Disease Associated Mutations in KIR Proteins Linked to Aberrant Inward Rectifier Channel Trafficking




Abstract
1. Introduction
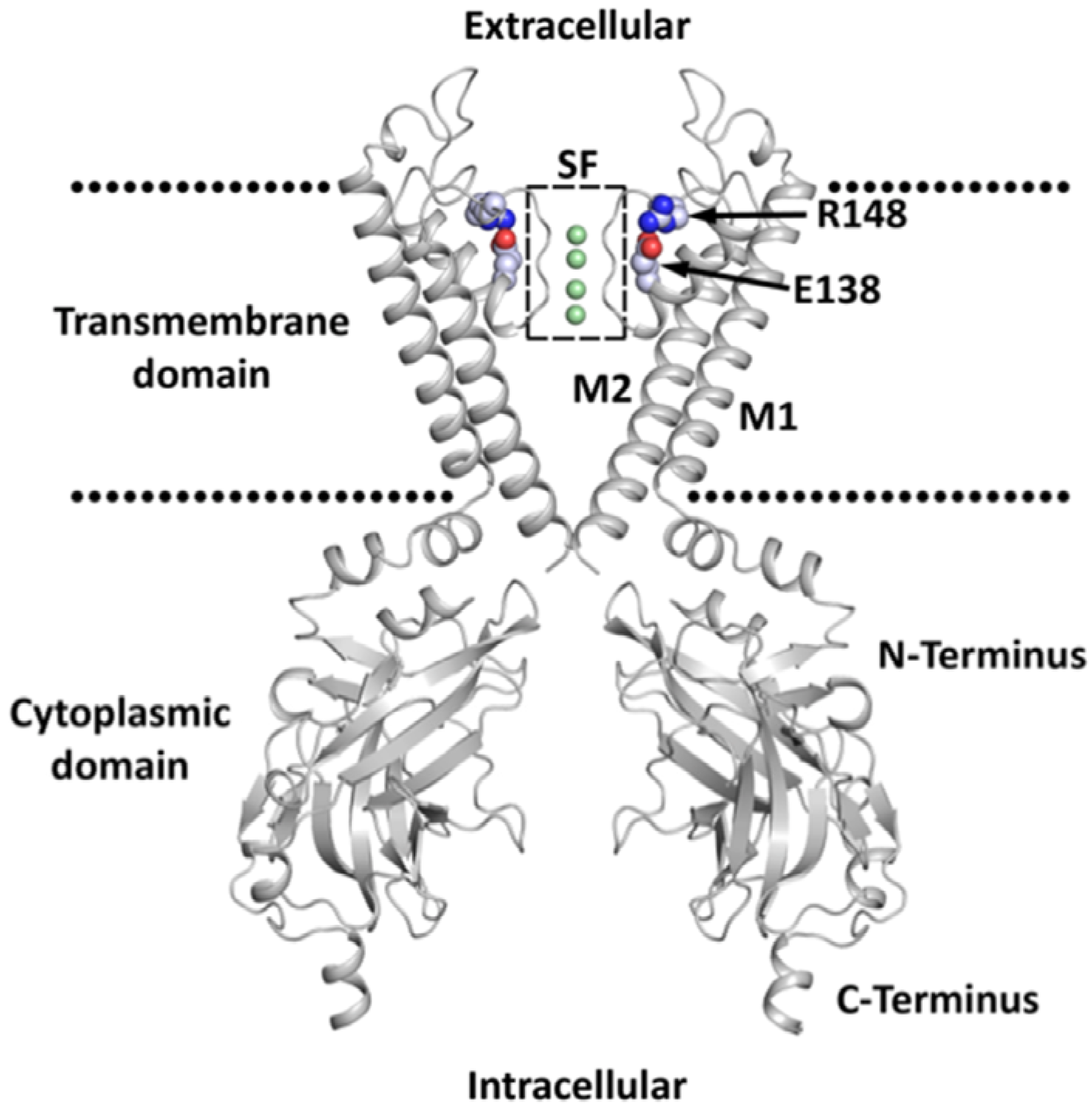
2. Classification, Structure, and Expression
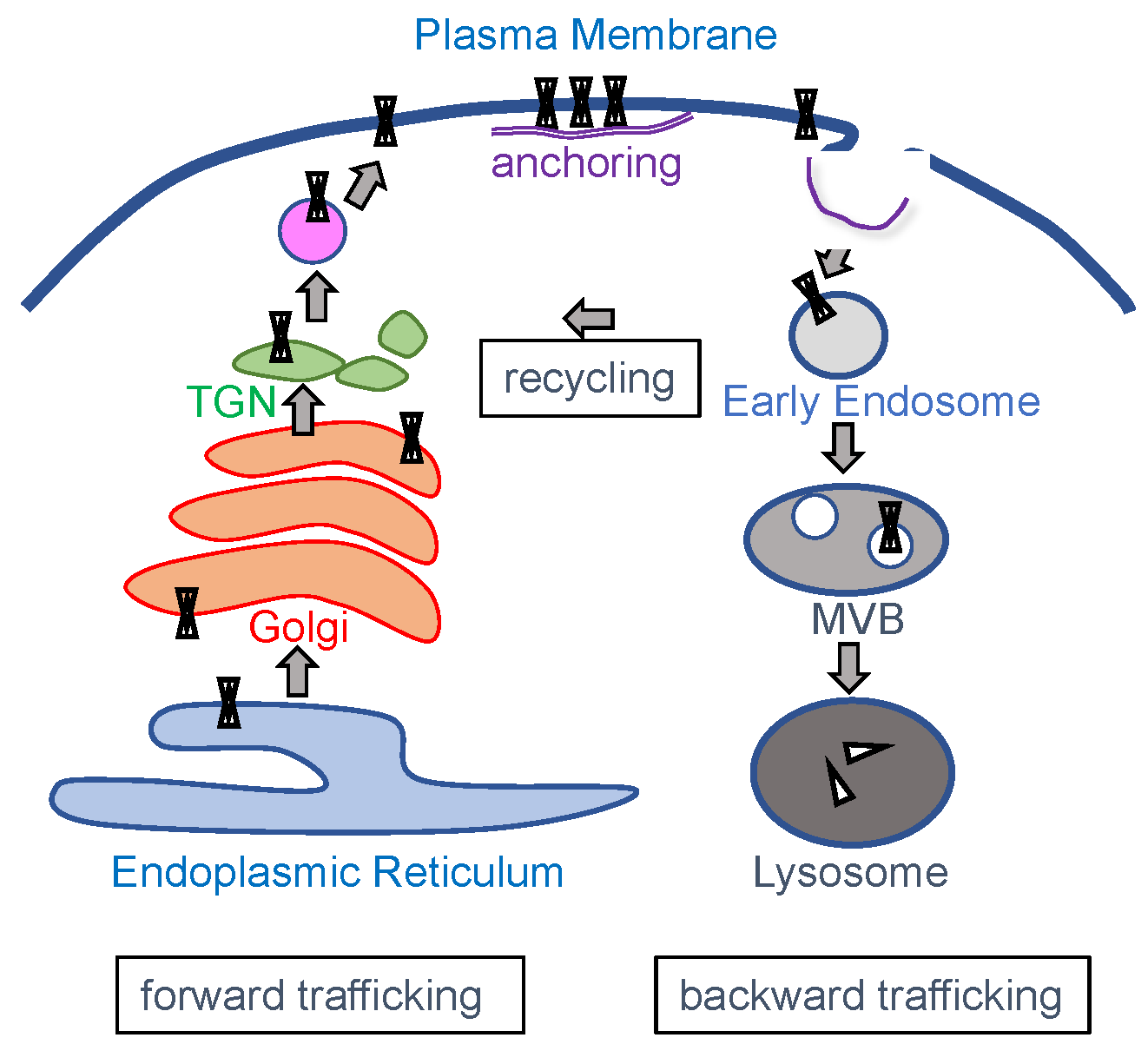
3. Channel Trafficking
4. Diseases and Syndromes Associated with KIR Channel Dysfunction
5. KIR Protein Alignment of Trafficking Associated Disease Mutations
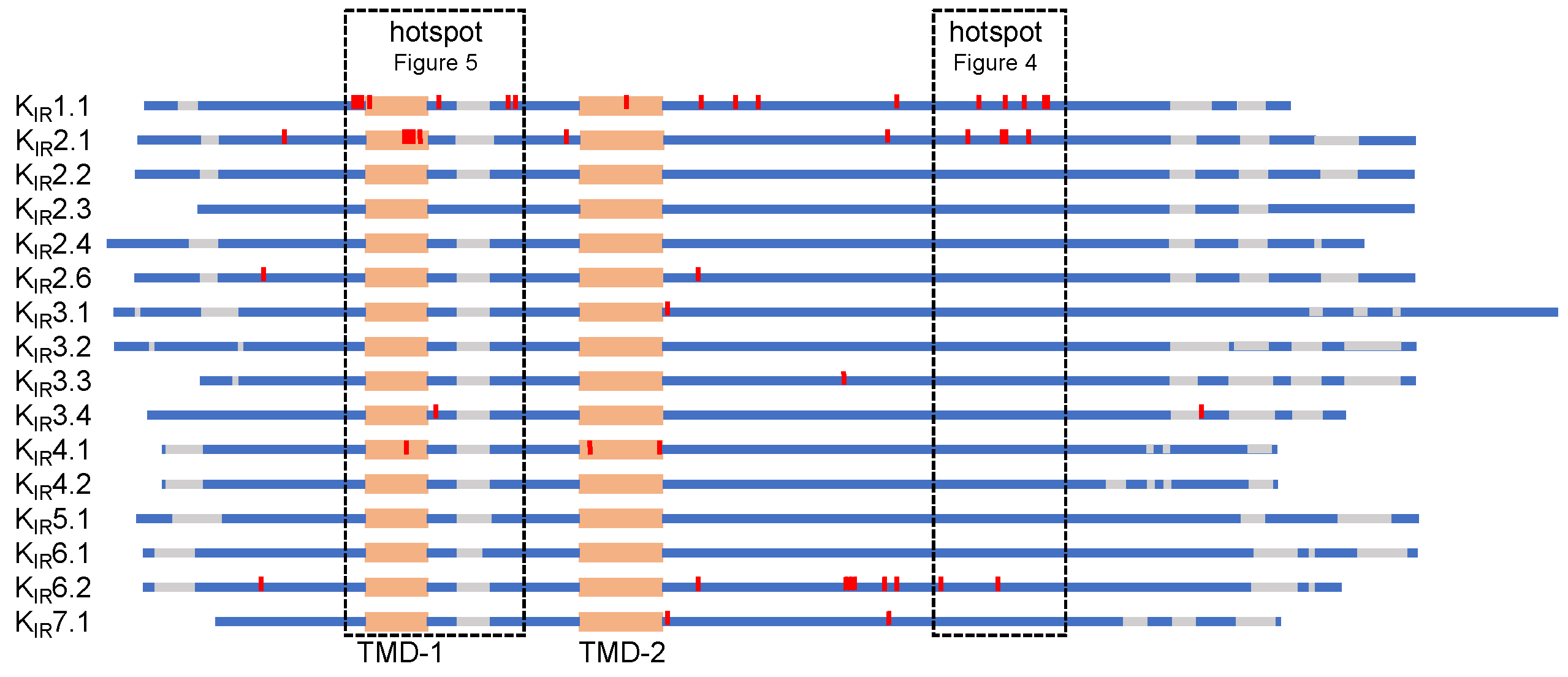
5.1. C-Terminal Trafficking Mutation Hotspot
5.2. Transmembrane Region 1 Mutation Hotspot
5.3. N-Terminal Golgi-Export Patch, KIR2.x ER Export, and KIR6.x ER Exit and Retention Signals
6. Structural Mapping of Trafficking Defect Causing Mutants
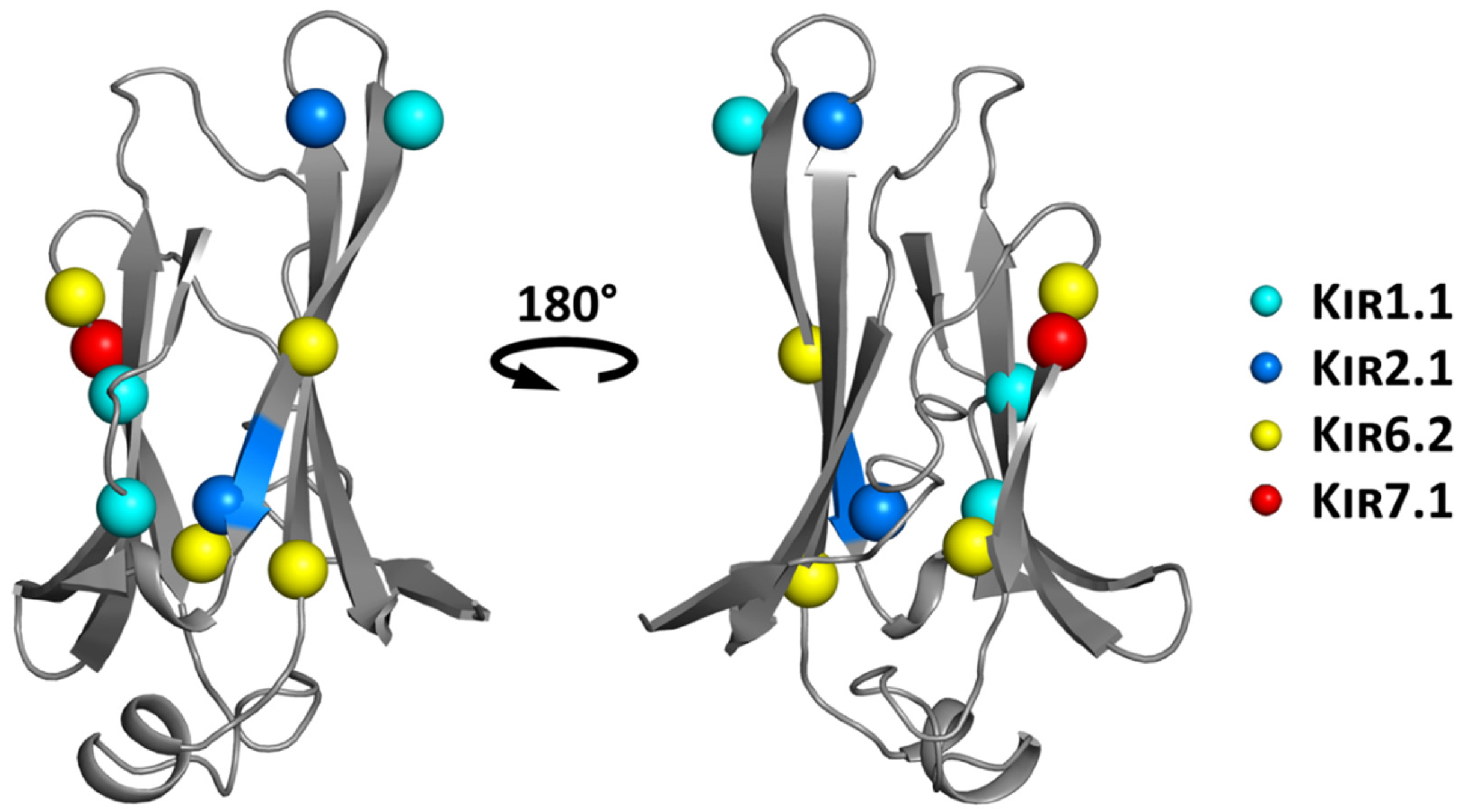
6.1. Structure-Based Hotspot in the IgLD Beta Barrel of the CTD
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Katz, B. Les constantes électriques de la membrane du muscle. Arch. Sci. Physiol. 1949, 2, 285–299. [Google Scholar]
- Matsuda, H.; Saigusa, A.; Irisawa, H. Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature 1987, 325, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Lopatin, A.N.; Makhina, E.N.; Nichols, C.G. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 1994, 372, 366–369. [Google Scholar] [CrossRef] [PubMed]
- De Boer, T.P.; Houtman, M.J.; Compier, M.; Van der Heyden, M.A. The mammalian KIR2. x inward rectifier ion channel family: Expression pattern and pathophysiology. Acta Physiol. 2010, 199, 243–256. [Google Scholar] [CrossRef]
- Wang, L.; Chiamvimonvat, N.; Duff, H.J. Interaction between selected sodium and potassium channel blockers in guinea pig papillary muscle. J. Pharmacol. Exp. Ther. 1993, 264, 1056–1062. [Google Scholar]
- Kokubun, S.; Nishimura, M.; Noma, A.; Irisawa, H. Membrane currents in the rabbit atrioventricular node cell. Pflügers Arch. 1982, 393, 15–22. [Google Scholar] [CrossRef]
- Yang, J.; Yu, M.; Jan, Y.N.; Jan, L.Y. Stabilization of ion selectivity filter by pore loop ion pairs in an inwardly rectifying potassium channel. Proc. Natl. Acad. Sci. USA 1997, 94, 1568–1572. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Gordon, E.A.; Wickman, K.; Velimirović, B.; Krapivinsky, L.; Clapham, D.E. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature 1995, 374, 135–141. [Google Scholar] [CrossRef]
- Lüscher, C.; Slesinger, P.A. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010, 11, 301–315. [Google Scholar] [CrossRef]
- Ma, D.; Zerangue, N.; Lin, Y.F.; Collins, A.; Yu, M.; Jan, Y.N.; Jan, L.Y. Role of ER export signals in controlling surface potassium channel numbers. Science 2001, 291, 316–319. [Google Scholar] [CrossRef]
- Stockklausner, C.; Ludwig, J.; Ruppersberg, J.P.; Klöcker, N. A sequence motif responsible for ER export and surface expression of Kir2.0 inward rectifier K+ channels. FEBS Lett. 2001, 493, 129–133. [Google Scholar] [CrossRef]
