Abstract
The integration of cell extrinsic and intrinsic signals is required to maintain appropriate cell physiology and homeostasis. Bone morphogenetic proteins (BMPs) are cytokines that belong to the transforming growth factor-β (TGF-β) superfamily, which play a key role in embryogenesis, organogenesis and regulation of whole-body homeostasis. BMPs interact with membrane receptors that transduce information to the nucleus through SMAD-dependent and independent pathways, including PI3K-AKT and MAPKs. Reactive oxygen species (ROS) are intracellular molecules derived from the partial reduction of oxygen. ROS are highly reactive and govern cellular processes by their capacity to regulate signaling pathways (e.g., NF-κB, MAPKs, KEAP1-NRF2 and PI3K-AKT). Emerging evidence indicates that BMPs and ROS interplay in a number of ways. BMPs stimulate ROS production by inducing NOX expression, while ROS regulate the expression of several BMPs. Moreover, BMPs and ROS influence common signaling pathways, including PI3K/AKT and MAPK. Additionally, dysregulation of BMPs and ROS occurs in several pathologies, including vascular and musculoskeletal diseases, obesity, diabetes and kidney injury. Here, we review the current knowledge on the integration between BMP and ROS signals and its potential applications in the development of new therapeutic strategies.
1. Introduction
Bone morphogenetic proteins (BMPs) were first described in the 1960s as osteoinductive soluble factors that belong to the transforming growth factor-β (TGF-β) superfamily [1]. BMPs are expressed in multiple cell types and participate in a wide variety of processes, including morphogenesis, cell differentiation and regulation of whole-body homeostasis. Several studies demonstrate that BMPs contribute to gastrulation and mesoderm patterning. In extraembryonic tissues, BMPs are critical for primitive streak formation [2] and to establish left-right pattering [3]. During organogenesis, BMP signaling promotes mesoderm differentiation into heart tissue. In the ectoderm, BMPs induce neural differentiation and are necessary for neural tube formation [4]. In the peripheral nervous system, BMPs induce neuronal lineage commitment and neuronal differentiation whereas in the progenitor cells of the Central Nervous System (CNS), BMPs suppress neuronal or oligodendroglial differentiation and promote astroglial formation [5]. BMPs are also involved in skeletogenesis [6] and bone remodeling after birth [7]. In fact, BMP signaling is required for chondrogenesis [8] and osteogenesis [9]. In kidney organogenesis, BMPs promote the elongation of the ureteric bud and regulate the proliferation and differentiation of the metanephric mesenchyme [10]. BMP signaling is also critical for the development of vascular system in embryos, and for the regulation of vascular homeostasis in adults and, when altered, being causative for several vascular dysfunctions. For instance, pulmonary arterial hypertension and hereditary hemorrhagic telangiectasia have been directly linked to impaired BMP signaling [11,12,13]. Finally, BMPs are crucial for a proper reproductive system function. In gonadal differentiation, BMP8 is important for spermatogenesis maintenance [14], while BMP15 is associated with granulosa cell proliferation in the ovary [15]. Due to BMP relevance in organ generation and maintenance, alterations in BMP signaling are associated with several pathologies.
BMPs interact with specific serine-threonine kinase receptors present in the cell membrane. Subsequently, the information is transduced to the nucleus through the SMAD pathway. Besides this signaling mode, several non-canonical, SMAD-independent BMP transducers have been identified, including small GTPases, phosphatidylinositol 3-kinase/AKT (PI3K/AKT) and distinct types of mitogen-activated protein kinases (MAPKs) [16]. In this context, recent evidence suggests that reactive oxygen species (ROS) can also act as second messengers for BMPs.
ROS are highly reactive molecules derived from the reduction of molecular oxygen as a consequence of cellular metabolism or the activity of specific enzymes, such as the NADPH oxidase (NOX) complexes [17]. Since ROS are highly unstable, they react with proteins, nucleic acids, lipids and other cellular components and often disrupt their cellular functions. Therefore, ROS were first considered as harmful molecules. Accordingly, oxidative stress is involved in multiple pathologies such as cardiac and neurodegenerative diseases, vascular disorders, diabetes, infections and cancer [18]. Even though oxidative stress occurs in several pathologies, numerous studies demonstrate that ROS signaling is also important in normal physiology and in the generation of proper redox biological responses [19]. For instance, in skeletal muscle, ROS are required for normal contraction, where they modulate muscle adaptations to exercise [20]. ROS also contribute to immune response activation and regulation [21] and mediate leucocyte adhesion to endothelial cells [22]. Indeed, the physiological role of ROS is based on their capacity to modulate several signaling pathways, including nuclear factor-kappa B (NF-κB), MAPKs, kelch-like ECH-associated protein 1 (KEAP1)-nuclear factor erythroid 2-related factor 2 (NRF2) and PI3K-AKT [19].
Although little is known about the relationship between BMPs and ROS, both signals share common transducing modules, and their dysregulation occur in several pathologies. In this review, we will describe the putative modes of signaling crosstalk between BMPs and ROS and analyze their implications in physiology and disease.
2. BMP Signals
BMPs structurally belong to the transforming growth factor-β (TGF-β) superfamily [23]. BMPs share seven highly conserved cysteines with other TGF-β family members. Six of them form three intramolecular disulfide bonds, and the seventh one creates a covalent disulfide bond with another monomer to form homo- or heterodimers [24]. BMPs are also sub-classified into several groups, including BMP2/4, BMP5/6/7/8, growth and differentiation factor (GDF)-5/6/7 and BMP9/10 [23]. BMPs diffuse away from the cell of origin or form stable complexes with their cleaved proto-domains.
Outside the cell, BMPs bind to BMP receptors (BMPRs) and initiate an intracellular transduction cascade. BMPRs are serine/threonine kinase receptors with a short extracellular domain, a single transmembrane domain and an intracellular serine/threonine kinase domain. They are classified in two groups: BMPR type I (ACVR1, ACVRL1, BMPR1A and BMPR1B) and BMPR type II (BMPR2, ACVR2 and ACVR2B) [25]. Different BMP subgroups display disparate type I and type II receptor binding specificities [23]. BMP binding induces the formation of a heteromeric complex between type I and type II receptors. Once the complex is established, constitutively active kinase activity present in type II receptors phosphorylates the GS domain (a glycine and serine-rich intracellular region located adjacent to the kinase domain) of type I receptors, an action that leads to its activation. The active complex phosphorylates and activates the SMAD family of transcription factors that transduce the signal to the nucleus [26]. Under basal conditions, receptor-regulated SMADs (R-SMAD) are anchored at the cell membrane through their interaction with several proteins. In the canonical pathway, type I receptors phosphorylate R-SMADs 1/5/8. R-SMAD phosphorylation releases them from the membrane and disrupts the auto-inhibitory interaction between their N- and C-terminal domains. Then, two active R-SMAD 1/5/8 molecules bind to the common partner SMAD4 and translocate to the nucleus, where they interact with several transcription factors and transcriptional co- activators/repressors [27,28] (Figure 1).
