Statistically Designed Medium Reveals Interactions between Metabolism and Genetic Information Processing for Production of Stable Human Serum Albumin in Pichia pastoris

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
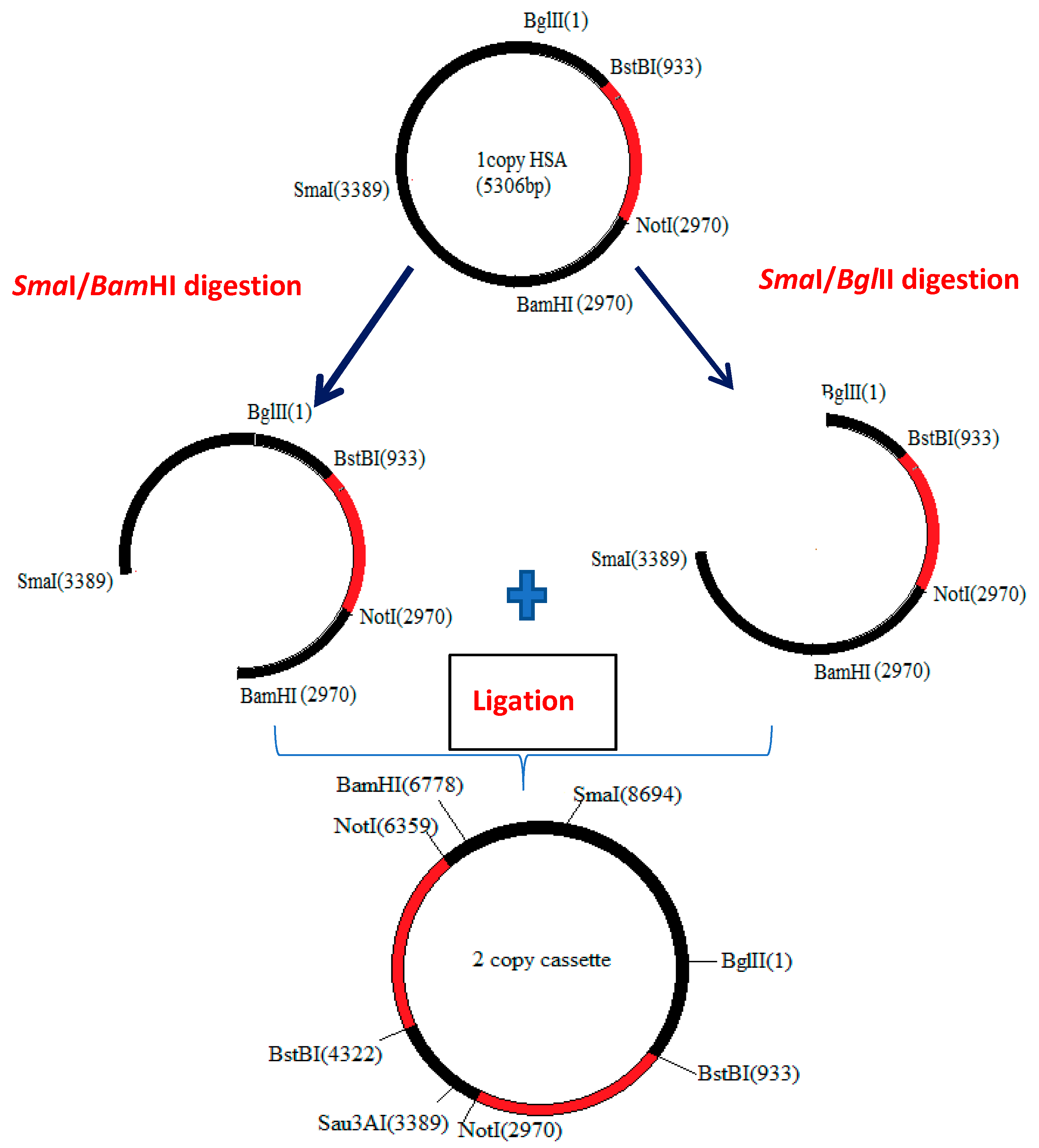
2.2. Strains and Construction of Single- and 2-Copy Expression Cassettes of HSA
2.3. Animal Cell Lines and Cell Proliferation Assay Kit
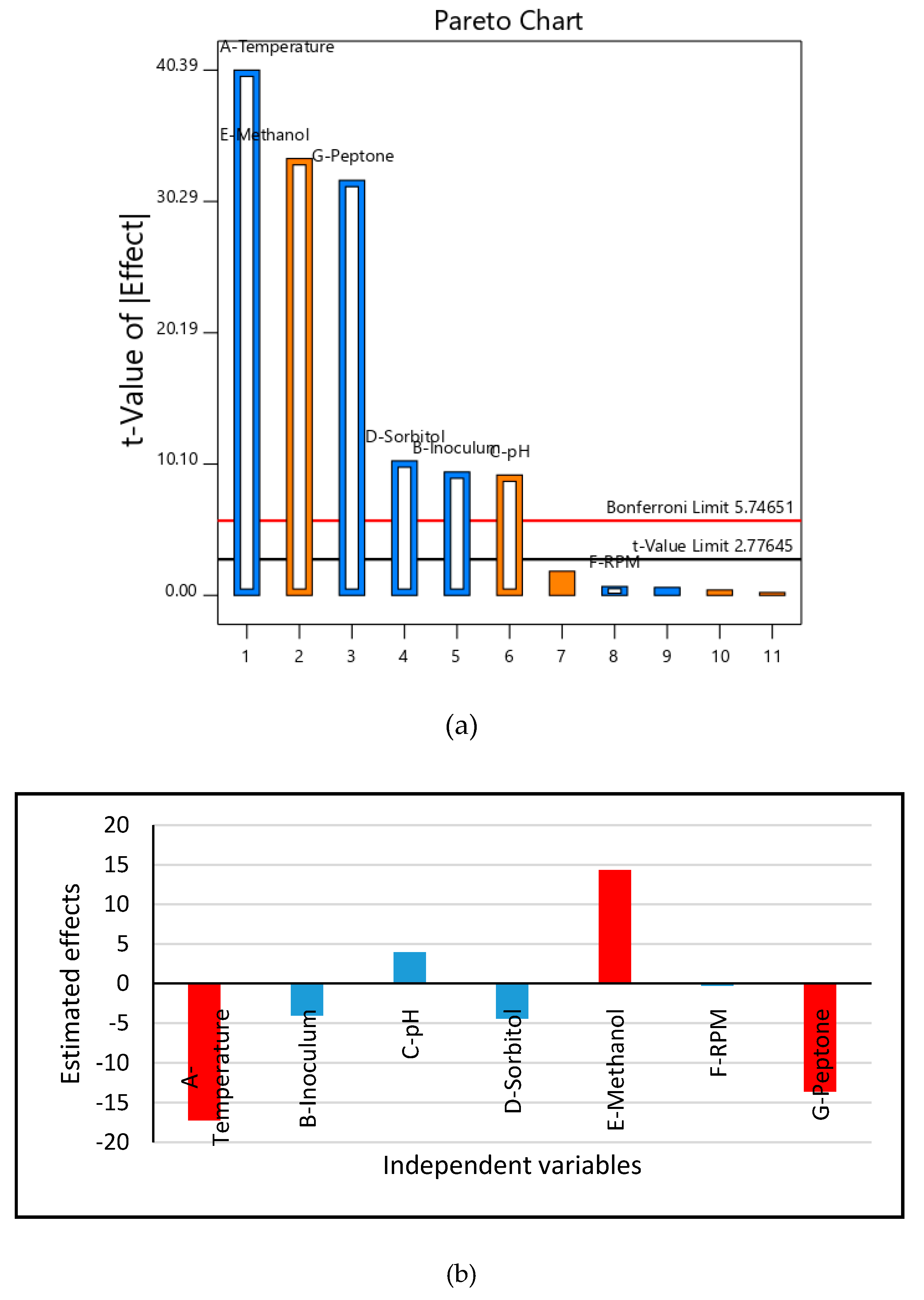
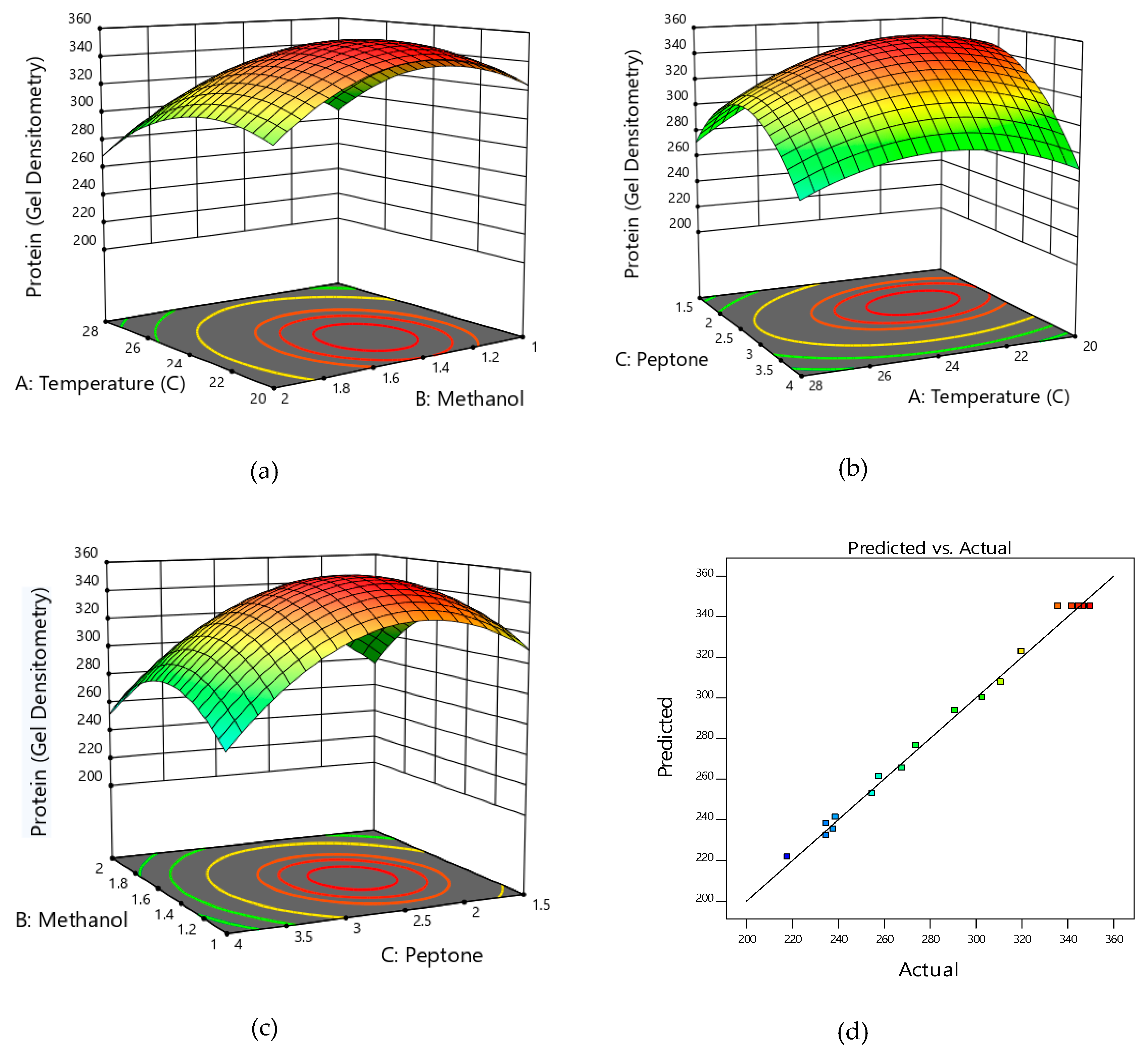
2.4. Cultivation of Recombinant HSA Producing P. pastoris on Unoptimized and Optimized Medium and Optimization of HSA Production through Response Surface Methodology
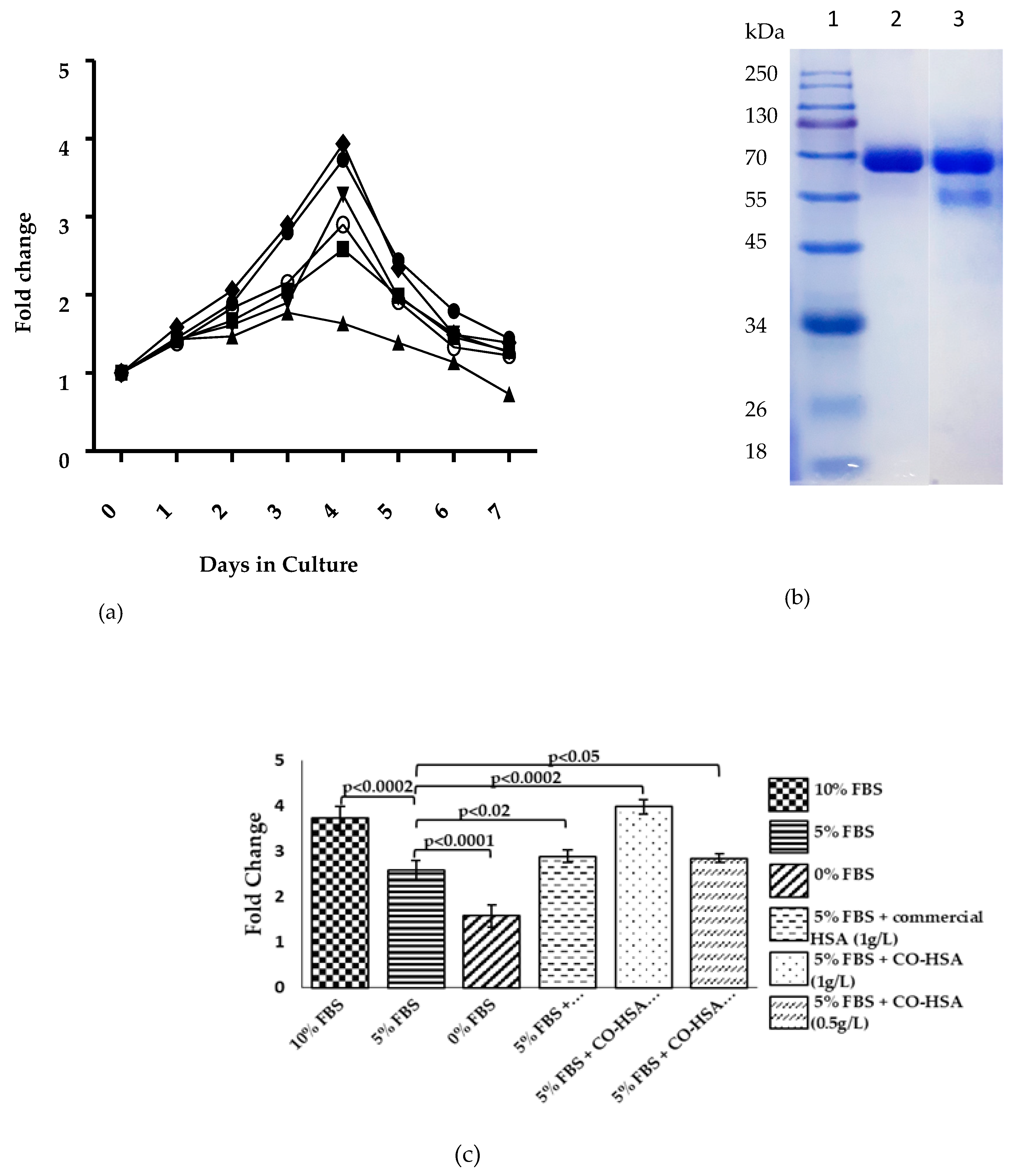
2.5. Purification and Cell Proliferation Assay of Recombinant HSA
2.6. Transcriptome Analysis
2.7. qPCR Validation of the Identified Target Genes
2.8. Analytical Methods
3. Results
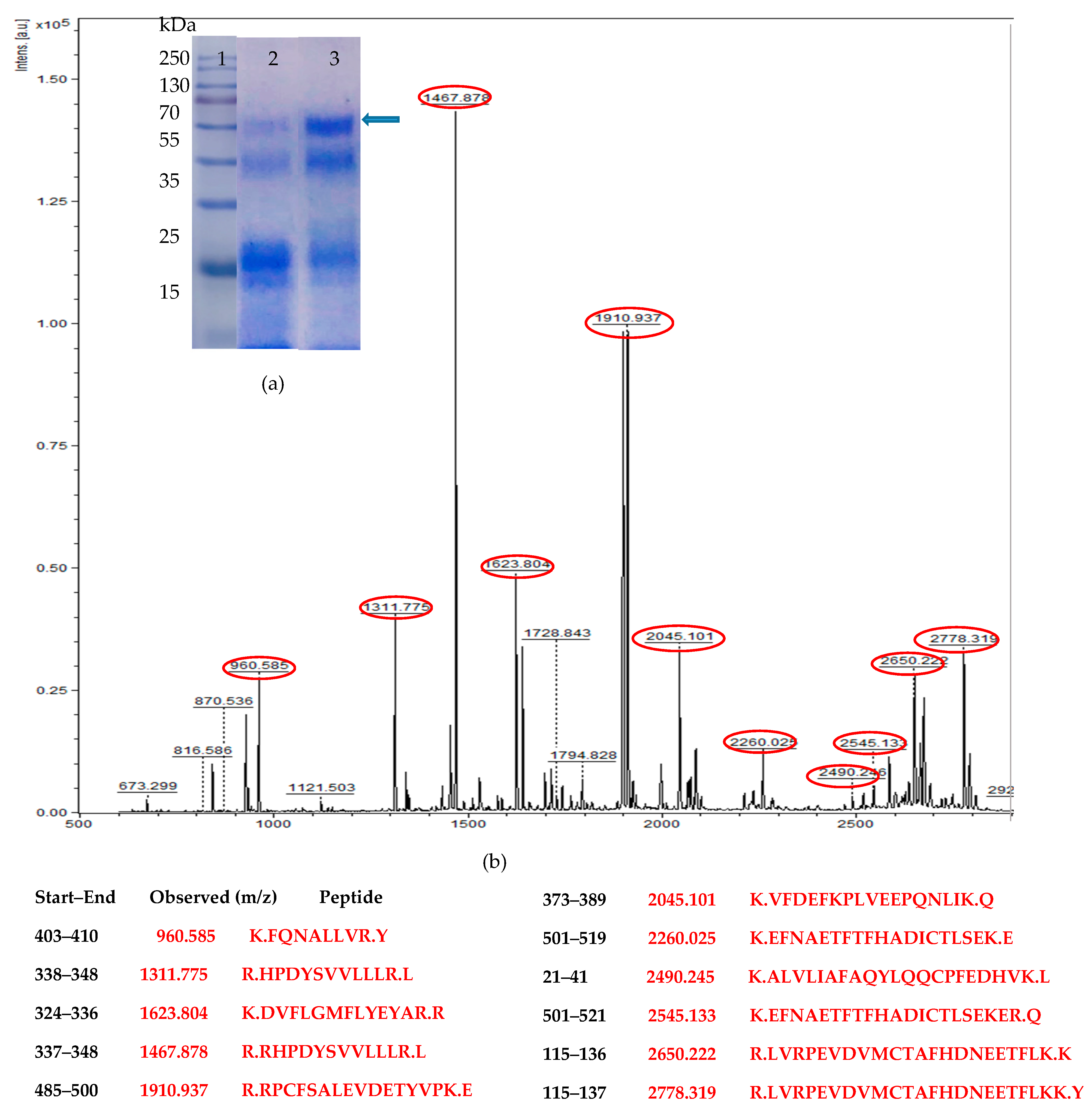
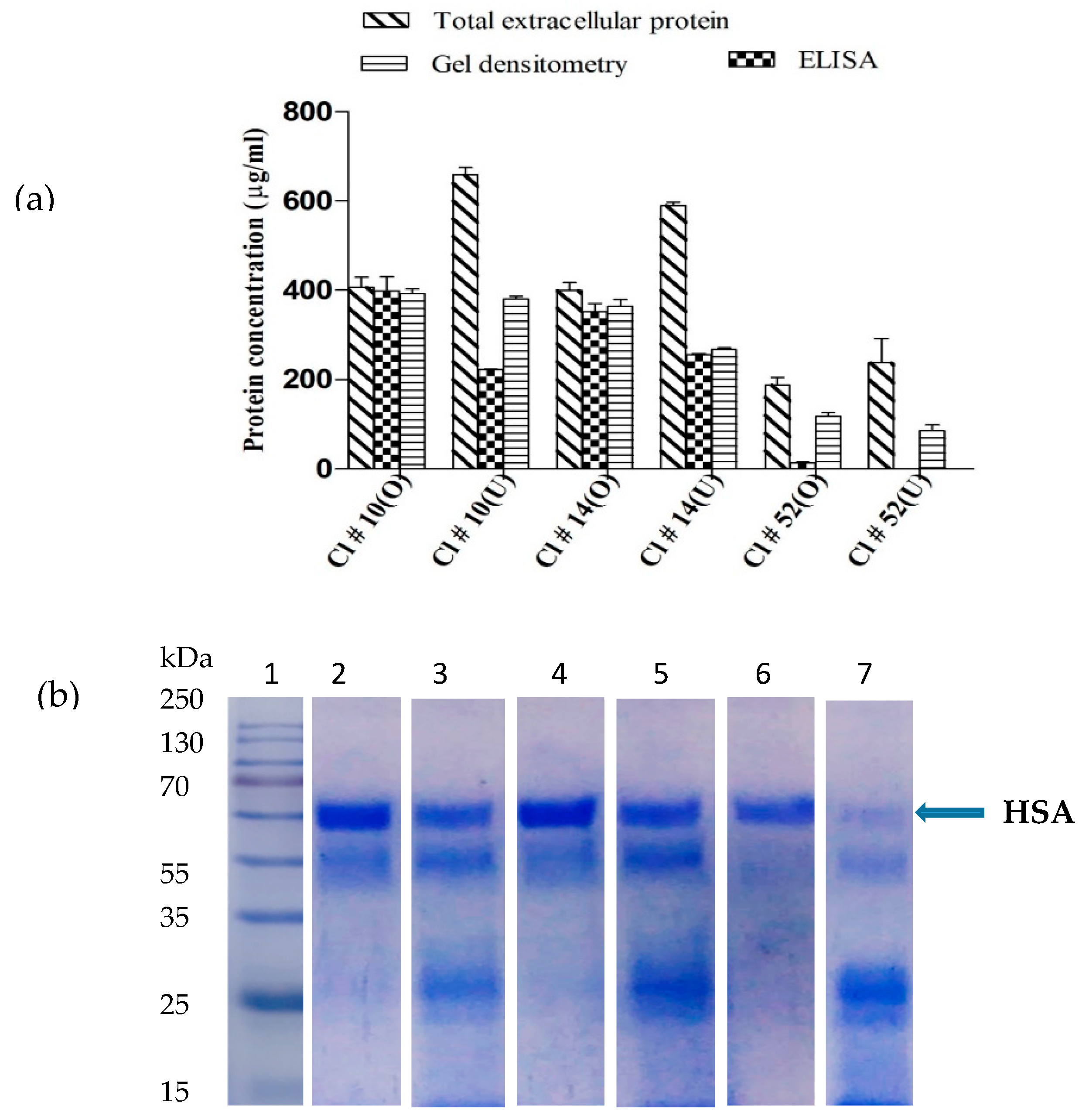
3.1. Codon Optimization of Native HSA and Production in P. pastoris
3.2. Medium Design and Evaluation of Secreted HSA
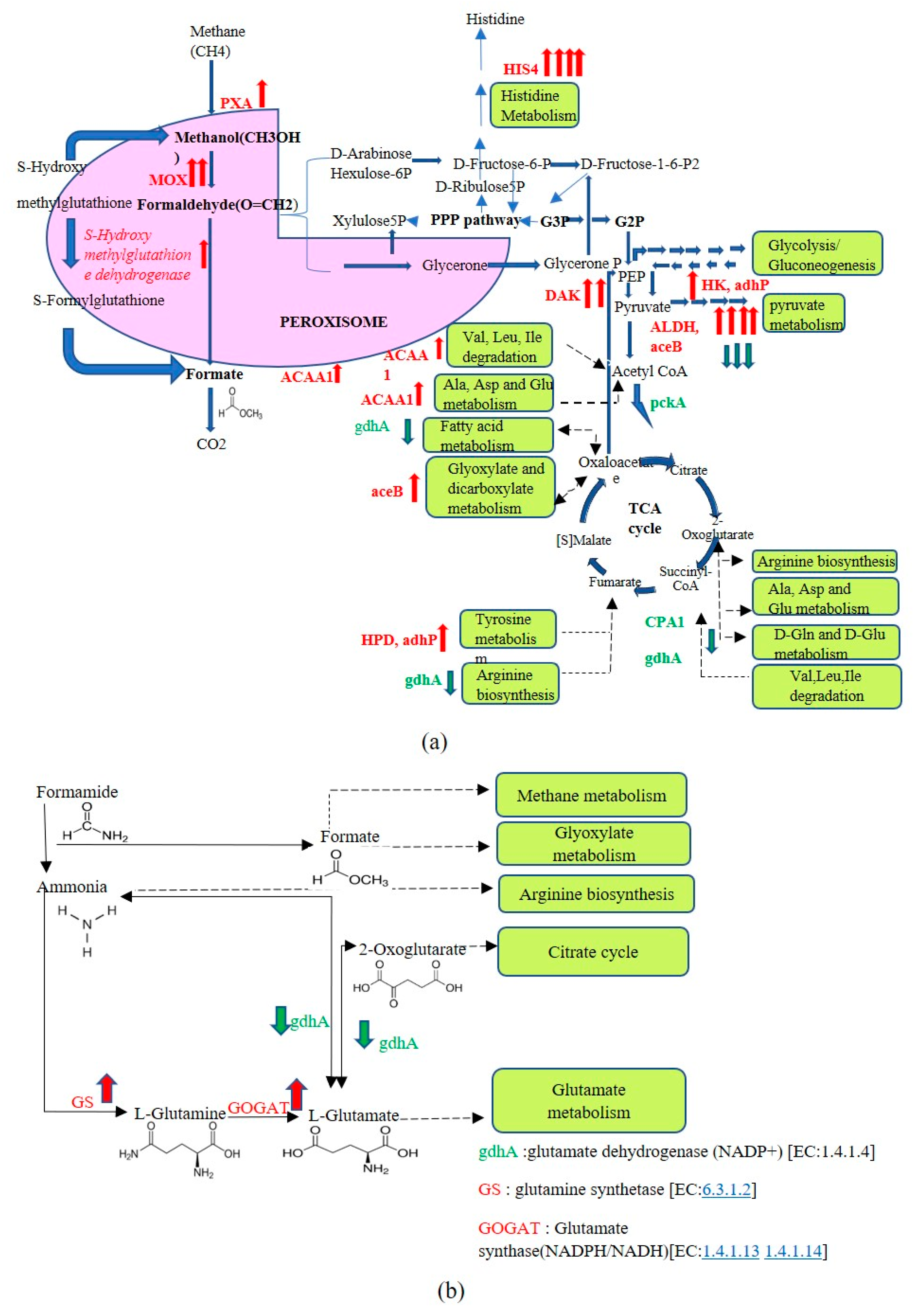
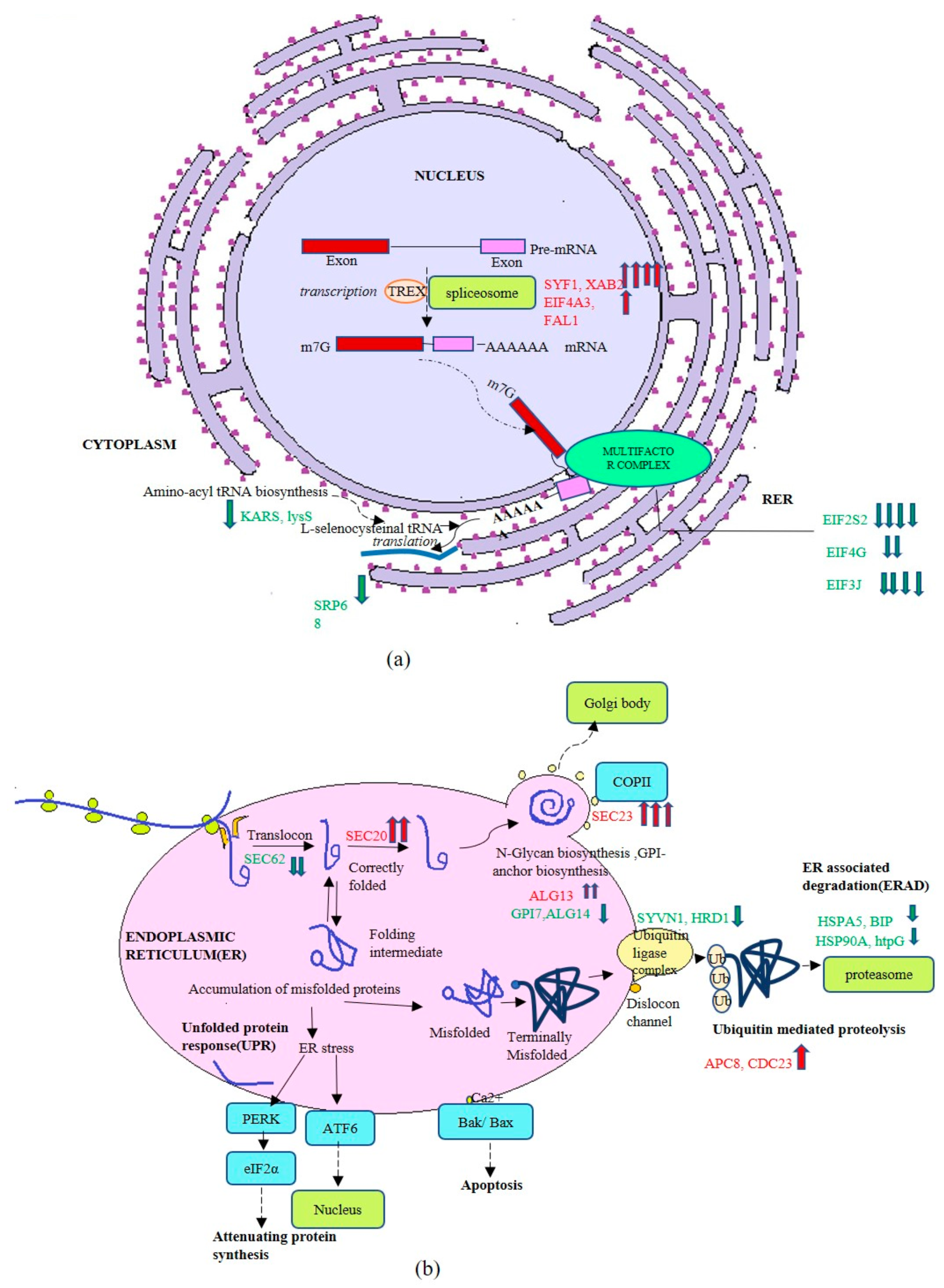
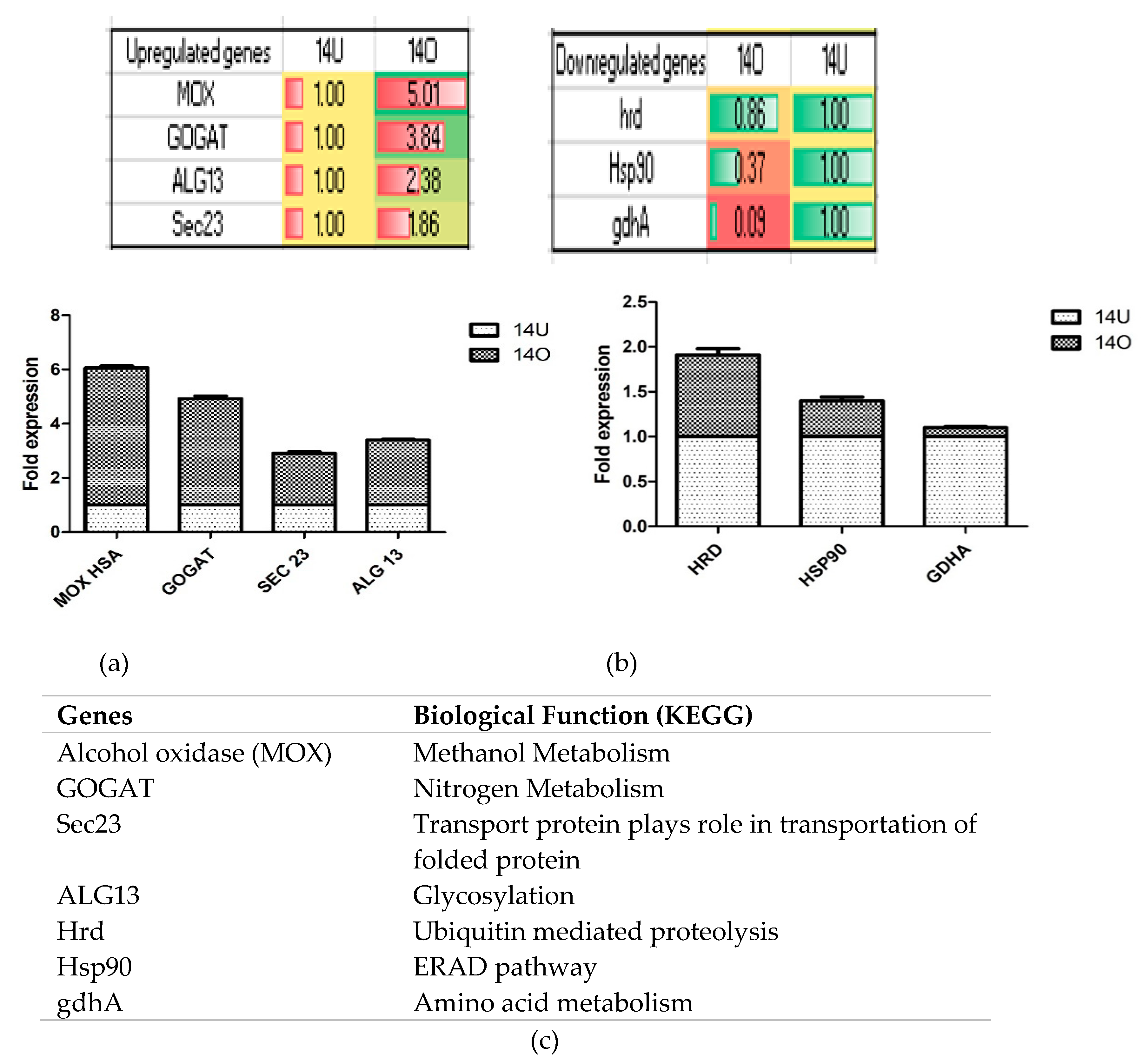
3.3. Transcriptome Profiling of Recombinant P. pastoris Cultivated on Optimized and Unoptimized Medium
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
Appendix C
Appendix D
References
- Rabert, C.; Weinacker, D.; Pessoa, A.; Ferias, J.G. Recombinant proteins for industrial uses: Utilization of Pichia pastoris expression system. Braz. J. Microbiol. 2013, 44, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Porro, D.; Gasser, B.; Fossati, T.; Maurer, M.; Branduardi, P.; Sauer, M.; Mattanovich, D. Production of recombinant proteins and metabolites in yeasts: When are these systems better than bacterial production systems? Appl. Microbiol. Biotechnol. 2011, 89, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Yu, C.Y.; Li, X.X.; Zhang, P.; Tang, J.; Yang, Q.; Fu, T.; Zhang, X.; Cui, X.; Tu, G.; et al. Therapeutic target database update 2018: Enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2018, 46, 1121–1127. [Google Scholar] [CrossRef]