- Ma, D.; Zerangue, N.; Raab-Graham, K.; Fried, S.R.; Jan, Y.N.; Jan, L.Y. Diverse trafficking patterns due to multiple traffic motifs in G protein-activated inwardly rectifying potassium channels from brain and heart. Neuron 2002, 33, 715–729. [Google Scholar] [CrossRef]
- Zerangue, N.; Schwappach, B.; Jan, Y.N.; Jan, L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 1999, 22, 537–548. [Google Scholar] [CrossRef]
- Bundis, F.; Neagoe, I.; Schwappach, B.; Steinmeyer, K. Involvement of Golgin-160 in cell surface transport of renal ROMK channel: Co-expression of Golgin-160 increases ROMK currents. Cell Physiol. Biochem. 2006, 17, 1–12. [Google Scholar] [CrossRef]
- Taneja, T.K.; Ma, D.; Kim, B.Y.; Welling, P.A. Golgin-97 Targets Ectopically Expressed Inward Rectifying Potassium Channel, Kir2.1, to the trans-Golgi Network in COS-7 Cells. Front. Physiol. 2018, 9, 1070. [Google Scholar] [CrossRef]
- Ma, D.; Taneja, T.K.; Hagen, B.M.; Kim, B.Y.; Ortega, B.; Lederer, W.J.; Welling, P.A. Golgi export of the Kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell 2011, 145, 1102–1115. [Google Scholar] [CrossRef]
- Li, X.; Ortega, B.; Kim, B.; Welling, P.A. A Common Signal Patch Drives AP-1 Protein-dependent Golgi Export of Inwardly Rectifying Potassium Channels. J. Biol. Chem. 2016, 291, 14963–14972. [Google Scholar] [CrossRef]
- Zeng, W.Z.; Babich, V.; Ortega, B.; Quigley, R.; White, S.J.; Welling, P.A.; Huang, C.L. Evidence for endocytosis of ROMK potassium channel via clathrin-coated vesicles. Am. J. Physiol. Renal Physiol. 2002, 283, 630–639. [Google Scholar] [CrossRef][Green Version]
- Mackie, T.D.; Kim, B.Y.; Subramanya, A.R.; Bain, D.J.; O’Donnell, A.F.; Welling, P.A.; Brodsky, J.L. The endosomal trafficking factors CORVET and ESCRT suppress plasma membrane residence of the renal outer medullary potassium channel (ROMK). J. Biol. Chem. 2018, 293, 3201–3217. [Google Scholar] [CrossRef]
- Kolb, A.R.; Needham, P.G.; Rothenberg, C.; Guerriero, C.J.; Welling, P.A.; Brodsky, JL. ESCRT regulates surface expression of the Kir2.1 potassium channel. Mol. Biol. Cell 2014, 25, 276–289. [Google Scholar] [CrossRef]
- Jansen, J.A.; de Boer, T.P.; Wolswinkel, R.; van Veen, T.A.; Vos, M.A.; van Rijen, H.V.M.; van der Heyden, M.A.G. Lysosome mediated Kir2.1 breakdown directly influences inward rectifier current density. Biochem. Biophys. Res. Commun. 2008, 367, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Varkevisser, R.; Houtman, M.J.; Waasdorp, M.; Man, J.C.; Heukers, R.; Takanari, H.; Tieland, R.G.; van Bergen En Henegouwen, P.M.; Vos, M.A.; van der Heyden, M.A. Inhibiting the clathrin-mediated endocytosis pathway rescues K(IR)2.1 downregulation by pentamidine. Pflugers Arch. 2013, 465, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.X.; Su, X.T.; Wu, P.; Gao, Z.X.; Wang, W.H.; Staub, O.; Lin, D.H. Kir5.1 regulates Nedd4-2-mediated ubiquitination of Kir4.1 in distal nephron. Am. J. Physiol. Renal Physiol. 2018, 315, F986–F996. [Google Scholar] [CrossRef] [PubMed]
