Synthesis of β-d-galactopyranoside-Presenting Glycoclusters, Investigation of Their Interactions with Pseudomonas aeruginosa Lectin A (PA-IL) and Evaluation of Their Anti-Adhesion Potential

 ,
,  , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Synthesis
2.2.1. General Method A for Azide-Alkyne Click Reaction
2.2.2. General Method B for Zemplén-Deacetylation
2.2.3. Compound 3
2.2.4. Compound 4
2.2.5. Compound 6
2.2.6. Compound 7
2.2.7. Compound 9
2.2.8. Compound 10
2.2.9. Compound 12
2.2.10. Compound 13
2.3. Lectin PA-IL Production and Purification
2.4. Hemagglutination Inhibition Assay (HIA)
2.5. Surface Plasmon Resonance (SPR)
2.6. Isothermal Titration Microcalorimetry (ITC)
2.7. Analytical Ultracentrifugation (AUC)
2.8. Pseudomonas Aeruginosa Cross-Linking Assay
2.9. Pseudomonas Aeruginosa Adherence Assay
2.9.1. Cell Labelling
2.9.2. Inhibition of Pseudomonas Aeruginosa Adhesion to Epithelial Cells
3. Results
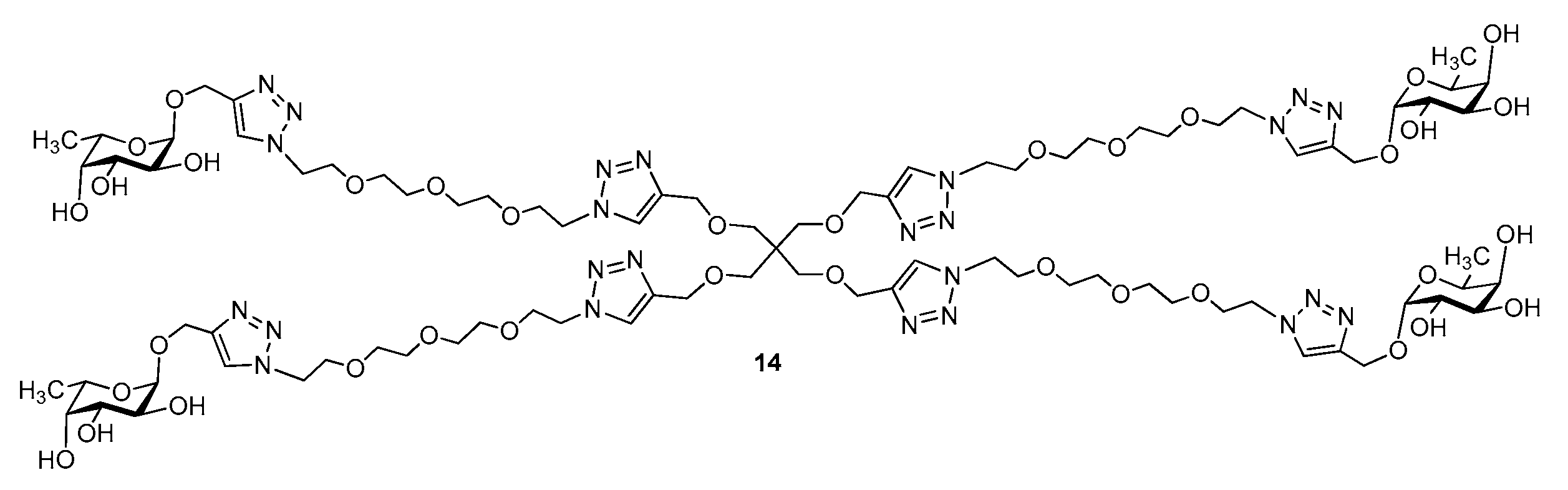
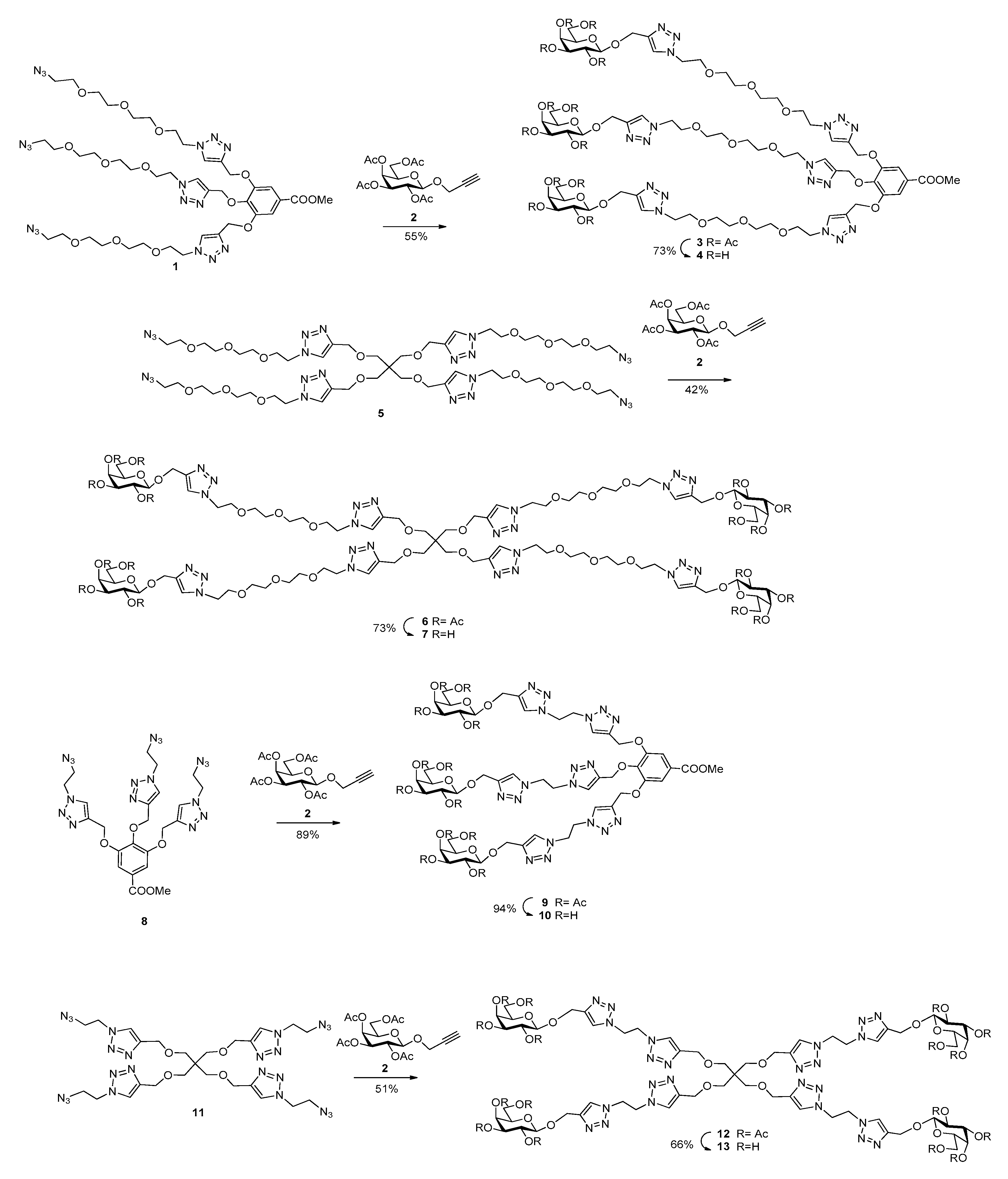
3.1. Synthesis of Multivalent Galactosides
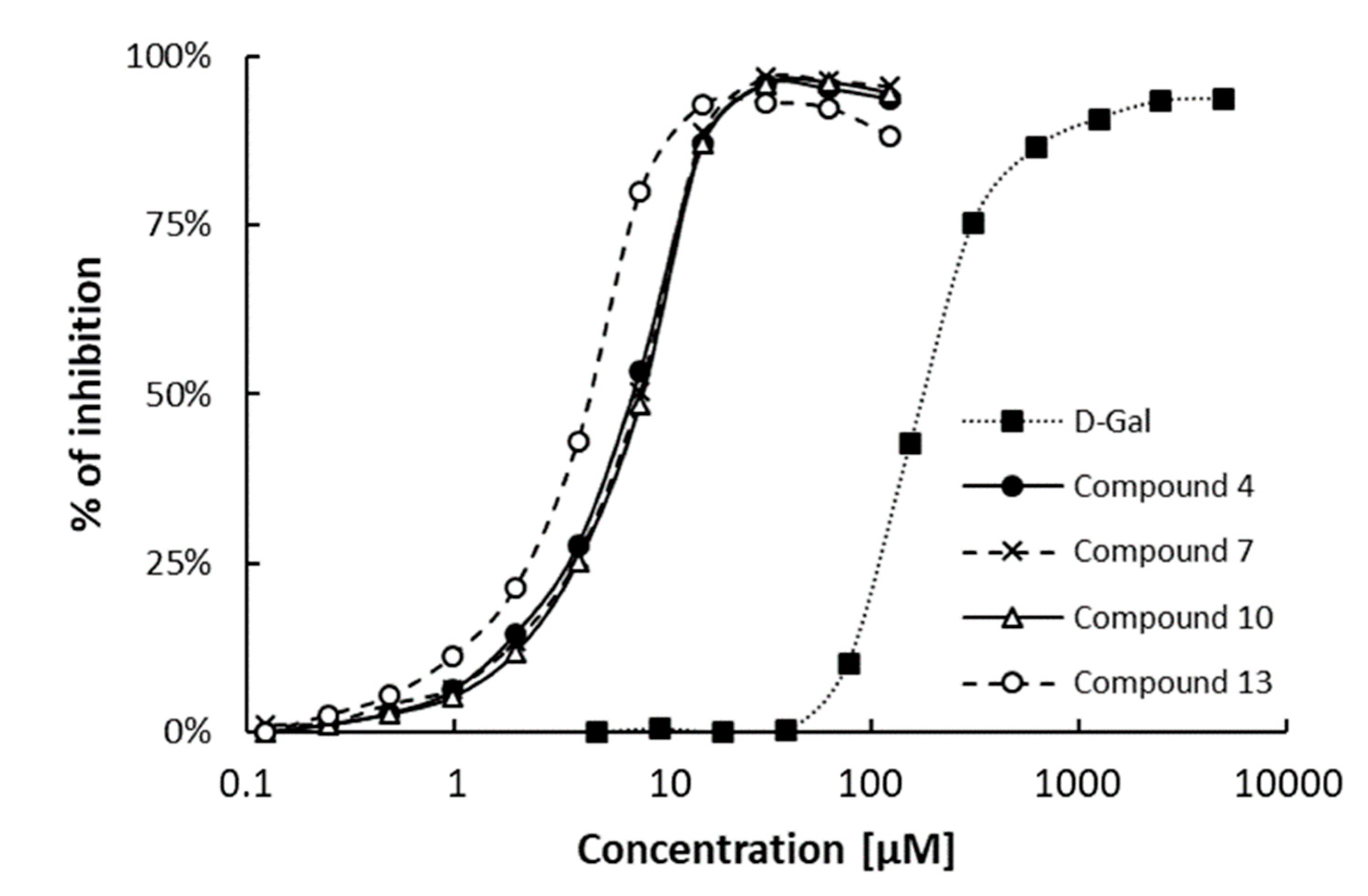
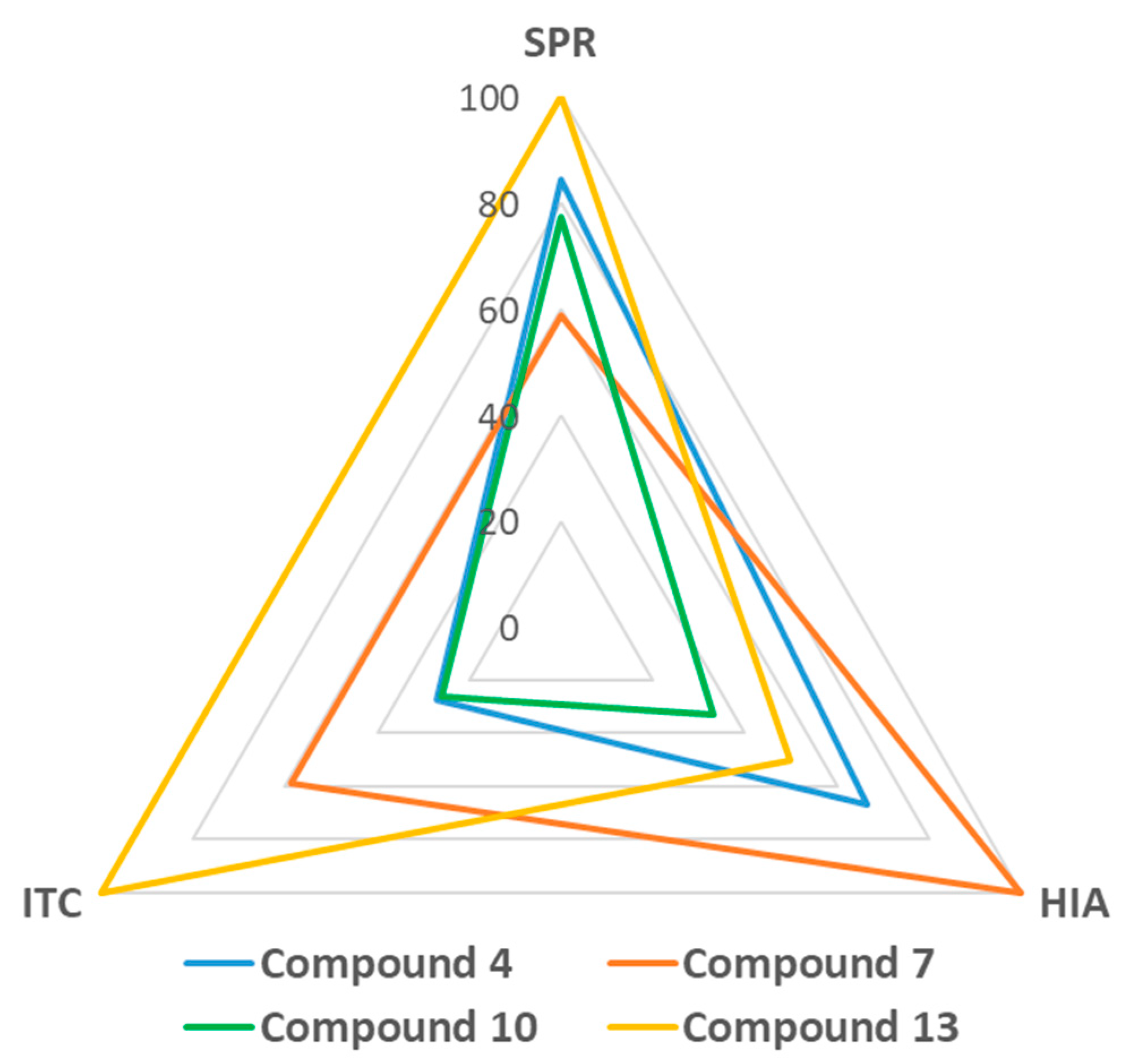
3.2. Inhibition of PA-IL with Multivalent Galactosylated Compounds
3.3. Thermodynamics of the Interactions
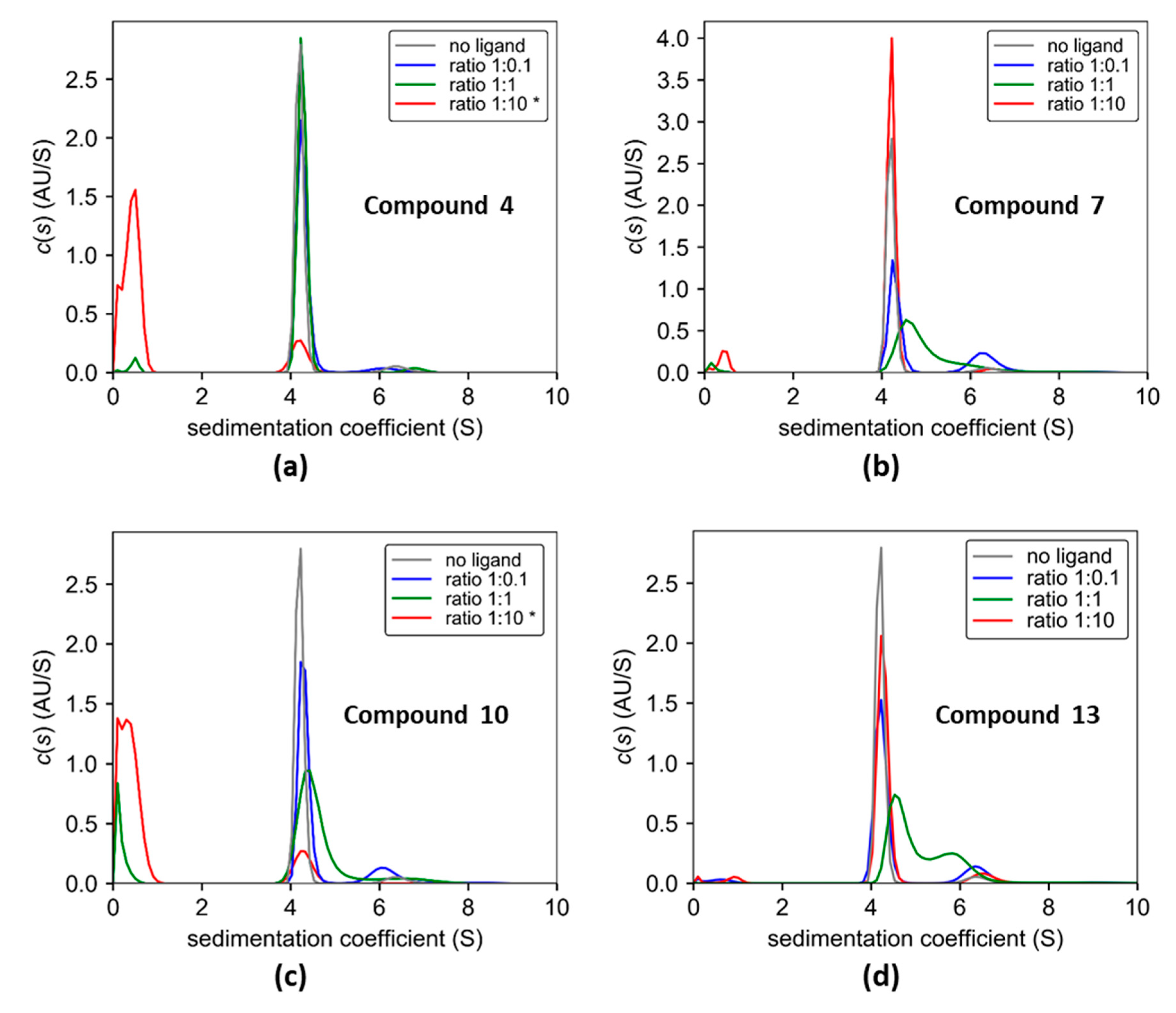
3.4. Cross-linking of Lectin PA-IL with Multivalent Galactosylated Inhibitors
3.5. Cross-linking of Pseudomonas Aeruginosa Cells with Multivalent Galactosylated Inhibitors
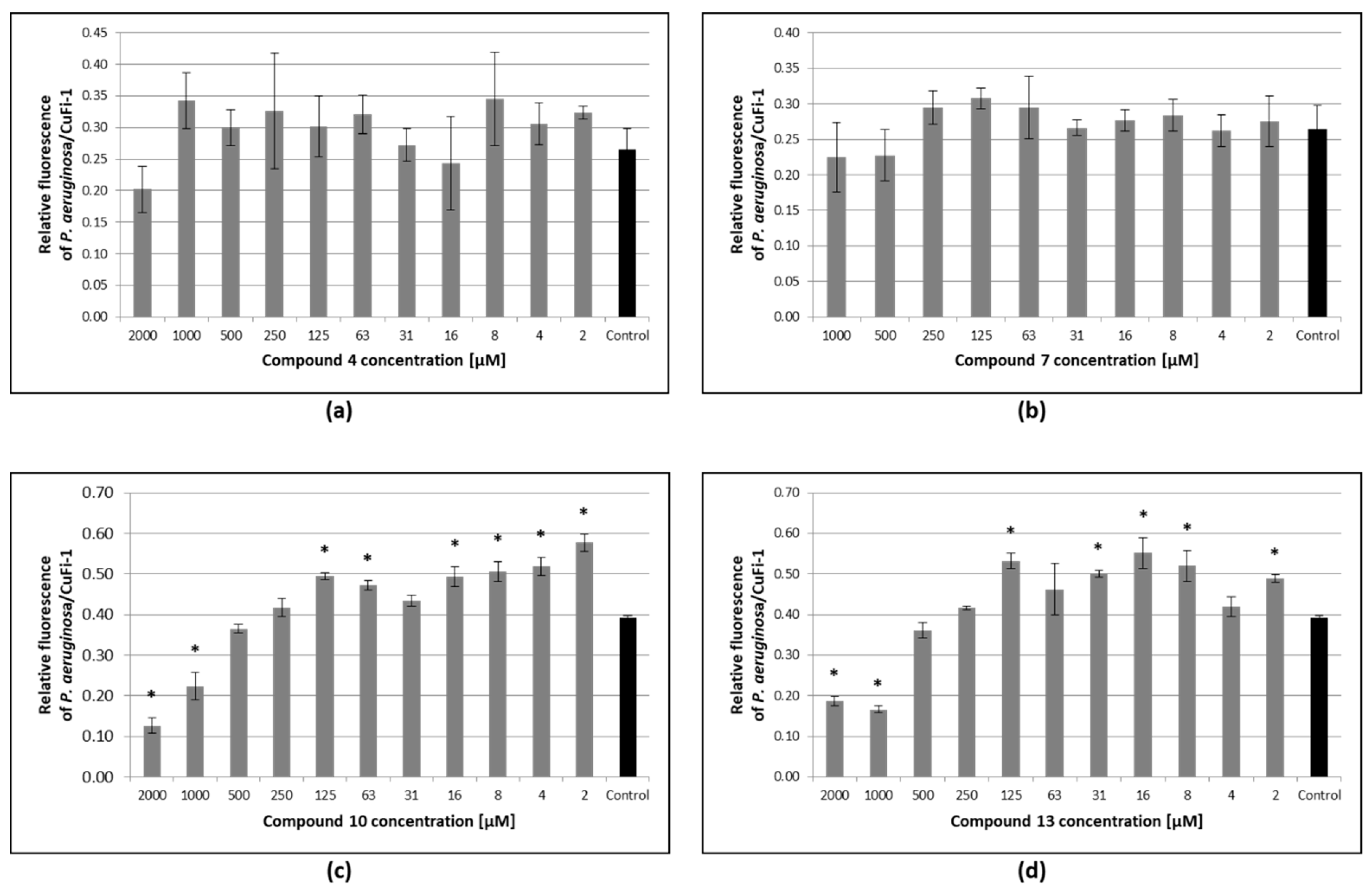
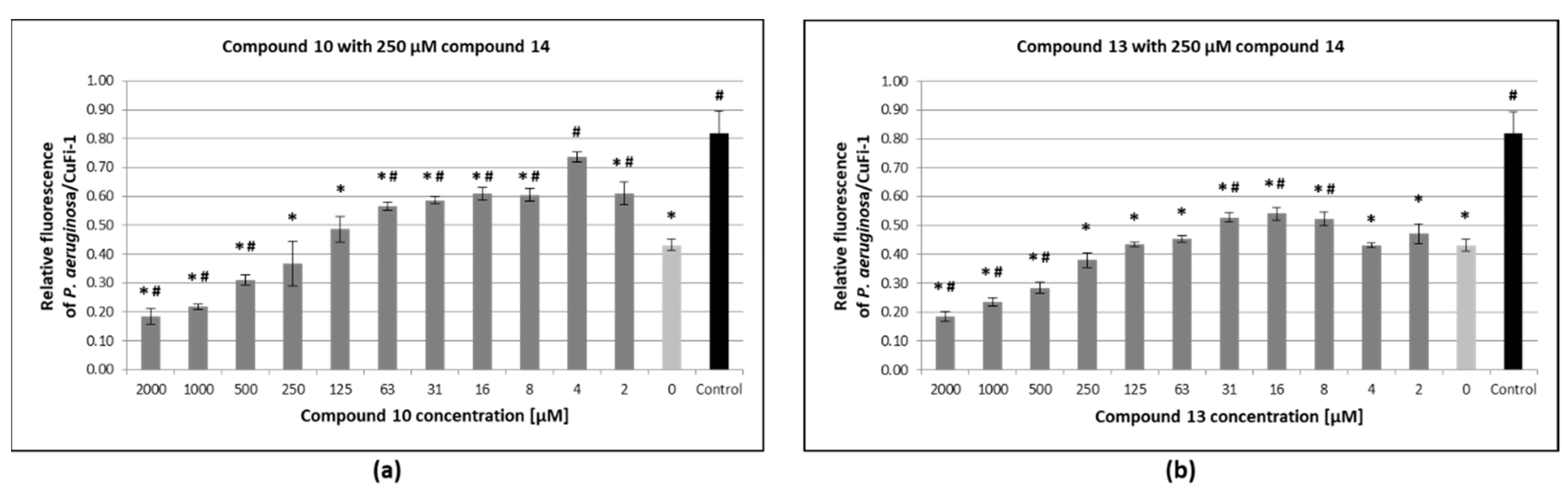
3.6. Inhibition of Pseudomonas Aeruginosa Adhesion to Epithelial Bronchial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sharon, N.; Lis, H. Lectins: Carbohydrate-specific proteins that mediate cellular recognition. Chem. Rev. 1998, 98, 637–674. [Google Scholar] [CrossRef]