- Xie, D.; Shao, Z.; Achkar, J.; Zha, W.; Frost, J.W.; Zhao, H. Microbial synthesis of triacetic acid lactone. Biotechnol. Bioeng. 2006, 93, 727–736. [Google Scholar] [CrossRef]
- Celik, P.; Calik, P.; Oliver, S.G. Fed-batch methanol feeding strategy for recombinant protein production by Pichia pastoris in the presence of co-substrate sorbitol. Yeast 2009, 26, 473–484. [Google Scholar] [CrossRef]
- D’Anjou, M.C.; Daugulis, A.J. A rational approach to improving productivity in recombinant Pichia pastoris fermentation. Biotechnol. Bioeng. 2000, 72, 1–11. [Google Scholar] [CrossRef]
- Zhong, Y.; Yang, L.; Yugang, G.; Fang, F.; Wang, D.; Li, R.; Jiang, M.; Kang, W.; Ma, J.; Sun, J. High- temperature cultivation of recombinant Pichia pastoris increases endoplasmic reticulum stress and decreases production of human interleukin-10. Microb. Cell Fact. 2014, 13, 163. [Google Scholar] [CrossRef]
- Rathore, A.S.; Winkle, H. Quality by design for biopharmaceuticals. Nat. Biotechnol. 2009, 27, 26–34. [Google Scholar] [CrossRef]
- Alexander, M.R.; Ambre, J.J.; Liskow, B.I.; Trost, D.C. Therapeutic use of albumin. J. Am. Med. Assoc. 1979, 241, 2527–2529. [Google Scholar] [CrossRef]
- Fanali, G.; diMasi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human Serum Albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Youn, J.K.; Shang, L.; Kim, M.I.; Jeong, C.M.; Chang, H.N.; Hahm, M.S.; Rhee, S.K.; Kang, H.A. Enhanced production of human serum albumin by Fed-Batch culture of Hansenula polymorpha with high purity oxygen. J. Microbiol. Biotechnol. 2010, 20, 1534–1538. [Google Scholar] [CrossRef] [PubMed]
- Lodi, T.; Neglia, B.; Donnini, C. Secretion of human serum llbumin by Kluyveromyces lactis overexpressing KIPDI1 and KIERO1. Appl. Environ. Microbiol. 2005, 71, 4359–4363. [Google Scholar] [CrossRef] [PubMed]
- Quirk, A.V.; Geisow, M.J.; Woodrow, J.R.; Burton, S.J. Production of recombinant human serum albumin from Saccharomyces cerevisiae. Biotechnol. Appl. Biochem. 1989, 11, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.K.; Li, Y.T.; Lu, C.F.; Huang, L.F. Enhancement of recombinant human serum albumin in transgenic rice cell culture system by cultivation strategy. New Biotechnol. 