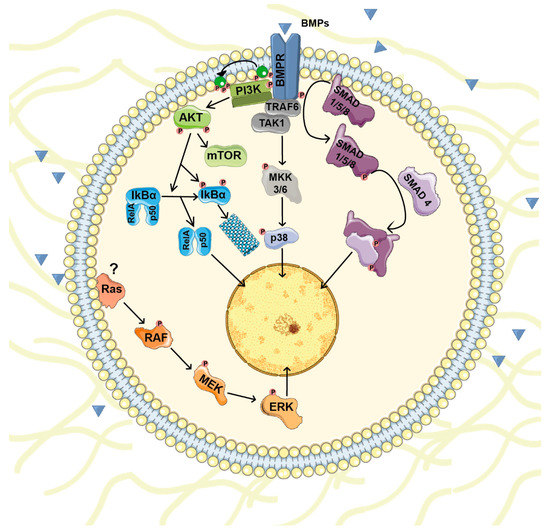
Figure 1.
BMP signaling pathways. Once BMP receptors (BMPR) are stimulated they form a complex and initiate an intracellular transduction cascade. In the canonical pathway, BMP receptors phosphorylate and activate the SMAD family of transcription factors that transduce the signal to the nucleus. Besides SMADs, BMPs modulate other intracellular signaling pathways (non-canonical BMP signaling) including PI3K/AKT and various types of MAPKs pathways.
Besides the canonical signaling through SMADs, BMPs modulate other intracellular signaling pathways with important roles in cell physiology. Non-canonical BMP signaling includes small GTPases, PI3K/AKT, lim kinase-1 (LIMK1) and various types of MAPKs [28]. For instance, BMPs regulate p38 activity through TGF-β-associated kinase 1 (TAK1) [29,30]. TAK1 is a member of the MAPKKK family whose activity is regulated by the E3 ubiquitin ligase TRAF6 in a receptor-kinase-independent manner. Upon BMP/BMPR complex formation, TRAF6 becomes poly-ubiquitinated and recruits and triggers TAK1 activation that induces p38 activity [31] (Figure 1).
BMP signaling is regulated by multiple mechanisms. Intracellularly, several miRNAs regulate the expression of BMPs, BMPRs and downstream effectors. Inhibitory Smads (such as Smad 6 and 7) and methylation of the promoters also regulate BMP signaling. Extracellularly, pseudo-receptors and BMPs antagonists control BMP availability [32,33]. More than 15 antagonists have been discovered which are classified in three subgroups based on the size of the cysteine knot, a common feature of BMPs. First, antagonists with an eight-member ring represented by the DAN family (including DAN, GREM2, Gremlin, Cerberus, Coco and Caronte. second, USAG-1, DANTE and twisted gastrulation (TSG) with a nine-member ring and finally, inhibitors with a ten-member rings including Chordin (CHD), Noggin (NOG), Ventroptin (CHRDL1), Follistatin (FST) and FLRG-follistatin-related gene (FSTL3) [32,34]. Finally, the binding of BMPs to several extracellular matrix (ECM) proteins also modulates their bioavailability [34].
3. ROS
ROS are radical and non-radical oxygen species that result from the partial reduction of oxygen [35]. Cellular ROS include the superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (OH·) that arise either as byproducts of aerobic metabolism or as defense mechanisms against xenobiotics or bacterial invasion [36]. Under physiological conditions, ROS act as signaling molecules that are important for adaptation to changes in nutrient supply and the oxidative environment [37]. ROS also contribute to regulate cell proliferation and differentiation. However, increased ROS levels or changes in their compartmentalization can dysregulate oxidation of proteins, lipids and nucleic acids [37]. Therefore, the ROS concentration is in a dynamic equilibrium and is modulated by cellular processes that produce and eliminate ROS.
A primary ROS source is O2−, which is generated in the mitochondria by complexes I, and III or in other cellular compartments by the action of several enzymes such as NOXs. Superoxide reacts with molecules to cause molecular damage or turn into H2O2 spontaneously or by the action of the enzyme superoxide dismutase (SOD). Other ROS sources are the mitochondrial enzymes monoamine oxidase and cytochrome b5ductase, as well as glycerol-3-phosphate dehydrogenase, aconitase, pyruvate dehydrogenase or α-ketoglutarate dehydrogenase [38].
Since high ROS levels generate cell damage, cells have developed several defense mechanisms to detoxify ROS. H2O2 is reduced to generate water by the action of catalase or glutathione peroxidase (GPx). Thioredoxins, peroxiredoxins and GPxs act as antioxidant enzymes by reversing the oxidation of cysteine residues. Finally, cells also have non-enzymatic antioxidants that include low-molecular-weight compounds, such as vitamins C and E, β-carotene, uric acid and glutathione (GSH) [36].
4. ROS-Mediated Cellular Signaling
ROS are highly reactive molecules with the capacity to target several transductor proteins (NF-κB, MAPKs, KEAP1-NRF2 and PI3K-AKT), ion channels and transporters (Ca2+ and mitochondrial permeability transition pore [mPTP]), and thus, impact on cell growth, differentiation, migration and death [37] (Figure 2).
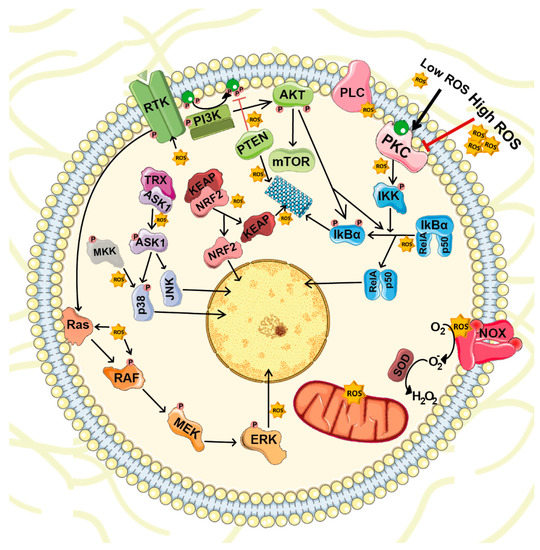
Figure 2.