- Sharon, N. Carbohydrates as future anti-adhesion drugs for infectious diseases. Biochim. Biophys. Acta 2006, 1760, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Cecioni, S.; Imberty, A.; Vidal, S. Glycomimetics versus Multivalent Glycoconjugates for the Design of High Affinity Lectin Ligands. Chem. Rev. 2015, 115, 525–561. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Høiby, N.; Molin, S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Microbiol. 2012, 10, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Gilboa-Garber, N. Inhibition of broad spectrum hemagglutinin from Pseudomonas aeruginosa by D-galactose and its derivatives. FEBS Lett. 1972, 20, 242–244. [Google Scholar] [CrossRef]
- Gilboa-Garber, N. Purification and properties of hemagglutinin from Pseudomonas aeruginosa and its reaction with human blood cells. Biochim. Biophys. Acta 1972, 273, 165–173. [Google Scholar] [CrossRef]
- Cioci, G.; Mitchell, E.P.; Gautier, C.; Wimmerová, M.; Sudakevitz, D.; Pérez, S.; Gilboa-Garber, N.; Imberty, A. Structural basis of calcium and galactose recognition by the lectin PA-IL of Pseudomonas aeruginosa. FEBS Lett. 2003, 555, 297–301. [Google Scholar] [CrossRef]
- Chemani, C.; Imberty, A.; de Bentzmann, S.; Pierre, M.; Wimmerová, M.; Guery, B.P.; Faure, K. Role of LecA and LecB lectins in Pseudomonas aeruginosa-induced lung injury and effect of carbohydrate ligands. Infect. Immun. 2009, 77, 2065–2075. [Google Scholar] [CrossRef]
- Diggle, S.P.; Stacey, R.E.; Dodd, C.; Cámara, M.; Williams, P.; Winzer, K. The galactophilic lectin, LecA, contributes to biofilm development in Pseudomonas aeruginosa. Environ. Microbiol. 2006, 8, 1095–1104. [Google Scholar] [CrossRef]