2015, 32, 328–334. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, Y.; Liu, J.; Lan, H.; Shao, M.; Yu, Y.; Quan, F.; Zhang, Y. Production of transgenic cattle highly expressing human serum albumin in milk by phiC31 integrase-mediated gene delivery. Transgenic Res. 2015, 24, 875–883. [Google Scholar] [CrossRef]
- Maccani, A.; Landes, N.; Stadlmayr, G.; Maresch, D.; Leitner, C.; Maurer, M.; Gasser, B.; Ernst, W.; Kunert, R.; Mattanovich, D. Pichia pastoris secretes recombinant proteins less efficiently than Chinese hamster ovary cells but allows higher space-time yields for less complex proteins. Biotechnol. J. 2014, 9, 526–537. [Google Scholar] [CrossRef]
- Zhu, W.; Gong, G.; Pan, J.; Han, S.; Zhang, W.; Hu, Y.; Xie, L. High level expression and purification of recombinant human serum albumin in Pichia pastoris. Protein Expr. Purif. 2018, 147, 61–68. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, R.; Wu, X. The proteolytic systems and heterologous proteins degradation in the methylotrophic yeast Pichia pastoris. Ann. Microbiol. 2007, 57, 553–560. [Google Scholar] [CrossRef]
- Looser, V.; Bruhlmann, B.; Bumbak, F.; Stenger, C.; Costa, A.; Camattari, A.; Fotiadis, D.; Kovar, K. Cultivation strategies to enhance productivity of Pichia pastoris: A review. Biotechnol. Adv. 2015, 33, 1177–1193. [Google Scholar] [CrossRef]
- Shi, X.; Karkut, T.; Chamankhah, M.; Alting-Mees, M.; Hemmingsen, S.M.; Hegedus, D. Optimal conditions for the expression of a single-chain antibody (scFv) gene in Pichia pastoris. Protein Expr. Purif. 2003, 28, 321–330. [Google Scholar] [CrossRef]
- Cai, H.; Chen, L.; Wan, L.; Zeng, L.; Yang, H.; Li, S.; Li, Y.; Cheng, J.; Lu, X. High-level expression of a functional humanized anti-CTLA4 single-chain variable fragment antibody in Pichia pastoris. Appl. Microbiol. Biotechnol. 2009, 82, 41–48. [Google Scholar] [CrossRef]
