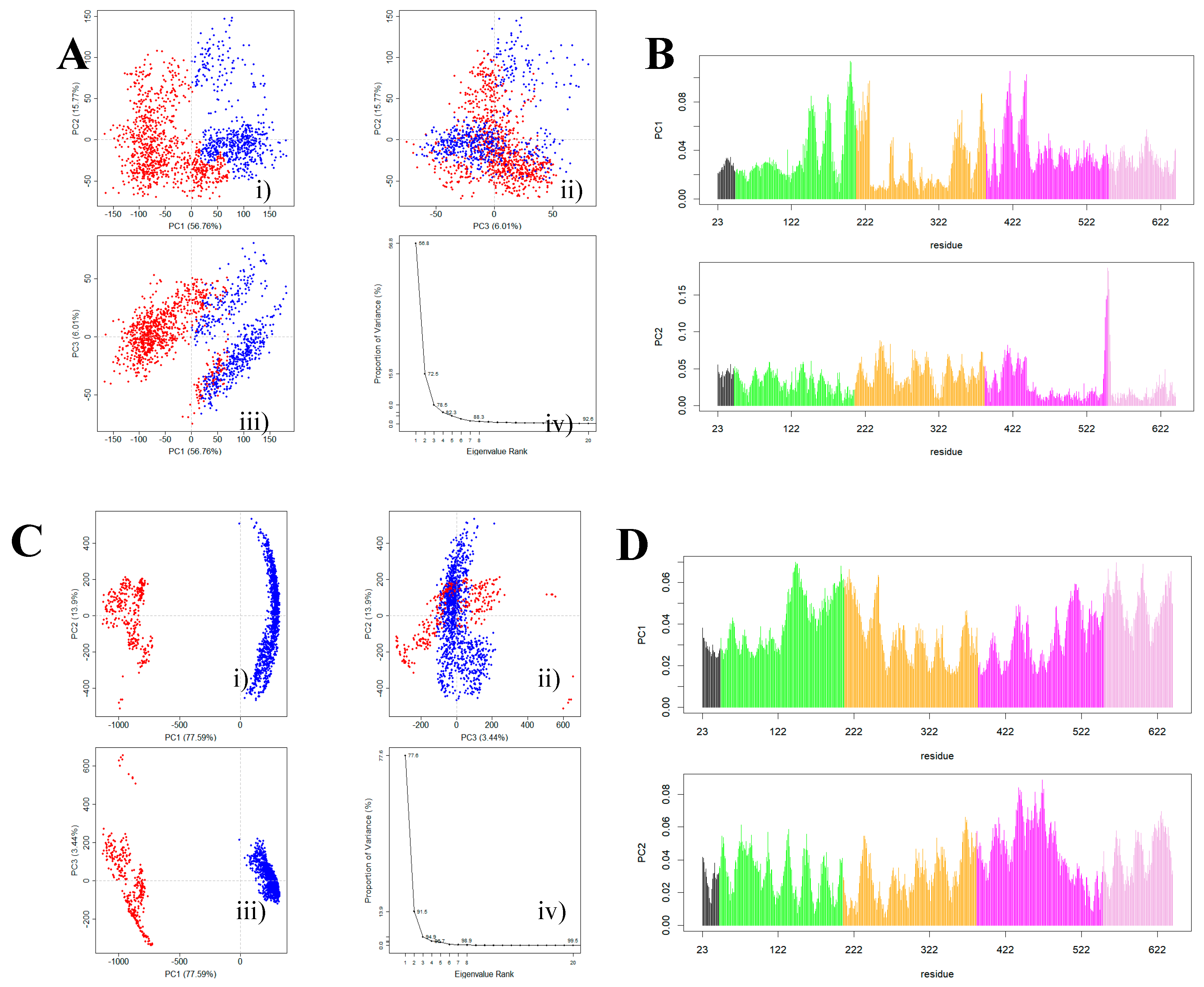
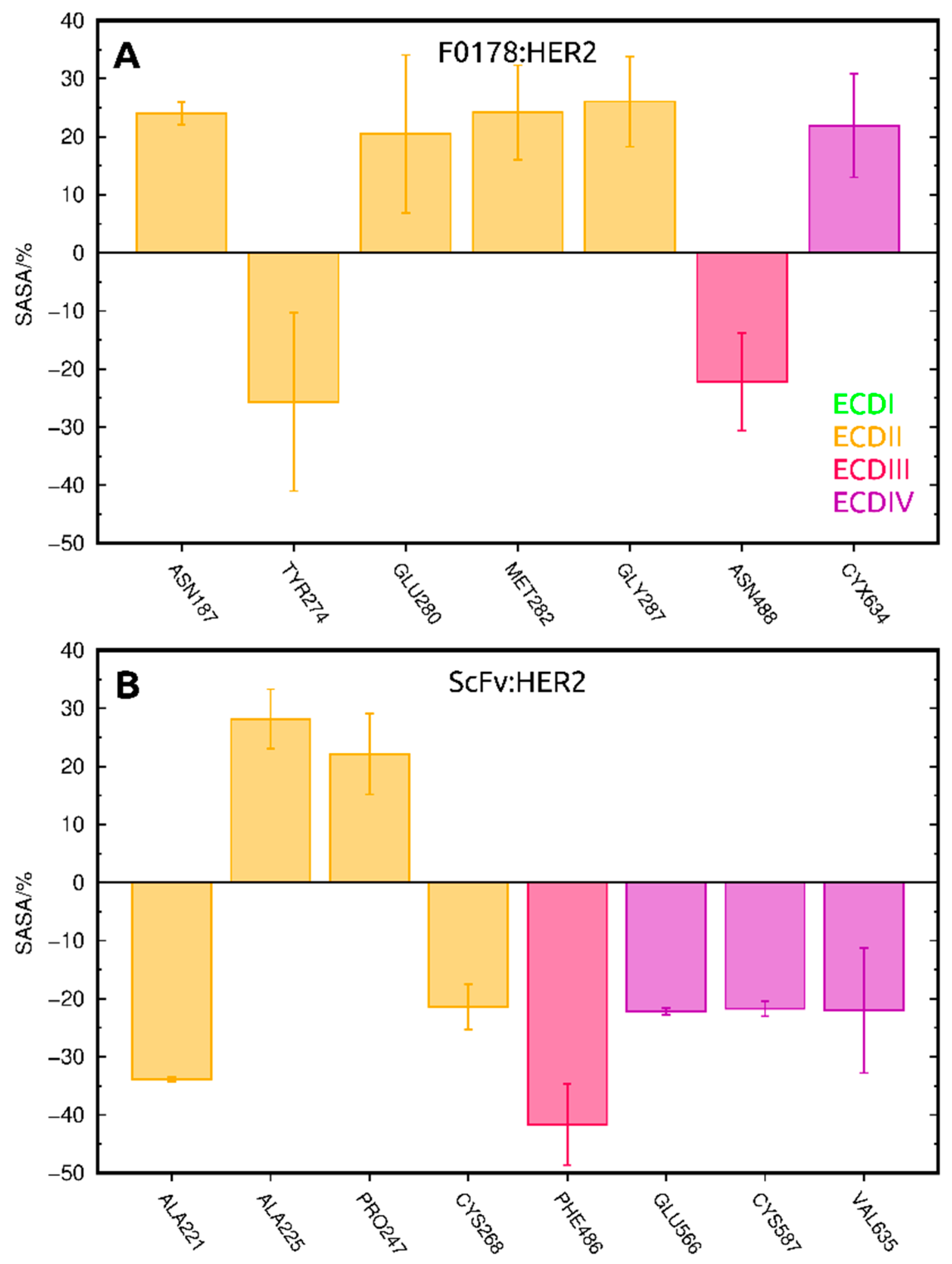
2.3.1. SASA Analysis of Interfacial Residues
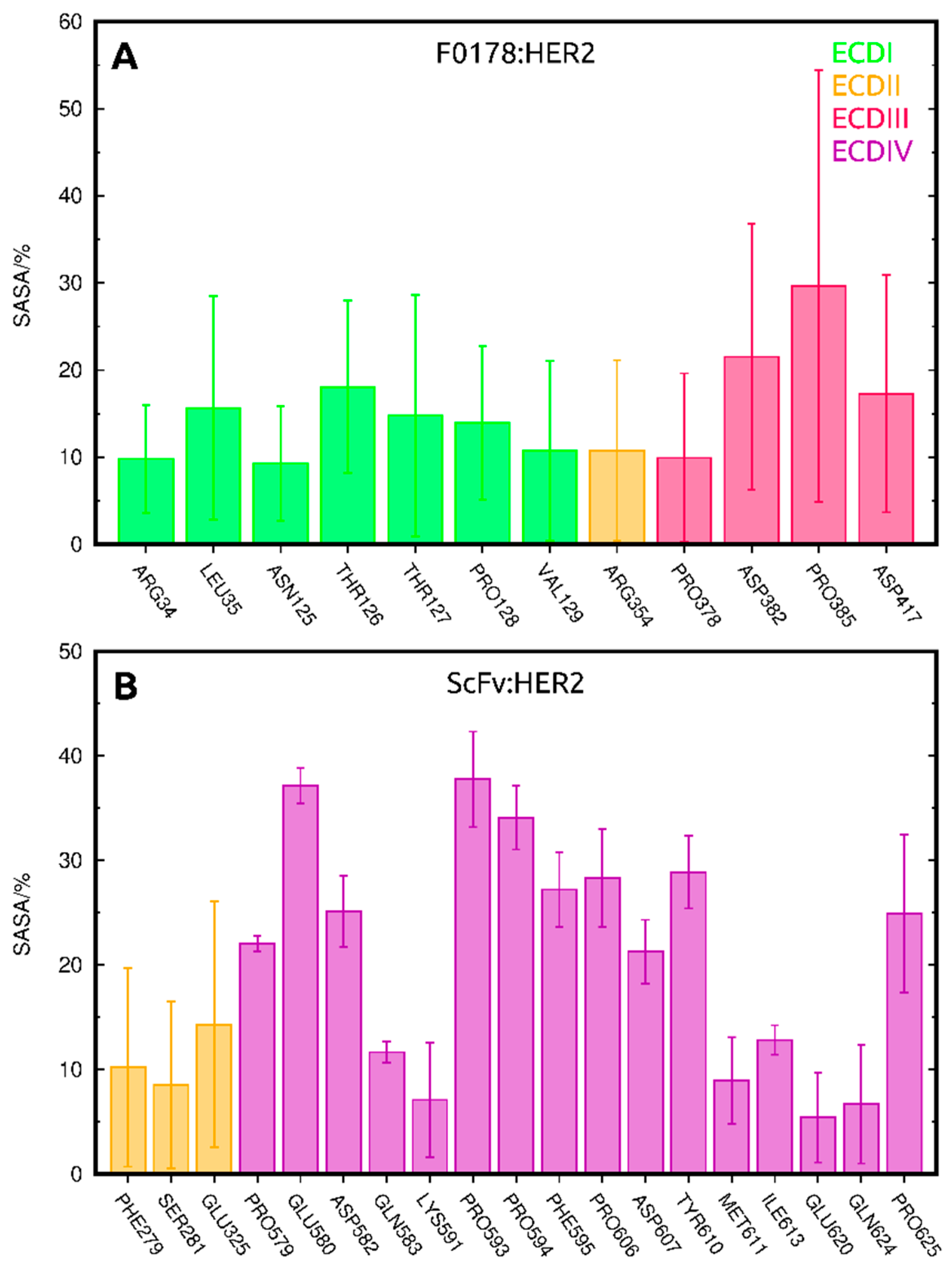
2.3.2. SASA Analysis of Allosterically Modulated Residues
2.4. Principal Component Analysis
2.5. Evolutionary Conservation of Interfacial Residues
3. Results
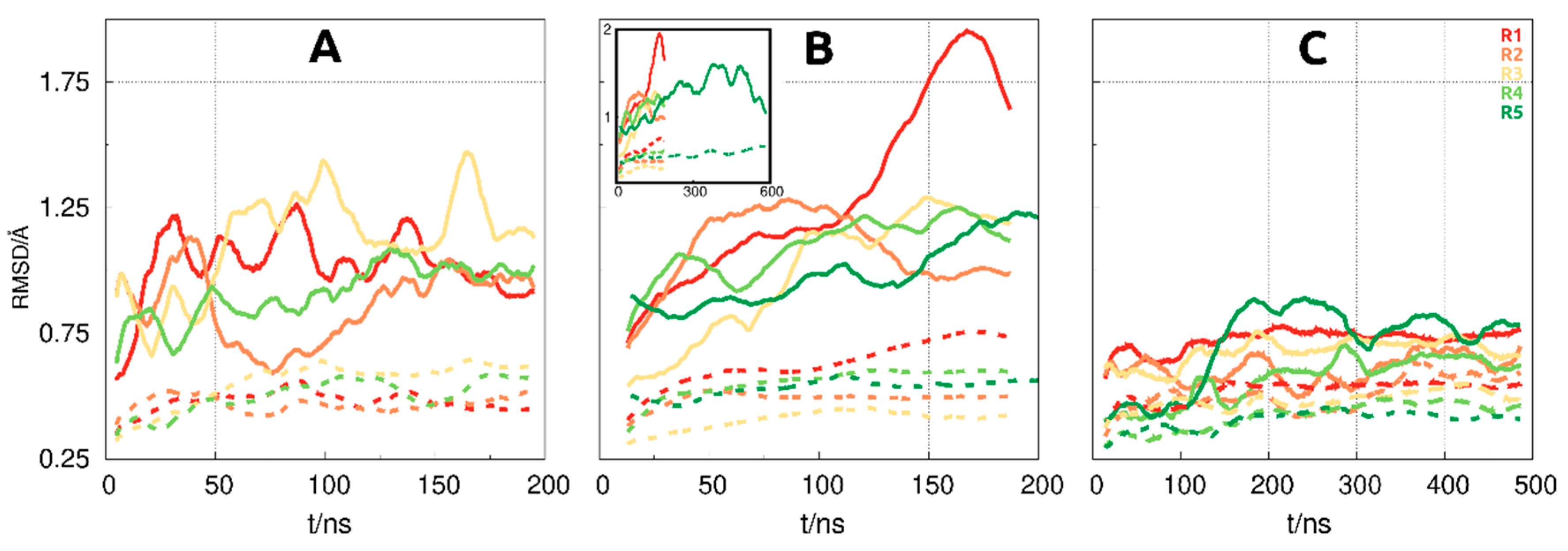
3.1. Reaching Equilibration in the MD Simulations
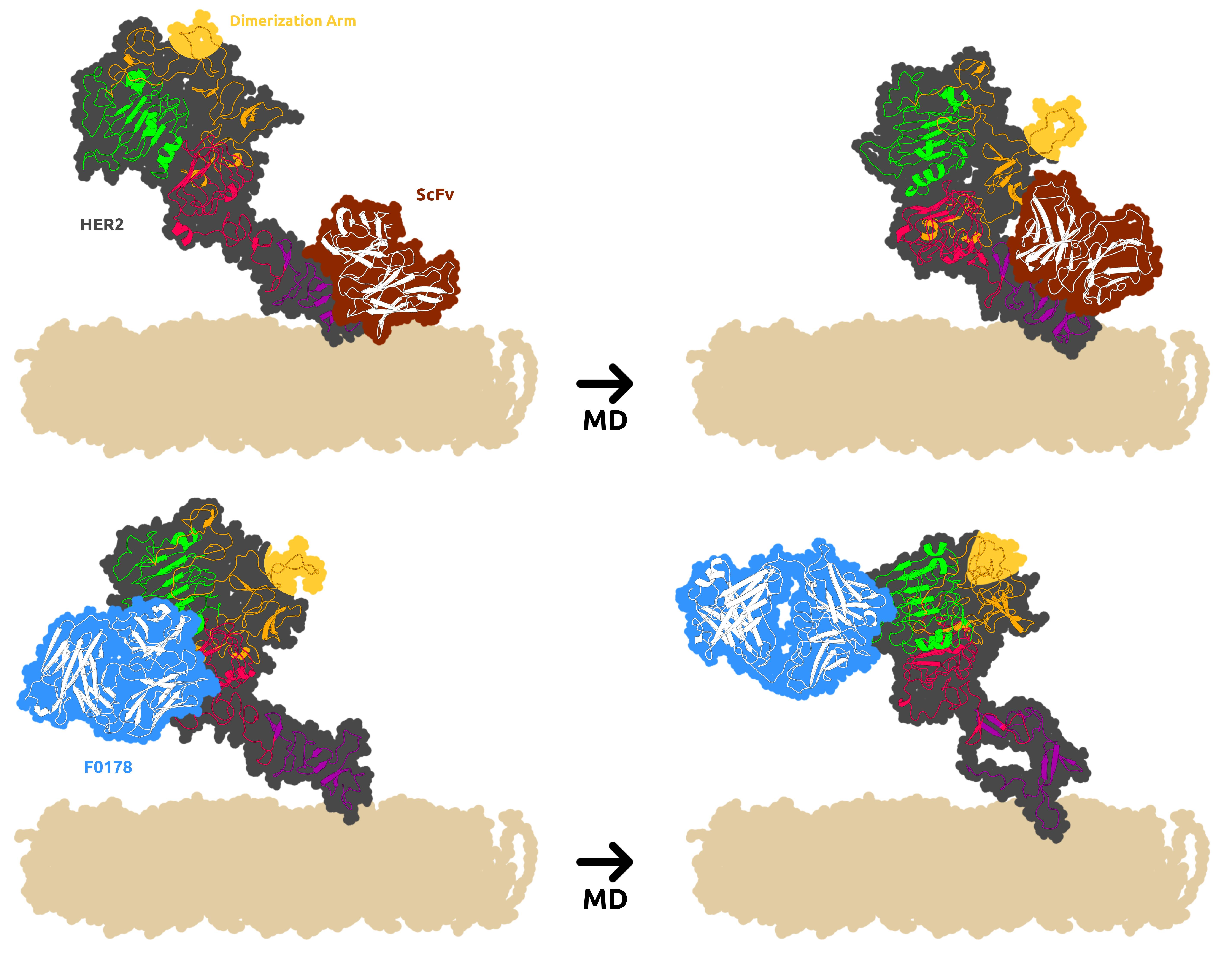
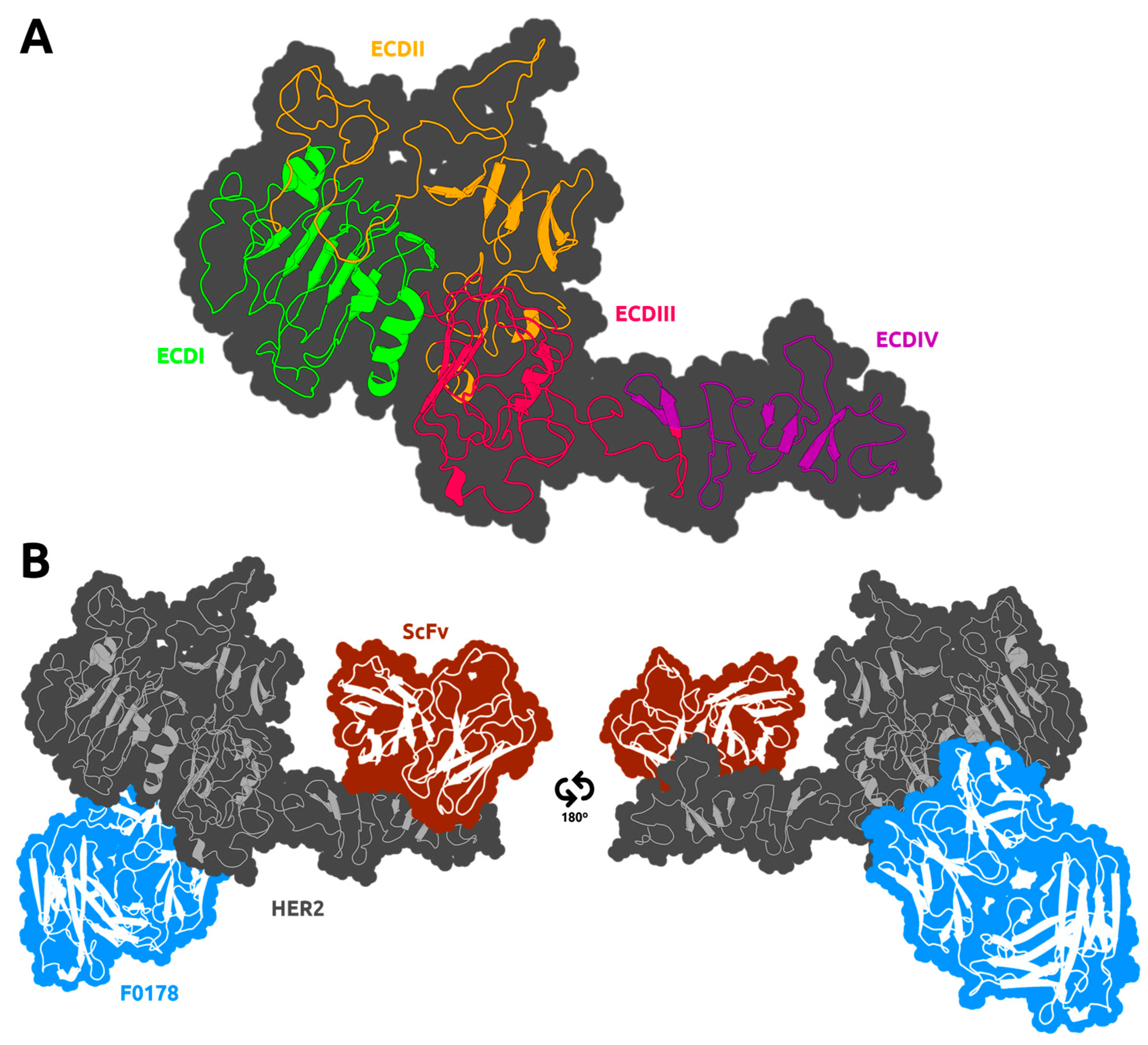
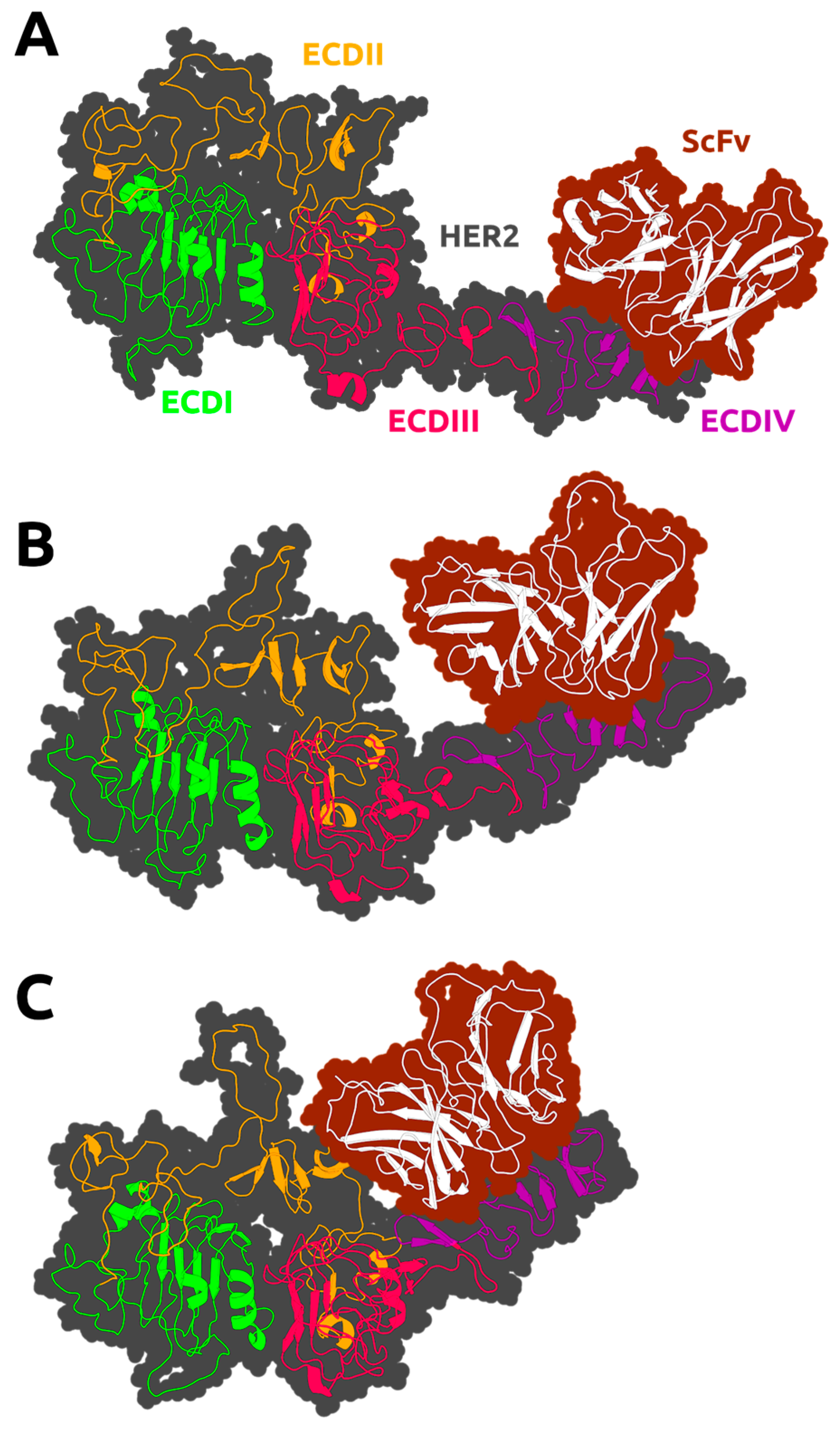
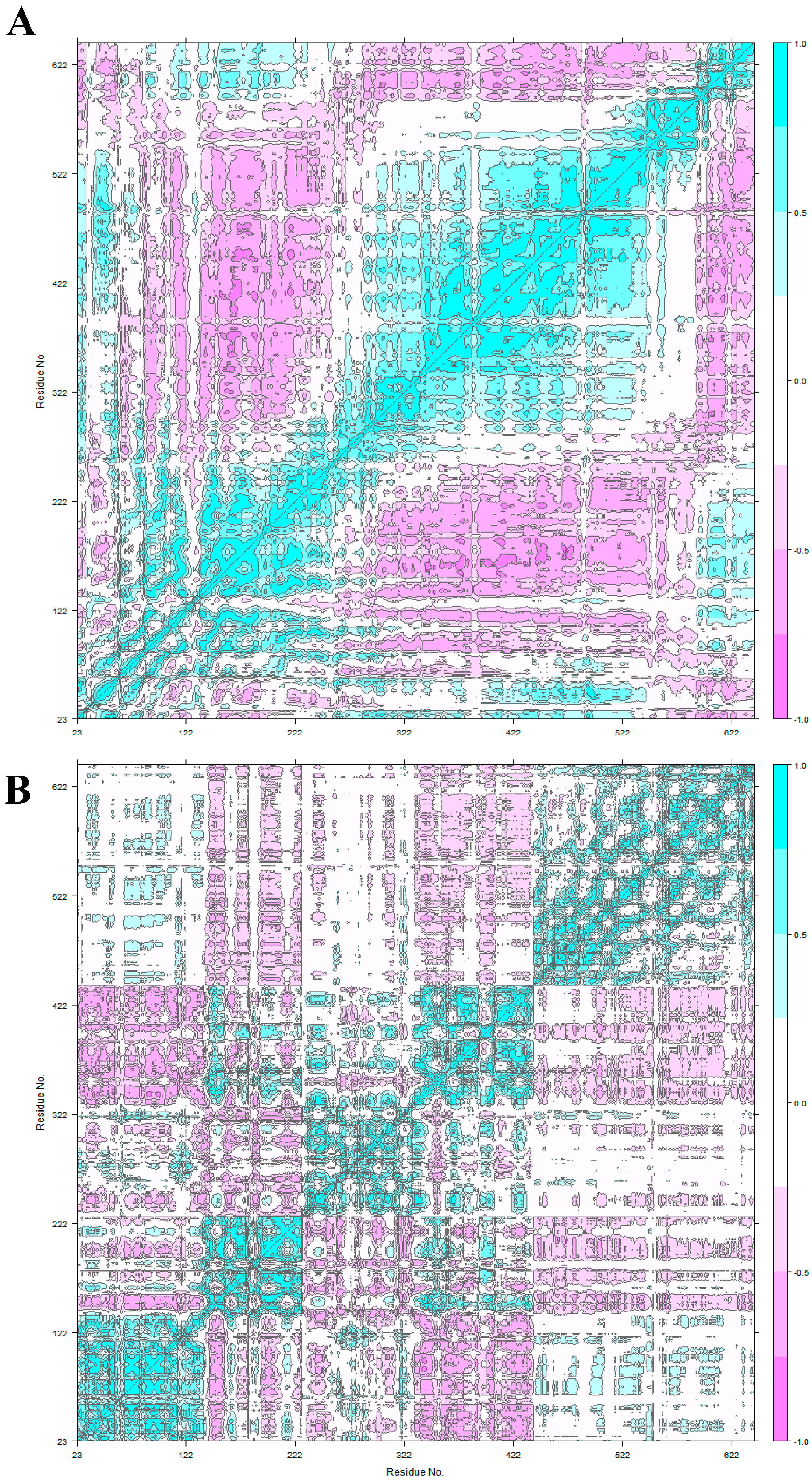
3.2. HER2 Conformational Reorganization
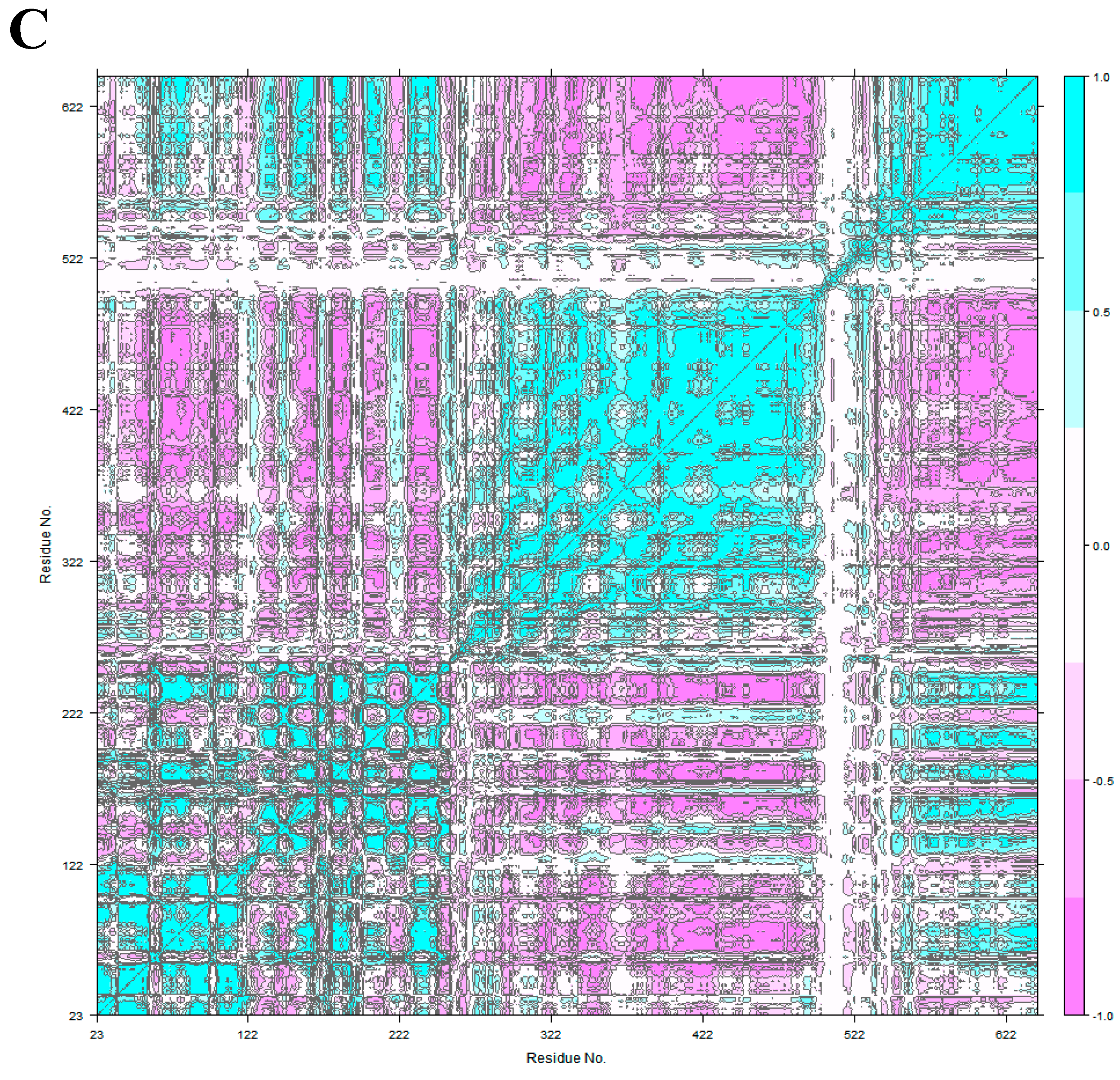
3.3. The Impact of ScFv and F0178 on the Conformational Space of HER2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, J.; Wang, Z.; Chen, Z.; Qiu, J.; Zhang, Y.; Wang, W.; Ma, Y.; Huang, N.; Cui, K.; et al. Cellular