- Bajolet-Laudinat, O.; Girod-de Bentzmann, S.; Tournier, J.M.; Madoulet, C.; Plotkowski, M.C.; Chippaux, C.; Puchelle, E. Cytotoxicity of Pseudomonas aeruginosa internal lectin PA-I to respiratory epithelial cells in primary culture. Infect. Immun. 1994, 62, 4481–4487. [Google Scholar]
- Cecioni, S.; Oerthel, V.; Iehl, J.; Holler, M.; Goyard, D.; Praly, J.P.; Imberty, A.; Nierengarten, J.F.; Vidal, S. Synthesis of dodecavalent fullerene-based glycoclusters and evaluation of their binding properties towards a bacterial lectin. Chemistry 2011, 17, 3252–3261. [Google Scholar] [CrossRef] [PubMed]
- Chabre, Y.M.; Giguère, D.; Blanchard, B.; Rodrigue, J.; Rocheleau, S.; Neault, M.; Rauthu, S.; Papadopoulos, A.; Arnold, A.A.; Imberty, A.; et al. Combining Glycomimetic and Multivalent Strategies toward Designing Potent Bacterial Lectin Inhibitors. Chem. Eur. J. 2011, 17, 6545–6562. [Google Scholar] [CrossRef] [PubMed]
- Kadam, R.U.; Bergmann, M.; Hurley, M.; Garg, D.; Cacciarini, M.; Swiderska, M.A.; Nativi, C.; Sattler, M.; Smyth, A.R.; Williams, P.; et al. A glycopeptide dendrimer inhibitor of the galactose-specific lectin LecA and of Pseudomonas aeruginosa biofilms. Angew. Chem. Int. Ed. 2011, 50, 10631–10635. [Google Scholar] [CrossRef] [PubMed]
- Soomro, Z.H.; Cecioni, S.; Blanchard, H.; Praly, J.P.; Imberty, A.; Vidal, S.; Matthews, S.E. CuAAC synthesis of resorcin[4]arene-based glycoclusters as multivalent ligands of lectins. Org. Biomol. Chem. 2011, 9, 6587–6597. [Google Scholar] [CrossRef] [PubMed]
- Cecioni, S.; Faure, S.; Darbost, U.; Bonnamour, I.; Parrot-Lopez, H.; Roy, O.; Taillefumier, C.; Wimmerová, M.; Praly, J.P.; Imberty, A.; et al. Selectivity among two lectins: Probing the effect of topology, multivalency and flexibility of “clicked” multivalent glycoclusters. Chem. Eur. J. 2011, 17, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, I.; Blanchard, B.; Borsali, R.; Imberty, A.; Kakuchi, T. Enhancement of plant and bacterial lectin binding affinities by three-dimensional organized cluster glycosides constructed on helical poly(phenylacetylene) backbones. ChemBioChem. 2010, 11, 2399–2408. [Google Scholar] [CrossRef]