- Chambers, S.P.; Swalley, S.E. Designing experiments for high-throughput protein expression. Methods Mol. Biol. 2009, 498, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Holmes, W.J.; Darby, R.A.; Wilks, M.D.; Smith, R.; Bill, R.M. Developing a scalable model of recombinant protein yield from Pichia pastoris: The influence of culture conditions, biomass and induction regime. Microb. Cell Fact. 2009, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Swalley, S.E.; Fulghum, J.R.; Chambers, S.P. Screening factors effecting a response in soluble protein expression: Formalized approach using design of experiments. Anal. Biochem. 2006, 351, 122–127. [Google Scholar] [CrossRef]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Wikstrom, C.; Wold, S. Design of experiments. J. Chemom. 2003, 15, 495–496. [Google Scholar]
- Bai, J.; Swartz, D.J.; Protasevich, I.I.; Brouillette, C.; Harrell, P.M.; Hildebrandt, E.; Gasser, B.; Mattanovich, D.; Ward, A.; Chang, G.; et al. A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbour Laboratory Press: Cold Spring Harbor, NY, USA, 2014. [Google Scholar]
- Meerloo, J.V.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 31, 237–245. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]
- Maity, N.; Thawani, A.; Sharma, A.; Gautam, A.; Mishra, S.; Sahai, V. Expression and control of codon optimized Granulocyte Colony Stimulating Factor in Pichia pastoris. Appl. Biochem. Biotechnol. 2016, 178, 159–172. [Google Scholar] [CrossRef]
- Gundry, R.L.; White, M.Y.; Murray, C.I.; Kane, L.A.; Fu, Q.; Stanley, B.A. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr. Protoc. Mol. Biol. 2009, 88. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon adaptation index - a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T.; Caceres, J.F.; Spector, D.L. The dynamics of a pre-mRNA splicing factor in living cells. Nature 1997, 387, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Asahina, H.; Citterio, E.; Rademakers, S.; Vermeulen, W.; Kamiuchi, S.; Yeo, J.P.; Khaw, M.C.; Saijo, M.; Kodo, N.; et al. XAB2, a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and transcription. J. Biol. Chem. 2000, 275, 34931–34937. [Google Scholar] [CrossRef] [PubMed]
- Spadiut, O.; Capone, S.; Krainer, F.; Glieder, A.; Herwig, C. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol. 2014, 32, 54–60. [Google Scholar] [CrossRef]