ROS-mediated cellular signaling. ROS are highly reactive molecules that act as second messengers inside the cell. Besides mitochondrial respiration, NOXs represent the major ROS source. Under physiological conditions, intracellular ROS target and stimulate several transductor proteins NF-κB, MAPKs, KEAP1-NRF2 and PI3K-AKT important for cell survival, proliferation and differentiation.
The NF-κB family of transcription factors are involved in multiple cellular processes such as immune and inflammatory responses, development, cellular growth and apoptosis. NF-κB dimers are bound to their inhibitors of κB (IκBs) and retained in the cytoplasm. Consequently, NF-κB activation depends on IκB degradation, which occurs after its phosphorylation by IκB kinase (IKK). Activators of the IKK complex include MAPKKKs such as MEKK1, MEKK3 and TAK1, which represent a common hub for several stimuli, including BMPs and ROS. Recent studies demonstrate that ROS can induce IκBα phosphorylation and degradation [39]. ROS also induce S-glutathionylation and IKK inhibition [40] and other upstream kinases, such as MKK1; these actions hamper NF-κB activation. Conversely, NF-κB can alter ROS levels by increasing the expression of antioxidant proteins such as Cu-Zn-SOD, Mn-SOD, GPx and glutathione S-transferase-Pi (GST-Pi) [37].
ROS can stimulate MAPK pathways at different levels. ROS activate growth factor receptors even without binding to their corresponding ligand [41]. ROS also mediate the oxidation of Cys124 in extracellular signal-regulated kinase (ERK), an action that results in its activation [42]. ROS can also activate the ERK pathway indirectly when superoxide induces phospholipase C-γ (PLC-γ) phosphorylation, which generates inositol trisphosphate (IP3) and diacylglycerol (DAG) [43]. c-Jun N-terminal kinase (JNK) is another MAPK activated by cytokines (tumor necrosis factor [TNF] and FAS) and several environmental stresses such as oxidative stress. In this case, MAPKKs phosphorylate JNK on critical threonine and tyrosine residues, changes that lead to its activation. Under unstressed conditions, apoptosis signal-regulating kinase 1 (ASK1), a MAPKKK in the pathway, is bound to thioredoxin (TRX), which inhibits its activity. TRX oxidation by ROS induces ASK1 release and activation [44]. The p38 MAPK pathway is also stimulated by cellular stress and cytokines, such as TNF-α and interleukin-1beta (IL-1β). ROS can induce p38 pathway in several ways. As mentioned previously, ROS can directly activate growth factor receptors. Oxidative stress also directly or indirectly affect MAPKKKs involved in p38 signaling, including MEKKs and MLK3 [37].
PI3K-AKT is also susceptible to ROS regulation. PI3K catalyzes phosphatidylinositol 3,4,5-trisphosphate (PIP3) synthesis that recruits it into the plasma membrane and activates proteins that contain the pleckstrin homology domains such as AKT and its upstream activator PDK1. Activation of AKT is induced by its phosphorylation in Thr308 and Ser473 [45]. AKT activity is counteracted by its dephosphorylation by protein phosphatase 2 (PP2A). PP2A is involved in the regulation of WNT, mTOR and MAP kinase pathways, cell cycle progression as well as DNA repair [46]. Inhibition of mTORC1 activates PP2A and subsequent DNA-PK. DNA-PK is required for the repair of DNA double-strand breaks (DSBs). Besides its role in DNA repair, DNA-PK also phosphorylates AKT in Ser473 kinase, under DNA damage conditions [47]. On the other hand, AKT activity can be also reduced through PIP3 dephosphorylation by phosphatase and tensin homolog (PTEN). ROS induce PTEN degradation [48] and may also promote oxidative inactivation of PP2A [49].
ROS also regulate protein kinase pathways through the oxidation of cysteine sulfhydryl groups that are present in several protein kinases, including protein kinase A (PKA) [50], protein kinase C (PKC) [51], protein kinase D (PKD) [52], receptor tyrosine kinases [53] and Ca/calmodulin-dependent protein kinase II (CaMKII) [54]. Recently, a study demonstrated that ROS oxidize Cys17 and 38 of the PKA regulatory subunits, changes that lead to the formation of an inter-subunit disulfide bond that activates PKA independently of cyclic AMP (cAMP) levels [55]. The PKC family contains membrane protein kinases composed of an N-terminal regulatory region and a C-terminal catalytic domain [56]. Both domains contain cysteine-rich motifs targeted by ROS and they play a dual role in PKC regulation. In the regulatory domain, these cysteines form two pairs of zinc fingers that inhibit PKC activity. Low ROS doses destroy the zinc finger conformation, thus activating PKC even in the absence of Ca2+ or phospholipids. However, high ROS doses react with cysteine residues in the catalytic domain, and these interactions lead to PKC inactivation [57,58]. Additionally, ROS enhance PKD activation in several ways: they induce the production of DAG, the activation of PKC and the phosphorylation of PKD at Tyr93, which facilitates its binding with PKC [59,60].
The KEAP1-NRF2 pathway is essential for the maintenance of redox balance. Under basal conditions, NRF2 remains in the cytosol associated with KEAP1, which recruits CUL3 that ubiquitinates NRF2, leading to its degradation. Increased ROS levels promote the dissociation of the NRF2-KEAP1 complex by the oxidation of key reactive cysteine residues (Cys151, Cys273 and Cys288) of KEAP1 and/or the activation of kinases such as PKC, PI3K or AMPK that phosphorylate NRF2 [61,62,63,64]. Both events reduce NRF2 degradation and facilitate its translocation to the nucleus. Once in the nucleus, NRF2 heterodimerizes with other transcription factors and binds to antioxidant response element (ARE) sequences to promote the expression of target genes [65]. Activation of these different pathways also contributes to ROS scavenging through the induction of NRF2. NF-κB acts as a transcription factor for NRF2 [66] and, as mentioned above, PKC and PI3K phosphorylate NRF2, an action that stabilizes the protein and promotes its nuclear translocation [62,63,64]. Simultaneously, ROS activate glycogen synthase kinase 3β (GSK3β), which leads to nuclear import of Src kinases that phosphorylate NRF2 at Tyr568 and promote its nuclear export [67].