- Cecioni, S.; Praly, J.P.; Matthews, S.E.; Wimmerová, M.; Imberty, A.; Vidal, S. Rational design and synthesis of optimized glycoclusters for multivalent lectin-carbohydrate interactions: Influence of the linker arm. Chem. Eur. J. 2012, 18, 6250–6263. [Google Scholar] [CrossRef]
- Kašaková, M.; Malinovská, L.; Klejch, T.; Hlaváčková, M.; Dvořáková, H.; Fujdiarová, E.; Rottnerová, Z.; Maťátková, O.; Lhoták, P.; Wimmerová, M.; et al. Selectivity of original C-hexopyranosyl calix[4]arene conjugates towards lectins of different origin. Carbohydr. Res. 2018, 469, 60–72. [Google Scholar] [CrossRef]
- Palmioli, A.; Sperandeo, P.; Polissi, A.; Airoldi, C. Targeting bacterial biofilm: A new LecA multivalent ligand with inhibitory activity. ChemBioChem 2019. [Google Scholar] [CrossRef]
- Flockton, T.R.; Schnorbus, L.; Araujo, A.; Adams, J.; Hammel, M.; Perez, L.J. Inhibition of Pseudomonas aeruginosa Biofilm Formation with Surface Modified Polymeric Nanoparticles. Pathogens 2019, 8, 55. [Google Scholar] [CrossRef]
- Hu, Y.; Beshr, G.; Garvey, C.J.; Tabor, R.F.; Titz, A.; Wilkinson, B.L. Photoswitchable Janus glycodendrimer micelles as multivalent inhibitors of LecA and LecB from Pseudomonas aeruginosa. Colloids Surf. B Biointerfaces 2017, 159, 605–612. [Google Scholar] [CrossRef]
- Lundquist, J.J.; Toone, E.J. The Cluster Glycoside Effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar] [CrossRef]
- Visini, R.; Jin, X.; Bergmann, M.; Michaud, G.; Pertici, F.; Fu, O.; Pukin, A.; Branson, T.R.; Thies-Weesie, D.M.; Kemmink, J.; et al. Structural Insight into Multivalent Galactoside Binding to Pseudomonas aeruginosa Lectin LecA. ACS Chem. Biol. 2015, 10, 2455–2462. [Google Scholar] [CrossRef] [PubMed]