- Leonoudakis, D.; Mailliard, W.; Wingerd, K.; Clegg, D.; Vandenberg, C. Inward rectifier potassium channel Kir2.2 is associated with synapse-associated protein SAP97. J. Cell Sci. 2001, 114, 987–998. [Google Scholar] [PubMed]
- Leonoudakis, D.; Conti, L.R.; Radeke, C.M.; McGuire, L.M.; Vandenberg, C.A. A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J. Biol. Chem. 2004, 279, 19051–19063. [Google Scholar] [CrossRef] [PubMed]
- Leonoudakis, D.; Conti, L.R.; Anderson, S.; Radeke, C.M.; McGuire, L.M.; Adams, M.E.; Froehner, S.C.; Yates, J.R., 3rd; Vandenberg, C.A. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J. Biol. Chem. 2004, 279, 22331–22346. [Google Scholar] [CrossRef]
- Pegan, S.; Tan, J.; Huang, A.; Slesinger, P.A.; Riek, R.; Choe, S. NMR studies of interactions between C-terminal tail of Kir2.1 channel and PDZ1,2 domains of PSD95. Biochemistry 2007, 46, 5315–5322. [Google Scholar] [CrossRef][Green Version]
- Brasko, C.; Hawkins, V.; De La Rocha, I.C.; Butt, A.M. Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Brain Struct. Funct. 2017, 222, 41–59. [Google Scholar] [CrossRef]
- Tanemoto, M.; Fujita, A.; Higashi, K.; Kurachi, Y. PSD-95 mediates formation of a functional homomeric Kir5.1 channel in the brain. Neuron 2002, 34, 387–397. [Google Scholar] [CrossRef]
- Horio, Y.; Hibino, H.; Inanobe, A.; Yamada, M.; Ishii, M.; Tada, Y.; Satoh, E.; Hata, Y.; Takai, Y.; Kurachi, Y. Clustering and enhanced activity of an inwardly rectifying potassium channel, Kir4.1, by an anchoring protein, PSD-95/SAP90. J. Biol. Chem. 1997, 272, 12885–12888. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Taffet, S.M.; Vikstrom, K.L.; Anumonwo, J.M. Regulation of cardiac inward rectifier potassium current (I(K1)) by synapse-associated protein-97. J. Biol. Chem. 2010, 285, 28000–28009. [Google Scholar] [CrossRef] [PubMed]
- Sampson, L.J.; Leyland, M.L.; Dart, C. Direct interaction between the actin-binding protein filamin-A and the inwardly rectifying potassium channel, Kir2.1. J. Biol. Chem. 2003, 278, 41988–41997. [Google Scholar] [CrossRef] [PubMed]
- Seyberth, H.W.; Weber, S.; Kömhoff, M. Bartter’s and Gitelman’s syndrome. Curr. Opin. Pediatr. 2017, 29, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.; Pieper, G.H.; Wilders, R. Andersen-Tawil syndrome: Clinical and molecular aspects. Int. J. Cardiol. 2013, 170, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hancox, J.C.; Whittaker, D.G.; Du, C.; Stuart, A.G.; Zhang, H. Emerging therapeutic targets in the short QT syndrome. Expert Opin. Ther. Targets 2018, 22, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Fialho, D.; Robert, C.G.; Emma, M. Periodic paralysis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 148, pp. 505–520. [Google Scholar]