Immunotherapy for Carcinoma Using Genetically Modified EGFR-Specific T Lymphocytes. Neoplasia 2013, 15, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Clark, G.; Wong, S.; Levin, W.; Ullrich, A.; McGuire, W. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.T.; Claret, F.X. Trastuzumab: Updated Mechanisms of Action and Resistance in Breast Cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—Mechanism of Action and Use in Clinical Practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.; Yang-Feng, T.; Liao, Y.C.; Chen, E.; Gray, A.; McGrath, J.; Seeburg, P.H.; Libermann, T.A.; Schlessinger, J.; Francke, U.; et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 1985, 230, 1132–1139. [Google Scholar] [CrossRef]
- Rubin, I.; Yarden, Y. The Basic Biology of HER2. Ann. Oncol. 2001, 12, 3–8. [Google Scholar] [CrossRef]
- Lv, Q.; Meng, Z.; Yu, Y.; Jiang, F.; Guan, D.; Liang, C.; Zhou, J.; Lu, A.; Zhang, G. Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer. Int. J. Mol. Sci. 2016, 17, 2095. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef]
- Gutierrez, C.; Schiff, R. HER2: Biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [PubMed]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef]
- Fu, W.; Wang, Y.; Zhang, Y.; Xiong, L.; Takeda, H.; Ding, L.; Xu, Q.; He, L.; Tan, W.; Bethune, A.N.; et al. Insights into HER2 signaling from step-by-step optimization of anti-HER2 antibodies. Mabs 2014, 6, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, R. HER2 therapy. HER2 (ERBB2): Functional diversity from structurally conserved building blocks. Breast Cancer Res. 2007, 9, 202. [Google Scholar] [CrossRef]
- Hu, S.; Sun, Y.; Meng, Y.; Wang, X.; Yang, W.; Fu, W.; Guo, H.; Qian, W.; Hou, S.; Li, B.; et al. Molecular architecture of the ErbB2 extracellular domain homodimer. Oncotarget 2015, 6, 1695–1706. [Google Scholar] [CrossRef]