- Boukerb, A.M.; Rousset, A.; Galanos, N.; Méar, J.B.; Thépaut, M.; Grandjean, T.; Gillon, E.; Cecioni, S.; Abderrahmen, C.; Faure, K.; et al. Antiadhesive Properties of Glycoclusters against Pseudomonas aeruginosa Lung Infection. J. Med. Chem. 2014, 57, 10275–10289. [Google Scholar] [CrossRef] [PubMed]
- Jančaříková, G.; Herczeg, M.; Fujdiarová, E.; Houser, J.; Kövér, K.E.; Borbás, A.; Wimmerová, M.; Csávás, M. Synthesis of α-L-fucopyranoside-presenting glycoclusters and investigation of their interaction with recombinant Photorhabdus asymbiotica lectin (PHL). Chem. Eur. J. 2018, 24, 4055–4068. [Google Scholar] [CrossRef] [PubMed]
- Ruhal, R.; Antti, H.; Rzhepishevska, O.; Boulanger, N.; Barbero, D.R.; Wai, S.N.; Uhlin, B.E.; Ramstedt, M. A multivariate approach to correlate bacterial surface properties to biofilm formation by lipopolysaccharide mutants of Pseudomonas aeruginosa. Colloids Surf. B Biointerfaces 2015, 127, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef]
- Brautigam, C.A. Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol. 2015, 562, 109–133. [Google Scholar] [CrossRef]
- Thai, L.S.; Malinovska, L.; Vašková, M.; Mező, E.; Kelemen, V.; Borbás, A.; Hodek, P.; Wimmerová, M.; Csávás, M. Investigation of the Binding Affinity of a Broad Array of l-Fucosides with Six Fucose-Specific Lectins of Bacterial and Fungal Origin. Molecules 2019, 24, 2262. [Google Scholar] [CrossRef]
- Csávás, M.; Malinovská, L.; Perret, F.; Gyurkó, M.; Illyés, Z.T.; Wimmerová, M.; Borbás, A. Tri- and tetravalent mannoclusters cross-link and aggregate BC2L-A lectin from Burkholderia cenocepacia. Carbohydr. Res. 2017, 437, 1–8. [Google Scholar] [CrossRef][Green Version]