5. NOXs
NOXs and dual oxidases (DUOXs) are membrane-bound enzymatic complexes that catalyze the transfer of one electron from NADPH to oxygen to generate ROS in response to a stimulus, such as growth factors, cytokines and calcium signals. In the human genome, seven NOX homologues have been identified: NOX1 to NOX5 and DUOX1 and DUOX2. These genes differ in their preferential tissue of expression, the type of ROS released and their regulatory mechanisms. NOXs produce either superoxide (NOX1-3 and NOX5) or H2O2 (NOX4 and DUOX1-2) in a NADPH-dependent manner [68]. NOXs are classified in three main groups according to the domains they contain. NOX1, NOX2, NOX3 and NOX4 contain the electron transfer center. NOX5 contains the catalytic domain and an amino-terminal calmodulin-like domain that acts as a regulatory domain. DUOXs contain the catalytic domain and a calmodulin-like domain connected by an α-helix to an amino-terminal peroxidase-homology domain.
Short-term NOX regulation involves regulatory subunits. NOX1, NOX2 and NOX3 are present as inactive transmembrane monomers, and interaction with other subunits is required for their activity. However, NOX4 is synthesized in a constitutively active form [69]. NOX1 to NOX4 interact with p22phox, which has a scaffold function. NOX5 does not interact with p22phox but requires homo- or multimerization for its stabilization [70] and depends on cytosolic calcium levels for its activation. To activate the NOX catalytic center, three signaling events must occur. First, the regulatory domain (p40phox, p47phox and p67phox), which is in the cytosol, is phosphorylated in the auto-inhibitory region. This process involves protein kinases, including PKC and AKT [71].
Then, phosphorylation of p47phox releases its binding to the SRC-homology 3 (SH3) domain, allowing p47phox to bind to p22phox (in the cell membrane) and to lipids provided by PI3K and phospholipase D [72,73]. Finally, it is required the presence of two guanine nucleotide-binding proteins, RAP1A and RAC2. RAP1A is a membrane protein, while RAC2 localizes in the cytosol in a dimeric complex with its inhibitor guanine nucleotide dissociation inhibitor (Rho-GDI). Upon its activation, RAC2 binds GTP and translocates to the membrane along with p40phox, p47phox and p67phox, that constitute the active complex [74].
NOX enzymes are also regulated at the transcriptional level. This regulation depends on the specific enzyme and cell type. For instance, NOX1 is strongly induced by the growth factors angiotensin II, platelet-derived growth factor (PDGF) and phorbol esters (in vascular smooth muscle) and keratinocyte growth factor-alpha (KGF-α) and interferon gamma (IFN-γ) in human colon cells. Comparatively, NOX4 is regulated by angiotensin II [75].
NOX enzymes participate in several biological processes. In neutrophils, NOXs generate high superoxide concentrations in the phagosome to enhance killing bacteria. In barrier cells, IFN-γ and lipopolysaccharide (LPS) induce NOX1 expression through Toll-like receptor-4 to help kill invading microorganisms. In addition to host defense, NOX-derived ROS contribute to signal transduction by regulating biological processes such as cell growth, differentiation, angiogenesis and senescence. For instance, NOX4 overexpression in tumors is associated with apoptosis and cell senescence [75]. NOX4 is also implicated in angiotensin-II-dependent signaling in vascular smooth-muscle cells [76] and in insulin signaling in adipose tissues [77]. DUOX enzymes are implicated in biosynthetic reactions that involve ECM proteins [75].
6. Interplay between BMPs and ROS Signaling
BMPs stimulate ROS production in several cell types. BMPs stimulate ROS production by activation and/or induction of NOX1-5 expression. For instance, in adult renal progenitors BMP2 induces NOX4 and NOX2 expression and activity [78]. ROS reciprocally regulate the expression of BMP2 in osteoblasts [79]. Both BMPs and ROS activate several common signaling pathways that should be coordinated (Figure 3). Consequently, unregulated changes in their expression or production or altered signaling could result in severe repercussions.
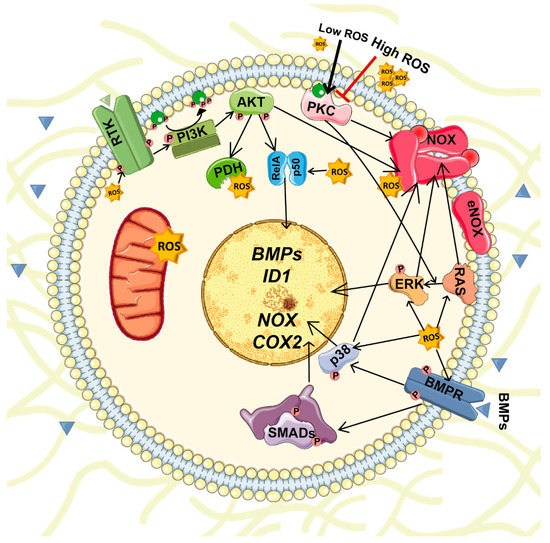
Figure 3.
Interplay between BMPs and ROS signaling. Production of BMPs and ROSs are reciprocally regulated. BMPs stimulate ROS production by activation and/or induction of NOX1-5 expression while ROS regulate the expression of several BMPs. Furthermore, both BMPs and ROS activate several common signaling pathways including PI3K-AKT, ERK, JNK and p38 MAPKs.
NOX1, NOX2 and NOX3 require p47phox and p67phox phosphorylation to facilitate translocation of the cytosolic subunits and formation of an active complex. The primary kinase involved in p47 phosphorylation is PKC (α, β, δ, and ζ). Other pathways, including ERK and p38, PKA and AKT, also contribute to NOX activation [80]. Therefore, BMPs can activate NOX enzymes by inducing their expression or through phosphorylation by the non-canonical signaling pathways above mentioned. For instance, in osteoblasts BMP2 enhances ROS production by activating NOX [79], and BMP2, BMP4 and BMP7 upregulate NOX4, NOX1 and NOX2 expression in osteoblasts, sympathetic neurons and monocytes [79,81,82]. Moreover, in podocytes, BMP2 increases the free cytosolic Ca2+concentration, and the expression of DNA-binding protein inhibitor 1 (ID1). Both events elevate NADPH-dependent ROS production [83].
Activation of non-canonical pathways by BMPs also induces cyclooxygenase 2 (COX2) gene expression [84], which is a source of ROS [85]. Moreover, different prostanoids produced by COX2 modulate NOX activity and regulate ROS production [85]. Finally, PI3K/AKT signaling, which can be activated by BMPs [30], contribute to elevate ROS levels by activating NOX, modulating mitochondrial bioenergetics and enhancing the metabolic rate. In this latter case, AKT phosphorylate GSK3β, which reduces its activity thus enhancing the activity of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase complexes. Both of these complexes generate superoxide and H2O2 [86]. Unlike other NOX isoforms, NOX4 is constitutively active. Thus, its regulation occurs mainly at the transcriptional level. Several cytokines, including BMP2, TGFβ and angiotensin II, can upregulate NOX4 expression through PKC and/or SMAD pathways [78,87].