- Mallem, M.; Warburton, S.; Li, F.; Shandil, I.; Nylen, A.; Kim, S.; Jiang, Y.; Meehl, M.; Anjou, M.; Stadheim, T.A.; et al. Maximizing recombinant human serum albumin production in Muts Pichia pastoris strain. Biotechnol. Prog. 2014, 30, 1488–1496. [Google Scholar] [CrossRef]
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein expression in Pichia pastoris: Recent achievements and perspectives for heterologous protein production. Appl. Microbiol. Biotechnol. 2014, 98, 5301–5317. [Google Scholar] [CrossRef]
- Mendez, C.M.; McClain, C.J.; Marsano, L.S. Albumin therapy in clinical practice. Nutr. Clin. Pract. 2005, 20, 314–320. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kuwae, S.; Ohya, T.; Ohyama, M.; Tomomitsu, K. High level secretion of recombinant human serum albumin by fed-batch fermentation of the methylotrophic yeast Pichia pastoris based on optimal methanol feeding strategy. J. Biosci. Bioeng. 2000, 90, 280–288. [Google Scholar] [CrossRef]
- Jafari, R.; Sundstrom, B.E.; Holm, P. Optimization of production of the anti-keratin 8 single chain Fv TS-218 in Pichia pastoris using design of experiments. Microb. Cell Fact. 2011, 10, 34. [Google Scholar] [CrossRef]
- Chang, S.W.; Shieh, C.J.; Lee, G.C.; Akoh, C.C.; Shaw, J.F. Optimized growth kinetics of Pichia pastoris and recombinant Candida rugosa LIP1 production by RSM. J. Microb. Biotechnol. 2006, 11, 28–40. [Google Scholar] [CrossRef]
- Khatri, N.K.; Hoffmann, F. Impact of methanol concentration on secreted protein production in oxygen-limited cultures of recombinant Pichia pastoris. Biotechnol. Bioeng. 2006, 93, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Potter, K.J.H.; Plantz, B.A.; Schlegel, V.L.; Smith, L.A.; Meagher, M.M. Pichia pastoris fermentation with mixed-feeds of glycerol and methanol: Growth kinetics and production improvement. J. Ind. Microbiol. Biotechnol. 2003, 30, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Berrios, J.; Flores, M.-O.; Barrera, A.D.; Altamirano, C.; Martinez, I.; Cabrera, Z. A comparative study of glycerol and sorbitol as co-substrates in methanol-induced cultures of Pichia pastoris: Temperature effect and scale-up simulation. J. Ind. Microbiol. Biotechnol. 