- Herczeg, M.; Mező, E.; Molnár, N.; Ng, S.K.; Lee, Y.C.; Dah-Tsyr Chang, M.; Borbás, A. Inhibitory Effect of Multivalent Rhamnobiosides on Recombinant Horseshoe Crab Plasma Lectin Interactions with Pseudomonas aeruginosa PAO1. Chem. Asian J. 2016, 11, 3398–3413. [Google Scholar] [CrossRef] [PubMed]
- Sumii, Y.; Hibino, H.; Saidalimu, I.; Kawahara, H.; Shibata, N. Design and synthesis of galactose-conjugated fluorinated and non-fluorinated proline oligomers: Towards antifreeze molecules. Chem. Commun. 2018, 54, 9749–9752. [Google Scholar] [CrossRef]
- Fox, J.M.; Zhao, M.; Fink, M.J.; Kang, K.; Whitesides, G.M. The Molecular Origin of Enthalpy/Entropy Compensation in Biomolecular Recognition. Annu. Rev. Biophys. 2018, 47, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Ogawa, H. Hemagglutination (Inhibition) Assay. In Lectins, 1st ed.; Hirabayashi, J., Ed.; Humana Press: New York, NY, USA, 2014; Volume 1200, pp. 47–52. [Google Scholar]
- Schlick, K.H.; Cloninger, M.J. Inhibition binding studies of glycodendrimer-lectin interactions using surface plasmon resonance. Tetrahedron 2010, 66, 5305–5310. [Google Scholar] [CrossRef]
- Gimeno, A.; Delgado, S.; Valverde, P.; Bertuzzi, S.; Berbís, M.A.; Echavarren, J.; Lacetera, A.; Martín-Santamaría, S.; Surolia, A.; Cañada, F.J.; et al. Minimizing the Entropy Penalty for Ligand Binding: Lessons from the Molecular Recognition of the Histo Blood-Group Antigens by Human Galectin-3. Angew. Chem. Int. Ed. 2019, 58, 7268–7272. [Google Scholar] [CrossRef]
- Imberty, A.; Wimmerová, M.; Mitchell, E.P.; Gilboa-Garber, N. Structures of the lectins from Pseudomonas aeruginosa: Insight into the molecular basis for host glycan recognition. Microbes Infect. 2004, 6, 221–228. [Google Scholar] [CrossRef]