- Bessman, N.J.; Bagchi, A.; Ferguson, K.M.; Lemmon, M.A. Complex relationship between ligand binding and dimerization in the epidermal growth factor receptor. Cell Rep. 2014, 9, 1306–1317. [Google Scholar] [CrossRef]
- Arkhipov, A.; Shan, Y.; Kim, E.T.; Dror, R.O.; Shaw, D.E. Her2 activation mechanism reflects evolutionary preservation of asymmetric ectodomain dimers in the human EGFR family. Elife 2013, 2, e00708. [Google Scholar] [CrossRef]
- Tai, W.; Mahato, R.; Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef]
- Banappagari, S.; Ronald, S.; Satyanarayanajois, S.D. A Conformationally Constrained Peptidomimetic Binds to the Extracellular Region of HER2 Protein. J. Biomol. Struct. Dyn. 2010, 28, 289–308. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Z.; Xiang, Z.; Li, D.; Hu, Z.; Cui, W.; Geng, L.; Fang, Q. Peptide probes derived from pertuzumab by molecular dynamics modeling for HER2 positive tumor imaging. PLoS Comput. Biol. 2017, 13, e1005441. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Wang, Z.; Yang, X.; Li, D.; Lian, W.; Xiang, Z.; Wang, W.; Bu, X.; Lai, W.; Hu, Z.; et al. Structure-based Design of Peptides with High Affinity and Specificity to HER2 Positive Tumors. Theranostics 2015, 5, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Pieper, U.; Eswar, N.; Braberg, H.; Madhusudhan, M.S.; Davis, F.P.; Stuart, A.C.; Mirkovic, N.; Rossi, A.; Marti-Renom, M.A.; Fiser, A.; et al. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004, 32, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.D.; Ultsch, M.; Lingel, A.; Schaefer, G.; Shao, L.; Birtalan, S.; Sidhu, S.S.; Eigenbrot, C. Structure of the Complex between HER2 and an