- Masotti, A.; Uva, P.; Davis-Keppen, L.; Basel-Vanagaite, L.; Cohen, L.; Pisaneschi, E.; Celluzzi, A.; Bencivenga, P.; Fang, M.; Tian, M.; et al. Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6. Am. J. Hum. Genet. 2015, 96, 295–300. [Google Scholar] [CrossRef]
- Horvath, G.A.; Zhao, Y.; Tarailo-Graovac, M.; Boelman, C.; Gill, H.; Shyr, C.; Lee, J.; Blydt-Hansen, I.; Drögemöller, B.I.; Moreland, J.; et al. Gain-of-function KCNJ6 Mutation in a Severe Hyperkinetic Movement Disorder Phenotype. Neuroscience 2018, 384, 152–164. [Google Scholar] [CrossRef]
- Korah, H.E.; Scholl, U.I. An Update on Familial Hyperaldosteronism. Horm. Metab. Res. 2015, 47, 941–946. [Google Scholar] [CrossRef]
- Bohnen, M.S.; Peng, G.; Robey, S.H.; Terrenoire, C.; Iyer, V.; Sampson, K.J.; Kass, R.S. Molecular Pathophysiology of Congenital Long QT Syndrome. Physiol. Rev. 2017, 97, 89–134. [Google Scholar] [CrossRef]
- Abdelhadi, O.; Iancu, D.; Stanescu, H.; Kleta, R.; Bockenhauer, D. EAST syndrome: Clinical, pathophysiological, and genetic aspects of mutations in KCNJ10. Rare Dis. 2016, 4, e1195043. [Google Scholar] [CrossRef]
- Nichols, C.G.; Singh, G.K.; Grange, D.K. KATP channels and cardiovascular disease: Suddenly a syndrome. Circ. Res. 2013, 112, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Tinker, A.; Aziz, Q.; Li, Y.; Specterman, M. ATP-Sensitive Potassium Channels and Their Physiological and Pathophysiological Roles. Compr. Physiol. 2018, 8, 1463–1511. [Google Scholar] [PubMed]
- Kumar, M.; Pattnaik, B.R. Focus on Kir7.1: Physiology and channelopathy. Channels 2014, 8, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Taneja, T.K.; Mankouri, J.; Karnik, R.; Kannan, S.; Smith, A.J.; Munsey, T.; Christesen, H.B.; Beech, D.J.; Sivaprasadarao, A. Sar1-GTPase-dependent ER exit of KATP channels revealed by a mutation causing congenital hyperinsulinism. Hum. Mol. Genet. 2009, 18, 2400–2413. [Google Scholar] [CrossRef]
- Bendahhou, S.; Donaldson, M.R.; Plaster, N.M.; Tristani-Firouzi, M.; Fu, Y.H.; Ptácek, L.J. Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil syndrome. J. Biol. Chem. 2003, 278, 51779–51785. [Google Scholar] [CrossRef]
- Ma, D.; Tang, X.D.; Rogers, T.B.; Welling, P.A. An andersen-Tawil syndrome mutation in Kir2. 1 (V302M) alters the G-loop cytoplasmic K+ conduction pathway. J. Biol. Chem. 2007, 282, 5781–5789. [Google Scholar] [CrossRef]
- Peters, M.; Ermert, S.; Jeck, N.; Derst, C.; Pechmann, U.; Weber, S.; Schlingmann, K.P.; Seyberth, H.W.; Waldegger, S.; Konrad, M. Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. 2003, 64, 923–932. [Google Scholar] [CrossRef]
- Choi, B.O.; Kim, J.; Suh, B.C.; Yu, J.S.; Sunwoo, I.N.; Kim, S.J.; Kim, G.H.; Chung, K.W. Mutations of KCNJ2 gene associated with Andersen-Tawil syndrome in Korean families. J. Hum. Genet. 2007, 52, 280–283. [Google Scholar] [CrossRef]
- Gloyn, A.L.; Pearson, E.R.; Antcliff, J.F.; Proks, P.; Bruining, G.J.; Slingerland, A.S.; Howard, N.; Srinivasan, S.; Silva, J.M.; Molnes, J.; et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 2004, 350, 1838–1849. [Google Scholar] [CrossRef]