Reciprocally, ROS contribute to the regulation of BMP expression. In particular, BMP2 and BMP4 are upregulated in conditions associated with oxidative stress [11]. For example, BMP2 autoregulation of its own expression is mediated by SMAD-dependent transcription and can also be modulated by NOX4-derived ROS in osteoblasts [79]. Further, increased H2O2 levels, produced by NOX activation or other conditions such as inflammation, induce NF-κΒ activation. Once active, NF-κΒ enhances BMP2 expression [11,88]. Therefore, although the mechanisms are mostly unknown, evidence suggests that there is a positive regulatory loop between ROS and BMP signals. Consequently, alteration of these regulatory loops can lead to oxidative stress and cell dysfunction and contribute to several pathologies.
The crosstalk between BMPs and ROS goes beyond the regulation of their own transcription and activation; it also affects downstream signaling pathways shared by these two molecules. As mentioned above, ROS activate ERK, JNK and p38 MAPKs [42], while BMPs activates TAK1 that acts upstream of JNK and p38 [89]. PI3K-AKT is another intracellular pathway where ROS and BMP signaling converge. BMP2 increases AKT phosphorylation in pancreatic cancer cells [90], while ROS reduces PI3K-AKT downregulation by targeting PTEN and PP2A in embryonic rat heart cells [91].
7. BMPs and ROS in Cell Specification
BMPs were identified as molecules capable of modulating mesenchymal cell specification and inducing bone development. In particular, BMPs regulate osteoblast differentiation through induction of bone-determining transcription factors [92]. ROS generation also influences bone cell differentiation. Undifferentiated mesenchymal stem cells (MSCs) display low levels of ROS and express high levels of antioxidant enzymes. This evidence is linked to MSC self-renewal, as physiological ROS upregulation is required for MSC differentiation. For instance, during MSC osteogenic differentiation, WNT/β-catenin activation increases ROS [93]. In osteoblast progenitor cells, BMP2 activates NOX4, which is highly expressed in pre-osteoblasts, and this activation results in ROS production. Superoxide anions generated by NOX4 are rapidly converted into H2O2, and ROS produced by NOX4 induce alkaline phosphatase expression and activate PI3K and SMAD 1/5 signal transduction pathways. MSCs also generate both permanent and transient cartilage. BMP signaling is critical for cartilage formation [94], and ROS generated by NOX2 and NOX4 are also essential for the early chondrogenesis stages. ROS also contribute to osteoclast differentiation. Osteoclasts arise from the fusion of multiple monocyte and macrophage precursors in the presence of receptor activator of NF-κB ligand (RANKL). RANKL stimulates ROS production, which is required for RANKL-induced activation of AKT, NF-κB and ERK pathways [95]. Moreover, ROS are necessary for the resorptive function of osteoclasts [96].
ROS and BMPs also participate in the cardiovascular system development. In embryonic stem cells, ROS promote differentiation toward both cardiomyogenic and vascular cell lineages through AMP-activated protein kinase (AMPK) pathway activation. ROS and NOX4 activation are also involved in cardiogenesis and cardiac myofibrillogenesis, since antioxidants and NOX4 silencing impair the cardiac differentiation of stem cell precursors. In early embryonic mouse heart, NOX4 is the main NOX isoform expressed. NOX4 generates ROS, an action that leads to p38 phosphorylation and monocyte enhancer factor 2C (MEF2C) nuclear translocation. Both events promote cardiomyocyte specification, transcription of sarcomeric proteins and myofibrillogenesis [97]. ROS also upregulate the expression of other cardiac-specific transcription factors and the expression of Bmp10 [97]. During embryonic heart formation, BMP10 prevents premature cell cycle withdrawal of mesenchymal cells and regulates the level of expression of several key cardiogenic transcriptional factors [98]. After birth, BMP2 upregulates NOX4 and enhance ROS-driven p38MAPK activation which promote cardiomyocyte differentiation in stem cells [99]. All these events contribute to the modulation of cardiac growth and function.
BMPs also affect dendritic formation in sympathetic neurons [100], where they promote dendritic growth in hippocampal [101], cortical [102] and retinal ganglion neurons [103]. In sympathetic neurons, BMP7 upregulates NOX2 expression, which is associated with an increase in oxygen consumption and ROS production. Newly synthesized ROS becomes restricted within certain cell compartments to avoid undesired cellular damage. In these sequestrations, they act as signaling molecules and upregulate the expression of differentiation genes [83]. Antioxidant treatment abolishes the effects of BMP7 on dendrite formation, data that demonstrates the importance of ROS in this phenomenon [81]. Additionally, BMP9 induces and maintains the neuronal cholinergic phenotypes in the central nervous system by inducing gene expression of choline acetyltransferase and vesicular acetylcholine transporter [104]. Interestingly, BMP9 levels and its bioavailability are reduced by redox-dependent proteolysis [105]. Consequently, increased extracellular ROS levels might be deleterious for neural differentiation. Although these data suggest a functional crosstalk between BMPs and ROS in the coordination of the differentiation of several cell types, further studies regarding the importance of the BMP and ROS interplay in the physiology of other tissues are necessary.
Cancer stem cells (CSC) constitute a small population of cells with the ability to proliferate indefinitely and give rise to the tumors [106]. In addition, differentiated progeny of CSCs create a niche that maintain both CSC and non-CSC components of the tumor hierarchy. BMPs have shown to be important not only for the differentiation of physiological stem cells but also for CSC. BMP2 have shown to induce CSC differentiation in osteosarcoma [107], while BMP4 induce differentiation of colorectal cancer stem cells [108], resulting in a reduction of tumorigenesis in both cases. In gliomas, where BMP2 expression is elevated, CSC secrete BMP antagonists, such as Gremlin1, to maintain their high proliferative capacity [109]. On the other hand, CSC, like normal stem cells have low intracellular levels of ROS, which contribute to their self-renewal capacity and resistance to chemotherapy. Several pathways are involved in controlling ROS levels in CSC. These pathways include PI3K/AKT/mTOR signaling pathway which regulate FOXO, HIF-1α, ATM, WNT, STAT and NF-κB pathways [110].