2017, 44, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.A.; White, C.E.; Meininger, D.P.; Komives, E.A. Generation of protease-deficient strains and their use in heterologous protein expression. Methods Mol. Biol. 1998, 103, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.J.; Li, Z.; Yu, R.S.; Wu, J.R.; Zheng, Z.Y.; Shi, Z.P.; Zhan, X.B.; Lin, C.C. Methanol/sorbitol co-feeding induction enhanced porcine interferon-α production by P. pastoris associated with energy metabolism shift. Bioprocess Biosyst. Eng. 2012, 35, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Hoy, Y.M.; Cameron, D.R.; Helle, S.S.; Duff, S.J.B. Production of a recombinant protein using Pichia pastoris grown in Kraft evaporator condensate from a Kraft pulp mill. Enzyme Microb. Technol. 2006, 38, 317–323. [Google Scholar] [CrossRef]
- Jahic, M.; Wallberg, F.; Bollok, M.; Garcia, P.; Enfors, S.-O. Temperature limited fed-batch technique for control of proteolysis in Pichia pastoris bioreactor cultures. Microb. Cell Fact. 2003, 2, 6. [Google Scholar] [CrossRef][Green Version]
- Gonchar, M.; Maidan, M.; Korpan, Y.; Sibirny, V.; Kotylak, Z.; Sibirny, Y. Metabolically engineered methylotrophic yeast cells and enzymes as sensor biorecognition elements. FEMS Yeast Res. 2002, 2, 307–314. [Google Scholar] [CrossRef]
- Heyland, J.; Fu, J.; Blank, L.M.; Schmid, A. Carbon metabolism limits recombinant protein production in Pichia pastoris. Biotechnol. Bioeng. 2011, 108, 1942–1953. [Google Scholar] [CrossRef]
- Jorda, J.; Jouhten, P.; Cámara, E.; Maaheimo, H.; Albiol, J.; Ferrer, P. Metabolic flux profiling of recombinant protein secreting Pichia pastoris growing on glucose: Methanol mixtures. J. Microb. Biotechnol. 2014, 24, 144–151. [Google Scholar] [CrossRef]
- Sauer, M.; Branduardi, P.; Gasser, B.; Valli, M.; Maurer, M.; Porro, D.; Mattanovich, D. Differential gene expression in recombinant Pichia pastoris analysed by heterologous DNA microarray hybridisation. Microb. Cell Fact. 2004, 3, 17. [Google Scholar] [CrossRef]