- Lin, Y.W.; Bushman, J.D.; Yan, F.F.; Haidar, S.; MacMullen, C.; Ganguly, A.; Stanley, C.A.; Shyng, S.L. Destabilization of ATP-sensitive potassium channel activity by novel KCNJ11 mutations identified in congenital hyperinsulinism. J. Biol. Chem. 2008, 283, 9146–9156. [Google Scholar] [CrossRef]
- Schulte, U.; Hahn, H.; Konrad, M.; Jeck, N.; Derst, C.; Wild, K.; Weidemann, S.; Ruppersberg, J.P.; Fakler, B.; Ludwig, J. pH gating of ROMK (K(ir)1.1) channels: Control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 15298–15303. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Häusler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. USA. 2009, 106, 5842–5847. [Google Scholar] [CrossRef] [PubMed]
- Limberg, M.M.; Zumhagen, S.; Netter, M.F.; Coffey, A.J.; Grace, A.; Rogers, J.; Böckelmann, D.; Rinné, S.; Stallmeyer, B.; Decher, N.; et al. Non dominant-negative KCNJ2 gene mutations leading to Andersen-Tawil syndrome with an isolated cardiac phenotype. Basic Res. Cardiol. 2013, 108, 353. [Google Scholar] [CrossRef] [PubMed]
- Fallen, K.; Banerjee, S.; Sheehan, J.; Addison, D.; Lewis, L.M.; Meiler, J.; Denton, J.S. The Kir channel immunoglobulin domain is essential for Kir1.1 (ROMK) thermodynamic stability, trafficking and gating. Channels 2009, 3, 57–68. [Google Scholar] [CrossRef]
- O’Donnell, B.M.; Mackie, T.D.; Subramanya, A.R.; Brodsky, J.L. Endoplasmic reticulum-associated degradation of the renal potassium channel, ROMK, leads to type II Bartter syndrome. J. Biol. Chem. 2017, 292, 12813–12827. [Google Scholar] [CrossRef]
- Károlyi, L.; Konrad, M.; Köckerling, A.; Ziegler, A.; Zimmermann, D.K.; Roth, B.; Wieg, C.; Grzeschik, K.H.; Koch, M.C.; Seyberth, H.W.; et al. Mutations in the gene encoding the inwardly-rectifying renal potassium channel, ROMK, cause the antenatal variant of Bartter syndrome: Evidence for genetic heterogeneity. International Collaborative Study Group for Bartter-like Syndromes. Hum. Mol. Genet. 1997, 6, 17–26. [Google Scholar]
- Fodstad, H.; Swan, H.; Auberson, M.; Gautschi, I.; Loffing, J.; Schild, L.; Kontula, K. Loss-of-function mutations of the K+ channel gene KCNJ2 constitute a rare cause of long QT syndrome. J. Mol. Cell. Cardiol. 2004, 37, 593–602. [Google Scholar] [CrossRef]
- Davies, N.P.; Imbrici, P.; Fialho, D.; Herd, C.; Bilsland, L.G.; Weber, A.; Mueller, R.; Hilton-Jones, D.; Ealing, J.; Boothman, B.R.; et al. Andersen-Tawil syndrome: New potassium channel mutations and possible phenotypic variation. Neurology 2005, 65, 1083–1089. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, J.H.; Rajawat, Y.S.; Jerng, H.; Rami, T.G.; Sanchez, X.; DeFreitas, G.; Carabello, B.; DeMayo, F.; Kearney, D.L.; et al. Functional and clinical characterization of a mutation in KCNJ2 associated with Andersen-Tawil syndrome. J. Med. Genet. 2006, 43, 653–659. [Google Scholar] [CrossRef]