8. BMPs and ROS in Pathology
8.1. ROS and BMPs in Vascular Diseases
Vascular vessels are structures integrated by endothelial, smooth muscle cells (SMCs) and fibroblasts that work in a coordinated manner to adapt to the changes in their environment. Vascular dynamics involve modifications in cell growth, death and migration, and the synthesis or degradation of the ECM. Vascular remodeling responds to physiological and pathological changes in haemodynamic conditions and is driven by the presence of local growth factors, vasoactive substances and hemodynamic stimulus [111].
ROS, as second messengers, induce vessel wall remodeling and influence SMC and endothelial cell growth and survival (Figure 4). Besides mitochondrial respiration, NOXs represent the major ROS source in vascular cells, and their dysregulation is associated with cardiovascular diseases [112]. The superoxide anion (O2−) is the major ROS molecule in the vascular wall. It inactivates nitric oxide (NO), a vascular relaxing factor, and thus impairs relaxation [113]. Several pathological conditions, such as inflammation or high blood pressure, activate NOX-derived H2O2 production via TNFα. In the endothelium, high pressure increases wall tension, which elevates the calcium concentration, activates PKC and stimulates NOX-dependent. O2− and H2O2 production [114]. Increased H2O2 levels promote IκB degradation, and the consequent NF-κΒ activation induces BMP2 expression [11,88]. Blood vessel smooth muscle and endothelial cells express BMP2 and BMP4 mRNA [88]; expression is higher in endothelial cells. Both BMP2 and BMP4 are upregulated in atherosclerotic lesions associated with oxidative stress, inflammation and hyperglycemia [115]. Moreover, plasma BMP2 levels positively correlate with calcium density of the plaque in hyperglycemia-induced vascular calcification [116].
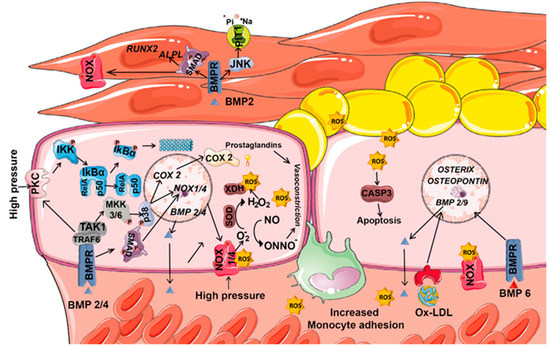
Figure 4.
Coordinated contribution of ROS and BMPs to vascular diseases. Vascular vessels are structures integrated by endothelial, smooth muscle cells (SMCs) and fibroblasts. In the endothelium, ROS are mainly produced by NOXs and its levels increase as a response to high pressure. In vascular vessels, ROS induce the expression of several BMPs, inactivates nitric oxide, and stimulate NF-κB pathway. By its part, BMPs activate MAPK pathways, increase NOX1/4 and COX2 expression and consequently enhance ROS generation. BMP6 and oxidized low-density lipoprotein (oxLDL) increase the expression of the osteoblast markers Osterix and Osteopontin in a NOX dependent manner. In SMCs, BMPs promotes the expression of the type III sodium-dependent phosphate cotransporter PIT1, increases the activity of NOX, and the levels of osteogenic markers (RUNX2 and ALPL).
In SMCs, BMP2 promotes the expression of the type III sodium-dependent phosphate cotransporter PIT1 via JNK pathway [117]. This effect is essential in matrix calcification induced by BMP-2. BMP2 also and increases the activity of NOX, which induces oxidative stress. The increase of NOX activity is mediated via activation of the bone morphogenetic protein receptor 2 and SMAD1 [118]. BMP2-mediated oxidative stress also induces endoplasmic reticulum (ER) stress that increases the function of GRP78, IRE1α, and XBP1. XBP1 is a transcription factor that binds to the Runx2 promoter and increases osteogenesis [118]. BMP2 also increases the levels of the osteogenic marker alkaline phosphatase (ALPL) and decreases the expression of the SMC marker SM22, changes that promote SMC transition to the osteochondrogenic phenotype and hence vascular calcification [117].
In endothelial cells, BMP2/4 activate PKC and MAPK pathways, which results in endothelial activation and increases monocyte adhesion. In these cells, MAPK activation is dependent on PKC and subsequent NOX-derived ROS generation [119]. In human coronary artery smooth muscle cells (HCSMC), BMP2 induce NOX via BMPR2 and SMAD1 [118]. In hypertensive patients, BMP2/4 upregulation is associated with impaired endothelial function in systemic blood vessels [120]. Moreover, BMP2/4 administration induces O2− generation, endothelial dysfunction and hypertension in systemic arteries but does not affect pulmonary blood vessels [121]. In fact, in pulmonary arteries resistant to atherogenesis, BMP4 does not induce ROS production. Consequently, high circulating BMP4 levels selectively induce systemic (but not pulmonary) hypertension. In the endothelium of hypertensive patients, BMP4 upregulates COX2 expression through a BMPR1A/p38 signaling pathway and stimulates NOX1 and NOX4 (but not NOX2), a phenomenon that increases ROS levels [122]. COX2 stimulation increases the release of constrictive prostaglandins, which promotes endothelium-dependent contraction. Induction of NOXs also increases the concentration of superoxide anions that inactivate NO and reduce endothelium dependent relaxations [123]. Besides, superoxide anions produced by NOX4 are converted to H2O2 by SOD. Then, H2O2 reacts with reduced iron ions, being converted into hydroxyl radicals that cause lipid peroxidation and protein carbonylation. Both modifications contribute to endothelial dysfunction [124]. Moreover, superoxide anions and H2O2 activate xanthine oxidase, which further promotes ROS production. All generated ROS initiate an inflammatory cascade and stimulates ICAM-1 expression leading to monocyte adhesion [82,125]. Thus, BMP4 inhibition has a potent anti-inflammatory and anti-atherogenic effect in coronary arteries [82].