- Camara, E.; Landes, N.; Albiol, J.; Gasser, B.; Mattanovich, D.; Ferrer, P. Increased dosage of AOX1 promoter-regulated expression cassettes leads to transcription attenuation of the methanol metabolism in Pichia pastoris. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wang, B.; Pan, L.; Ye, Y.; He, M.; Han, S.; Zheng, S.; Wang, X.; Lin, Y. Comprehensive structural end RNA sequencing. BMC Genomics 2012, 13, 738. [Google Scholar] [CrossRef]
- Van Zutphen, T.; Baerends, R.J.; Susanna, K.A.; De Jong, A.; Kuipers, O.P.; Veenhuis, M.; Vander Klei, I.J. Adaptation of Hansenula polymorpha to methanol: A transcriptome analysis. BMC Genomics 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vanz, A.L.; Nimtz Rina, U. Decrease of UPR- and ERAD- related proteins in Pichia pastoris during methanol-induced secretory insulin precursor production in controlled fed-batch cultures. Microb. Cell Fact. 2014, 13, 23. [Google Scholar] [CrossRef]
- Liu, P.; Huang, M.; Guo, M.; Qian, J.; Lin, W.; Chu, J.; Zhuang, Y.; Zhang, S. Combined 13C-assisted metabolomics and metabolic flux analysis reveals the impacts of glutamate on the central metabolism of high β-galactosidase-producing Pichia pastoris. Bioresour. Bioprocess. 2016, 3, 47. [Google Scholar] [CrossRef]
- Pena, D.A.; Gasser, B.; Zanghellini, J.; Steiger, M.G.; Matttanovich, D. Metabolic engineering of Pichia pastoris. Metabol. Eng. 2018, 50, 2–15. [Google Scholar] [CrossRef]
- McManus, C.J.; May, G.E.; Spealman, P.; Shteyman, A. Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res. 2014, 24, 422–430. [Google Scholar] [CrossRef]
- Qi, F.; Wang, C.; Liu, Y.; Kaleem, I.; Li, Q.; Li, C. Transcriptional profiling of protein expression related genes of Pichia pastoris under simulated microgravity. PLoS ONE 2011, 6, 1–11. [Google Scholar] [CrossRef][Green Version]
- Gardner, R.G.; Shearer, A.G.; Hampton, R.Y. In vivo action of the HRD ubiquitin ligase complex: Mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol. Cell Biol. 2001, 21, 4276–4291. [Google Scholar] [CrossRef]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Tsai, B.; Arvan, P. New insights into the physiological role of Endoplasmic Reticulum-assisted degradation. Trends Cell Biol. 2017, 27, 430–440. [Google Scholar] [CrossRef]