- Eckhardt, L.L.; Farley, A.L.; Rodriguez, E.; Ruwaldt, K.; Hammill, D.; Tester, D.J.; Ackerman, M.J.; Makielski, J.C. KCNJ2 mutations in arrhythmia patients referred for LQT testing: A mutation T305A with novel effect on rectification properties. Heart Rhythm. 2007, 4, 323–329. [Google Scholar] [CrossRef]
- Tani, Y.; Miura, D.; Kurokawa, J.; Nakamura, K.; Ouchida, M.; Shimizu, K.; Ohe, T.; Furukawa, T. T75M-KCNJ2 mutation causing Andersen-Tawil syndrome enhances inward rectification by changing Mg2+ sensitivity. J. Mol. Cell. Cardiol. 2007, 43, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Snider, K.E.; Becker, S.; Boyajian, L.; Shyng, S.L.; MacMullen, C.; Hughes, N.; Ganapathy, K.; Bhatti, T.; Stanley, C.A.; Ganguly, A. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J. Clin. Endocrinol. Metab. 2013, 98, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Mohnike, K.; Wieland, I.; Barthlen, W.; Vogelgesang, S.; Empting, S.; Mohnike, W.; Meissner, T.; Zenker, M. Clinical and genetic evaluation of patients with KATP channel mutations from the German registry for congenital hyperinsulinism. Horm. Res. Paediatr. 2014, 81, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Decher, N.; Renigunta, V.; Zuzarte, M.; Soom, M.; Heinemann, S.H.; Timothy, K.W.; Keating, M.T.; Daut, J.; Sanguinetti, M.C.; Splawski, I. Impaired interaction between the slide helix and the C-terminus of Kir2.1: A novel mechanism of Andersen syndrome. Cardiovasc. Res. 2007, 75, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Oberoi, S.; Tristani-Firouzi, M.; Etheridge, S.P.; Quitania, L.; Kramer, J.H.; Miller, B.L.; Fu, Y.H.; Ptácek, L.J. Andersen-Tawil syndrome: Prospective cohort analysis and expansion of the phenotype. Am. J. Med. Genet. A 2006, 140, 312–321. [Google Scholar] [CrossRef]
- Ballester, L.Y.; Benson, D.W.; Wong, B.; Law, I.H.; Mathews, K.D.; Vanoye, C.G.; George, A.L. Jr. Trafficking-competent and trafficking-defective KCNJ2 mutations in Andersen syndrome. Hum. Mutat. 2006, 27, 388. [Google Scholar] [CrossRef]
- Williams, D.M.; Lopes, C.M.; Rosenhouse-Dantsker, A.; Connelly, H.L.; Matavel, A.; O-Uchi, J.; McBeath, E.; Gray, D.A. Molecular basis of decreased Kir4.1 function in SeSAME/EAST syndrome. J. Am. Soc. Nephrol. 2010, 21, 2117–2129. [Google Scholar] [CrossRef]
- Takeda, I.; Takahashi, T.; Ueno, H.; Morino, H.; Ochi, K.; Nakamura, T.; Hosomi, N.; Kawakami, H.; Hashimoto, K.; Matsumoto, M. Autosomal recessive Andersen–Tawil syndrome with a novel mutation L94P in Kir2.1. Neurol. Clin. Neurosci. 2013, 1, 131–137. [Google Scholar] [CrossRef]
- Kuß, J.; Stallmeyer, B.; Goldstein, M.; Rinné, S.; Pees, C.; Zumhagen, S.; Seebohm, G.; Decher, N.; Pott, L.; Kienitz, M.C.; et al. Familial Sinus Node Disease Caused by a Gain of GIRK (G-Protein Activated Inwardly Rectifying K+ Channel) Channel Function. Circ. Genom. Precis. Med. 2019, 12, e002238. [Google Scholar] [CrossRef]