In atherosclerosis mice models, BMP6 and oxidized low-density lipoprotein (oxLDL) synergistically regulate the expression of Osterix (Sp7) and Osteopontin. This process requires NOX activation and ROS production to induce osteogenesis and chondrogenesis in endothelial cells [126]. In the same way, oxLDL requires the activation of the BMP pathway for ROS production, and oxLDL induces the expression of other BMPs such as BMP2 or BMP9 [127]. Their combination with oxLDL or H2O2 potentiates canonical BMP signaling [126]. Similar effects are obtained in situations where BMP type II receptors are downregulated. Loss of BMPR2 decreases JNK signaling and enhances BMP9-induced mineralization. Both events contribute to the endothelial to mesenchymal transition (EMT) and vascular calcification [128]. Moreover, in models of cardiomyocyte hypertrophy generated by pressure overload or angiotensin II infusion, Bmp4 expression increases and induces cardiomyocyte hypertrophy, apoptosis, and cardiac fibrosis in a NOX4- and ROS-dependent manner [129]. The pro-apoptotic activation of caspase-3 by BMP2 is triggered by NOX-derived ROS and downstream activation of p38 and JNK [120]. Consequently, BMP4 could be used as a predictor of vascular dysfunction and a pharmacological target to prevent vascular dysfunction [122].
In parallel, drugs that modulate oxidative responses are being investigated as an effective treatment for cardiovascular diseases. For instance, molecules that induce NRF2 transcription inhibit oxLDL production, hence decrease inflammatory cytokine generation and attenuate endothelial injury [130]. Activators of the PI3K/AKT signaling pathway are also useful in the treatment of cardiovascular disease, since they prevent ROS accumulation in the vessels and acts as a vasodilator [131].
8.2. ROS and BMPs in Obesity and Diabetes
Emerging evidence suggests that diabetes leads to depletion of the cellular antioxidant system, increases ROS levels and triggers oxidative stress that is involved in the complications associated with this disease [132]. The ROS sources in diabetes are still controversial. In vitro studies show that hyperglycemia enhances mitochondrial respiration and NOX activity. However, the main in vivo ROS source depends on the tissue and the type of diabetes. In type 1 diabetes mellitus (T1DM), mitochondrial ROS is implicated in cardiac complications, whereas NOX-derived ROS are associated with kidney and vascular injury [133,134]. However, in type 2 diabetes mellitus (T2DM), mitochondrial ROS are associated with kidney [135] and retina injury [136], whereas an increase in NOX activity is implicated in vascular tissue damage [137].
Obesity is associated with chronic inflammation and myocardial dysfunction. In the ventriculus of obese mice, Nox1 and Nox2 expression increases [138]. A similar pattern occurs for the expression of fibrotic markers, whereas anti-fibrotic markers BMP2 and active SMAD1/5 are significantly elevated upon reduction of obesity [138]. BMP pathway inhibition in obese mice reduces atherosclerosis and vascular calcification and improves lipid metabolism [127]. For instance, BMP inhibitors reduce apolipoprotein B100 secretion and thus lower the levels of LDL-cholesterol and improve liver steatosis [127]. In the liver, BMP inhibition reduces the levels of hepcidin, an iron regulator. Reduction of hepcidin by BMP inhibition reduces intracellular iron and oxidative stress [139]. In vivo mice models confirm that BMP pathway inhibition limits foam cell formation and atherosclerosis by reducing macrophage intracellular iron and lipid efflux capacity [139]. However, in hypertrophic obesity, upregulation of Gremlin, an antagonist of BMP4/7, impairs BMP4-induced beige/brown adipogenesis, inhibit the capacity of BMP4 to induce commitment and differentiation of preadipocytes and contribute to the dysregulation of the adipose tissue. Silencing Grem1 and/or adding BMP4 during white adipogenic differentiation reactivated beige/brown markers [140].
In adipose tissue, NOX4-generated H2O2 modulates adipogenesis and adipose tissue function. Although ROS, as well as BMP4, mediate preadipocyte differentiation into adipocytes, in mature cells, elevated ROS levels induce adipocyte dysfunction. NOX4-derived ROS from adipocytes in the early stages of obesity initiate the recruitment of immune cells, including macrophages, and this phenomenon worsens insulin resistance and adipose tissue inflammation [141]. All these modifications converge in NOX upregulation and activation of NF-κB and p38 MAPK pathways that induce pro-inflammatory gene expression as well as excessive ROS formation in macrophages. Additionally, mitochondria-derived ROS partially maintain insulin resistance and adipose tissue inflammation during the late stages of obesity [141].
One of the main complications in diabetes is endothelial dysfunction. In T1DM, angiotensin II signaling activates NOX1 and induces tetrahydrobiopterin (BH4) deficiency and consequent endothelial nitric oxide synthase (eNOS) uncoupling [142,143]. However, in T2DM animal models, angiotensin II signaling is apparently not the main cause of endothelial dysfunction. Dyslipidemia contributes to elevate BMP4 levels in these animals. BMP4 can also activate NOX1 and induce BH4 deficiency. Further, BMP4 activates COX2 and vascular cell adhesion molecule 1 (VCAM-1) and induces inflammation in T2DM [143]. Diabetes is also associated with medial vascular calcification, also known as Mönckeberg’s calcification. This condition correlates with glucose levels in blood and the presence of calcium crystals in the medial layer [144]. Hyperglycemia and diabetes are strong activators of BMP signaling, and BMP2 and BMP4 are associated with atherogenesis in hyperglycemia. Moreover, in diabetic models, BMP4 disturbs blood flow and activates arterial NOXs, phenomena that promote ROS-induced inflammation, endothelial dysfunction and hypertension; BMP2 induces endothelial calcifications via WNT-MSX2 [145,146,147].
In the kidney, mesangial cells are the major source of glucose-dependent ROS generation. In the glomerular mesangial cells, oxidative stress caused by hyperglycemia activates the PKCζ isoform, which enhances TGFβ expression and increases ECM production. Overproduction of these components promotes morphological changes in the glomeruli. These changes hamper renal function and produce diabetic nephropathy, a serious complication in diabetes [148]. In recent years, recombinant BMP7 emerged as a potential therapy against diabetic nephropathy. BMP7 inhibits the phosphorylation and activation of PKCζ and JNK and reduces ROS generation and ECM synthesis. BMP7 also rescues podocytes from high-glucose-induced apoptosis by regulating SMAD5 and p38 phosphorylation [149]. Interestingly, high glucose levels induce the expression of Gremlin, a BMP antagonist. In mesangial cells, Gremlin induces transdifferentiation of tubular epithelial cells to a fibroblast-like phenotype [150]. In retinal pericytes and in the vascular walls, gremlin antagonizes the antiproliferative effects of BMPs. This action contributes to increased vascular endothelial proliferation and ultimately proliferative retinopathies [151]. Oxidative stress is another key event in the pathogenesis of diabetic retinopathy. In the retina of diabetic patients, the activity of antioxidant enzymes is diminished. The increased concentration of ROS, besides directly affecting cellular structures, activate other metabolic pathways that are detrimental to the development of diabetic retinopathy including PKC [152]. Consequently, modulation of BMP signals and ROS scavenging might serve as targets for high-glucose-associated pathologies.