- Herrmann, G.; Yang, L.; Wu, H.; Song, Z.; Wang, H.; Høiby, N.; Ulrich, M.; Molin, S.; Riethmüller, J.; Döring, G. Colistin-tobramycin combinations are superior to monotherapy concerning the killing of biofilm Pseudomonas aeruginosa. J. Infect. Dis. 2010, 202, 1585–1592. [Google Scholar] [CrossRef]
- Hauber, H.P.; Schulz, M.; Pforte, A.; Mack, D.; Zabel, P.; Schumacher, U. Inhalation with fucose and galactose for treatment of Pseudomonas aeruginosa in cystic fibrosis patients. Int. J. Med. Sci. 2008, 5, 371–376. [Google Scholar] [CrossRef]
- Kubíčková, B.; Hadrabová, J.; Vašková, L.; Mandys, V.; Stiborová, M.; Hodek, P. Susceptibility of airways to Pseudomonas aeruginosa infection: Mouse neuraminidase model. Monatsh. Chem. 2017, 148, 1993–2002. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | MIC | Potency 2 | Valency | β 3 |
|---|---|---|---|---|
| d-galactose 1 | 6.25 mM | 1 | 1 | 1 |
| Compound 4 | 48.83 µM | 128 | 3 | 42.7 |
| Compound 7 | 24.41 µM | 256 | 4 | 64 |
| Compound 10 | 97.66 µM | 64 | 3 | 21.3 |
| Compound 13 | 48.83 µM | 128 | 4 | 32 |
| Inhibitor | IC50 | Potency 2 | Valency | β 3 |
|---|---|---|---|---|
| d-galactose 1 | 187.1 µM | 1 | 1 | 1 |
| Compound 4 | 6.8 µM | 27.5 | 3 | 9.2 |
| Compound 7 | 7.3 µM | 25.6 | 4 | 6.4 |
| Compound 10 | 7.4 µM | 25.3 | 3 | 8.4 |
| Compound 13 | 4.3 µM | 43.5 | 4 | 10.9 |
| Inhibitor | Valency | n | Ka [104 M−1] |
|---|---|---|---|
| d-galactose | 1 | 0.78 ± 0.05 | 0.68 ± 0.038 |
| Compound 4 | 3 | 0.51 ± 0.01 | 128 ± 15.1 |
| Compound 7 | 4 | 0.32 ± 0.01 | 281 ± 23.5 |
| Compound 10 | 3 | 0.53 ± 0.01 | 122 ± 12.8 |
| Compound 13 | 4 | 0.21 ± 0.01 | 470 ± 54.5 |
| Inhibitor | ΔH [kJ/mol] | −TΔS [kJ/mol] | ΔG [kJ/mol] |
| d-galactose | −37.8 ± 2.92 | 15.9 ± 2.12 | −21.9 ± 1.23 |
| Compound 4 | −80.7 ± 1.01 | 45.9 ± 5.99 | −34.8 ± 4.11 |
| Compound 7 | −110.0 ± 0.82 | 72.7 ± 6.65 | −36.8 ± 3.09 |