- Derst, C.; Wischmeyer, E.; Preisig-Müller, R.; Spauschus, A.; Konrad, M.; Hensen, P.; Jeck, N.; Seyberth, H.W.; Daut, J.; Karschin, A. A hyperprostaglandin E syndrome mutation in Kir1. 1 (renal outer medullary potassium) channels reveals a crucial residue for channel function in Kir1. 3 channels. J. Biol. Chem. 1998, 273, 23884–23891. [Google Scholar] [CrossRef][Green Version]
- Cheng, C.J.; Sung, C.C.; Wu, S.T.; Lin, Y.C.; Sytwu, H.K.; Huang, C.L.; Lin, S.H. Novel KCNJ5 mutations in sporadic aldosterone-producing adenoma reduce Kir3.4 membrane abundance. J. Clin. Endocrinol. Metab. 2015, 100, E155–E163. [Google Scholar] [CrossRef] [PubMed]
- Gélinas, R.; El Khoury, N.; Chaix, M.A.; Beauchamp, C.; Alikashani, A.; Ethier, N.; Boucher, G.; Villeneuve, L.; Robb, L.; Latour, F.; et al. Characterization of a Human Induced Pluripotent Stem Cell-Derived Cardiomyocyte Model for the Study of Variant Pathogenicity: Validation of a KCNJ2 Mutation. Circ. Cardiovasc. Genet. 2017, 10, e001755. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Lin, S.H.; Lo, Y.F.; Yang, S.S.; Hsu, Y.J.; Cannon, S.C.; Huang, C.L. Identification and functional characterization of Kir2.6 mutations associated with non-familial hypokalemic periodic paralysis. J. Biol. Chem. 2011, 286, 27425–27435. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Ren, F.; Zangerl-Plessl, E.M.; Heyman, S.; Stary-Weinzinger, A.; Yuan, P.; Nichols, C.G. Structural basis of control of inward rectifier Kir2 channel gating by bulk anionic phospholipids. J. Gen. Physiol. 2016, 148, 227–237. [Google Scholar] [CrossRef]
- Tanemoto, M.; Abe, T.; Uchida, S.; Kawahara, K. Mislocalization of K+ channels causes the renal salt wasting in EAST/SeSAME syndrome. FEBS Lett. 2014, 588, 899–905. [Google Scholar] [CrossRef]
- Pattnaik, B.R.; Tokarz, S.; Asuma, M.P.; Schroeder, T.; Sharma, A.; Mitchell, J.C.; Edwards, A.O.; Pillers, D.A. Snowflake vitreoretinal degeneration (SVD) mutation R162W provides new insights into Kir7.1 ion channel structure and function. PLoS ONE 2013, 8, 71744. [Google Scholar] [CrossRef]
- Sergouniotis, P.I.; Davidson, A.E.; Mackay, D.S.; Li, Z.; Yang, X.; Plagnol, V.; Moore, A.T.; Webster, A.R. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am. J. Hum. Genet. 2011, 89, 183–190. [Google Scholar] [CrossRef]
- McDonald, S.K.; Levitz, T.S.; Valiyaveetil, F.I. A Shared Mechanism for the Folding of Voltage-Gated K+ Channels. Biochemistry 2019, 58, 1660–1671. [Google Scholar] [CrossRef]
- Cho, H.C.; Tsushima, R.G.; Nguyen, T.T.; Guy, H.R.; Backx, P.H. Two critical cysteine residues implicated in disulfide bond formation and proper folding of Kir2. 1. Biochemistry 2000, 39, 4649–4657. [Google Scholar] [CrossRef]
- Hristova, K.; Wimley, W.C. A look at arginine in membranes. J. Membr. Biol. 2011, 239, 49–56. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene | Syndrome/Disease Character (OMIM)1 | Main Affected System(s) | Recent Review |