Antibody Paratope Formed by Side Chains from Tryptophan and Serine. J. Mol. Biol. 2010, 402, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35 (Suppl. 2), 522–525. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Berendsen, H.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Abdi, H.; Williams, L.J. Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, X.; Han, K. Molecular dynamics simulation of SRP GTPases: Towards an understanding of the complex formation from equilibrium fluctuations. Proteins Struct. Funct. Bioinform. 2010, 78, 2222–2237. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, 344–350. [Google Scholar] [CrossRef]
- Pupko, T.; Bell, R.E.; Mayrose, I.; Glaser, F.; Ben-Tal, N. Rate4Site: An algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 2002, 18, 71–77. [Google Scholar] [CrossRef]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; UniProt, C. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Pietras, R.J.; Arboleda, J.; Reese, D.M.; Wongvipat, N.; Pegram, M.D.; Ramos, L.; Gorman, C.M.; Parker, M.G.; Sliwkowski, M.X.; Slamon, D.J. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene 1995, 10, 2435–2446. [Google Scholar] [PubMed]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
R. Magalhães, P.; Machuqueiro, M.; G. Almeida, J.; Melo, A.; D. S. Cordeiro, M.N.; Cabo Verde, S.; H. Gümüş, Z.; S. Moreira, I.; D. G. Correia, J.; Melo, R. Dynamical Rearrangement of Human Epidermal Growth Factor Receptor 2 upon Antibody Binding: Effects on the Dimerization. Biomolecules 2019, 9, 706. https://doi.org/10.3390/biom9110706
R. Magalhães P, Machuqueiro M, G. Almeida J, Melo A, D. S. Cordeiro MN, Cabo Verde S, H. Gümüş Z, S. Moreira I, D. G. Correia J, Melo R. Dynamical Rearrangement of Human Epidermal Growth Factor Receptor 2 upon Antibody Binding: Effects on the Dimerization. Biomolecules. 2019; 9(11):706. https://doi.org/10.3390/biom9110706
Chicago/Turabian StyleR. Magalhães, Pedro, Miguel Machuqueiro, José G. Almeida, André Melo, M. Natália D. S. Cordeiro, Sandra Cabo Verde, Zeynep H. Gümüş, Irina S. Moreira, João D. G. Correia, and Rita Melo. 2019. "Dynamical Rearrangement of Human Epidermal Growth Factor Receptor 2 upon Antibody Binding: Effects on the Dimerization" Biomolecules 9, no. 11: 706. https://doi.org/10.3390/biom9110706
APA StyleR. Magalhães, P., Machuqueiro, M., G. Almeida, J., Melo, A., D. S. Cordeiro, M. N., Cabo Verde, S., H. Gümüş, Z., S. Moreira, I., D. G. Correia, J., & Melo, R. (2019). Dynamical Rearrangement of Human Epidermal Growth Factor Receptor 2 upon Antibody Binding: Effects on the Dimerization. Biomolecules, 9(11), 706. https://doi.org/10.3390/biom9110706