| Compound 10 | −68.2 ± 0.75 | 33.4 ± 3.87 | −34.7 ± 3.64 |
| Compound 13 | −134.0 ± 1.45 | 95.7 ± 12.15 | −38.1 ± 4.42 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malinovská, L.; Le, S.T.; Herczeg, M.; Vašková, M.; Houser, J.; Fujdiarová, E.; Komárek, J.; Hodek, P.; Borbás, A.; Wimmerová, M.; et al. Synthesis of β-d-galactopyranoside-Presenting Glycoclusters, Investigation of Their Interactions with Pseudomonas aeruginosa Lectin A (PA-IL) and Evaluation of Their Anti-Adhesion Potential. Biomolecules 2019, 9, 686. https://doi.org/10.3390/biom9110686
Malinovská L, Le ST, Herczeg M, Vašková M, Houser J, Fujdiarová E, Komárek J, Hodek P, Borbás A, Wimmerová M, et al. Synthesis of β-d-galactopyranoside-Presenting Glycoclusters, Investigation of Their Interactions with Pseudomonas aeruginosa Lectin A (PA-IL) and Evaluation of Their Anti-Adhesion Potential. Biomolecules. 2019; 9(11):686. https://doi.org/10.3390/biom9110686
Chicago/Turabian StyleMalinovská, Lenka, Son Thai Le, Mihály Herczeg, Michaela Vašková, Josef Houser, Eva Fujdiarová, Jan Komárek, Petr Hodek, Anikó Borbás, Michaela Wimmerová, and et al. 2019. "Synthesis of β-d-galactopyranoside-Presenting Glycoclusters, Investigation of Their Interactions with Pseudomonas aeruginosa Lectin A (PA-IL) and Evaluation of Their Anti-Adhesion Potential" Biomolecules 9, no. 11: 686. https://doi.org/10.3390/biom9110686
APA StyleMalinovská, L., Le, S. T., Herczeg, M., Vašková, M., Houser, J., Fujdiarová, E., Komárek, J., Hodek, P., Borbás, A., Wimmerová, M., & Csávás, M. (2019). Synthesis of β-d-galactopyranoside-Presenting Glycoclusters, Investigation of Their Interactions with Pseudomonas aeruginosa Lectin A (PA-IL) and Evaluation of Their Anti-Adhesion Potential. Biomolecules, 9(11), 686. https://doi.org/10.3390/biom9110686