- Li, Z.J.; Xiong, F.; Lin, Q.S.; d’Anjou, M.; Daugulis, A.J.; Yang, D.S.C.; Hew, C.L. Low-temperature increases the yield of biologically active herring antifreeze protein in Pichia pastoris. Protein Expr. Purif. 2001, 21, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Gasser, B.; Maurer, M.; Rautio, J.; Sauer, M.; Bhattacharyya, A.; Saloheimmo, M.; Penttila, M.; Mattanovich, D. Monitoring of transcriptional regulation in Pichia pastoris under protein production conditions. BMC Genomics 2007, 8, 179. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence of Primers |
|---|---|
| FP Hsp90 | 5′CTGAGGAAGATGACAAGAAGCC3′ |
| RP Hsp90 | 5′GAAGTGCTTAACAGCCAATGGG3′ |
| FP gdhA | 5′TTTCTCCGACTCTCCAGTCAAGTAC3′ |
| RP gdhA | 5′AGCCTCCAAAGTAGAACCCATG3′ |
| FP HRD | 5′GAACAGTCATCATCCCGAGAG3′ |
| RP HRD | 5′CTTTGTCCATACGAGAGAGATC3′ |
| FP MOX | 5′CAGAGGTTCCGCTTCTGATTAC3′ |
| RP MOX | 5′TTGGTACTCAGAAGCCCTCAAG3′ |
| FP ALG13 | 5′GCTGATTCGAACACATAGAAGG3′ |
| RP ALG13 | 5′GTCGGGTTTTATATCCCTGTG3′ |
| FP Sec23 | 5′AACAATACGCTTTTGCTTGCTCC3′ |
| RP Sec23 | 5′ATCACATTGGCCTTTCTCTTGG3′ |
| FP GOGAT | 5′GCTGCTATGGGAG CTGATGAA3′ |
| RP GOGAT | 5′GCAACGGCCAAG AATCATGTA3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maity, N.; Mishra, S. Statistically Designed Medium Reveals Interactions between Metabolism and Genetic Information Processing for Production of Stable Human Serum Albumin in Pichia pastoris. Biomolecules 2019, 9, 568. https://doi.org/10.3390/biom9100568
Maity N, Mishra S. Statistically Designed Medium Reveals Interactions between Metabolism and Genetic Information Processing for Production of Stable Human Serum Albumin in Pichia pastoris. Biomolecules. 2019; 9(10):568. https://doi.org/10.3390/biom9100568
Chicago/Turabian StyleMaity, Nitu, and Saroj Mishra. 2019. "Statistically Designed Medium Reveals Interactions between Metabolism and Genetic Information Processing for Production of Stable Human Serum Albumin in Pichia pastoris" Biomolecules 9, no. 10: 568. https://doi.org/10.3390/biom9100568
APA StyleMaity, N., & Mishra, S. (2019). Statistically Designed Medium Reveals Interactions between Metabolism and Genetic Information Processing for Production of Stable Human Serum Albumin in Pichia pastoris. Biomolecules, 9(10), 568. https://doi.org/10.3390/biom9100568