|---|---|---|---|---|
| KIR1.1 | KCNJ1 | Bartter syndrome, type 2 (241200) | Kidney; head; face; ear; eye; vascular; gastrointestinal; skeleton; skeletal muscle; CNS; platelets | [33] |
| KIR2.1 | KCNJ2 | Andersen syndrome (170390) Familial atrium fibrillation 9 (613980) Short QT syndrome 3 (609622) | Head; face; ear; eye; teeth; heart; skeleton; CNS | [34,35] |
| KIR2.2 | KCNJ12 | Non-described | ||
| KIR2.3 | KCNJ4 | Non-described | ||
| KIR2.4 | KCNJ14 | Non-described | ||
| KIR2.6 | KCNJ18 | Thyrotoxic hypokalemic periodic paralysis (613239) | Cardiovascular; skeletal muscle; CNS; eye | [36] |
| KIR3.1 | KCNJ3 | Non-described | ||
| KIR3.2 | KCNJ6 | Keppen–Lubinsky Syndrome (614098) | CNS; head; skin; skeleton; eye, face | No review available |
| KIR3.3 | KCNJ9 | Non-described | ||
| KIR3.4 | KCNJ5 | Familial hyperaldosteronism 3 (613677) Long QT syndrome 13 (613485) | Cardiovascular; kidney; skeletal muscle | [39,40] |
| KIR4.1 | KCNJ10 | Digenic enlarged vestibular aqueduct (600791) EAST/SESAME syndrome (612780) | Ear (hearing); vascular; kidney; CNS | [41] |
| KIR4.2 | KCNJ15 | Non-described | ||
| KIR5.1 | KCNJ16 | Non-described | ||
| KIR6.1 | KCNJ8 | Cantú syndrome (239850) | Head; face; cardiovascular; skeleton; hair; CNS | [42] |
| KIR6.2 | KCNJ11 | Transient neonatal diabetes mellitus 3 (610582) Permanent neonatal diabetes with or without neurologic features (606176) Familial hyperinsulinemic hypoglycemia 2 (601820) Maturity-onset diabetes of the young 13 (616329) Susceptible to diabetes mellitus 2 (125853) | Pancreas (beta-cells); CNS | [43] |
| KIR7.1 | KCNJ13 | Leber congenital amaurosis 16 (614186) Snowflake vitreoretinal degeneration (193230) | Eye (retina) | [44] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zangerl-Plessl, E.-M.; Qile, M.; Bloothooft, M.; Stary-Weinzinger, A.; van der Heyden, M.A.G. Disease Associated Mutations in KIR Proteins Linked to Aberrant Inward Rectifier Channel Trafficking. Biomolecules 2019, 9, 650. https://doi.org/10.3390/biom9110650
Zangerl-Plessl E-M, Qile M, Bloothooft M, Stary-Weinzinger A, van der Heyden MAG. Disease Associated Mutations in KIR Proteins Linked to Aberrant Inward Rectifier Channel Trafficking. Biomolecules. 2019; 9(11):650. https://doi.org/10.3390/biom9110650
Chicago/Turabian StyleZangerl-Plessl, Eva-Maria, Muge Qile, Meye Bloothooft, Anna Stary-Weinzinger, and Marcel A. G. van der Heyden. 2019. "Disease Associated Mutations in KIR Proteins Linked to Aberrant Inward Rectifier Channel Trafficking" Biomolecules 9, no. 11: 650. https://doi.org/10.3390/biom9110650
APA StyleZangerl-Plessl, E.-M., Qile, M., Bloothooft, M., Stary-Weinzinger, A., & van der Heyden, M. A. G. (2019). Disease Associated Mutations in KIR Proteins Linked to Aberrant Inward Rectifier Channel Trafficking. Biomolecules, 9(11), 650. https://doi.org/10.3390/biom9110650