8.3. ROS and BMPs in Kidney Injury
Sepsis, ischemia and nephrotoxicity are causative of acute kidney injury (AKI), which promotes structural damage and loss of kidney function. When treated quickly, the damage is reversible and kidney function is restored. However, in some cases AKI can lead to chronic kidney disease (CKD), where kidney damage worsens over time and kidney failure could occur [153]. Other conditions such as diabetes, hypertension or heart disease can also cause CKD. A characteristic of renal chronic diseases is the accumulation of excessive ECM and the presence of myofibroblast within the interstitium [154].
In the kidney, BMP2 is expressed by metanephric mesenchymal cells, mesangial cells in the glomerulus and differentiated podocytes. Moreover, in vivo AKI models demonstrate Bmp2 upregulation in adult renal progenitor cells, where it induces Sma, Col1a1 and Fibronectin expression, all of which drive a myofibroblastic transition. Furthermore, in this cell type BMP2 promotes NOX4 expression and activity. NOX4 is also necessary for TGFβ-induced kidney myofibroblast activation and to promote the transition of fibroblasts to myofibroblasts [78]. In podocytes, BMP2 increases the Ca2+ concentration and stimulates PKC activity, which is involved in the activation of NOX family members. In podocytes, BMP2 also up-regulates ID1, an inductor of NOX activity [83]. Thus, BMP2 stimulates ROS production through NOX enzyme activation in adult renal progenitors and podocytes. Elevated ROS levels induced by BMP promote the EMT, contribute to increased ECM production and hence impair kidney function.
On the other hand, BMP7 negatively regulates TGFβ1 signaling and suppresses inflammation, apoptosis and EMT. Moreover, BMP7 has antifibrotic activity, which prevents ureteral obstruction and diabetic nephropathy [155]. In chronic kidney injury, BMP7 can reverse TGFβ1-induced EMT and repair severe damage to renal tubular epithelial cells, actions that reverse chronic kidney injury [156]. Therefore, BMP7 signaling agonists play a protective role against renal fibrosis in kidney injury models and are postulated as a potential therapy in renal diseases [157,158]. In a similar way, silencing BMP7 antagonists, such as Gremlin, can induce therapeutic effects. For instance, in biopsies from patients with diabetic nephropathy, the levels of Gremlin are upregulated and correlated with elevated serum creatinine levels and tubulointerstitial fibrosis [159]. Inhibition of Gremlin allows the efficient binding of BMP7 to its receptor and improves kidney injury [160]. Furthermore, elevation of the antioxidant system by NRF2 activators appears to be beneficial in renal fibrosis [161,162].
8.4. ROS and BMPs in Musculoskeletal Diseases
Sarcopenia is a syndrome characterized by progressive and generalized loss of skeletal muscle mass and function. Histologically, sarcopenic muscles are characterized by the presence of adipose infiltration, fibrotic tissue and a decline in type II muscle fiber satellite cell content [163]. The cell fate decision between adipocyte and myoblast depends on BMP7 levels. In Myf5+ progenitors, BMP7 represses the expression of the adipogenic inhibitors Necdin and preadipocyte factor 1 (Pref1) and induces the expression of Prdm16, which activates the brown adipogenesis program and blocks the induction of myoblast-specific genes such as Myf5 and Myod1 [164]. Consequently, when BMP7 levels are high, Myf5+ progenitors differentiate into brown adipocytes, whereas if BMP7 expression decreases, Myf5+ progenitors can differentiate into the myogenic lineage [165].
In MSCs, chronic oxidative stress leads to S100 calcium-binding protein B (S100B) accumulation, increased NF-κB transcriptional activity and reduced expression of the promyogenic and anti-adipogenic mircoRNA-133 (miR-133). miR-133 reduction upregulates PRDM-16, which stimulates brown adipogenesis. In parallel, NF-κB activity also upregulates BMP7 expression that stimulates the myoblast-brown adipocyte transition in an autocrine/paracrine manner [166]. Thus, ROS-mediated BMP7 upregulation might promote sarcopenia, and its inhibition can be beneficial for this disease.
Osteonecrosis arises from the death of bone cells due to decreased blood flow. When osteonecrosis occurs next to the joints, it can lead to osteoarthritis (OA). In bone cells, the hypoxia produced by osteonecrosis increases the production of free oxygen radicals, including O2− and H2O2. Increased BMP2 expression occurs in necrotic lesions [167] and damaged cartilage from OA patients linked to higher ROS production [168]. Moreover, serum BMP2 levels correlate with OA severity, hence it has been suggested that its levels could also be used as a biomarker for this disease [169,170]. Interestingly, after ischemic osteonecrosis, hypoxic chondrocytes within the cartilage transcriptionally activate the BMP2 promoter. BMP2 stimulates proteoglycan synthesis, induces vascularization, promotes endochondral osteogenesis and has anabolic effects on chondrocyte metabolism and function [168,169]. In osteoarthritic cartilage, the increased concentration of BMPs is followed by an up-regulation of BMP antagonists, such as Gremlin and Follistatin. Gremlin1 expression is elevated since the early development of OA and its levels correlate with the progression of the disease [171,172]. Gremlin1 is also highly expressed in synovia and synovial fluids of patients with RA (Rheumatoid Arthritis) and its levels correlate with the concentration of proinflammatory cytokines. Upregulation of Gremlin1 induce the activation of ERK1/2, AKT, and increased expression of BCL2 while it reduces the levels of BAX. Both facts increase the survival, and capacity of migration of fibroblast-like synoviocytes [173] Treatment with Gremlin1 markedly inhibited terminal hypertrophic differentiation with no effects on the chondrogenesis [174] in conclusion, Gremlin1 could be a good therapeutical target in RA, capable of demoting hyperplastic synovitis [173].
9. Conclusions
Many biochemical, pharmacological and genetic studies confirm the relevance of ROS and BMPs in numerous aspects of cell biology and pathophysiology. Evidence also supports that both signals influence each other: BMP-dependent regulation of ROS production, and vice-versa, increases in the expression of distinct BMPs or modification on the expression of BMP antagonists in response to ROS generated by oxidative stress or NOX activity. However, many questions are still open and much more remains to be learnt regarding the relevance in the interplay between ROS and expression of specific BMPs and their antagonists in the context of normal tissue homeostasis and in pathological conditions. It should be possible to determine the effects of genetic alterations in the BMP signaling on cellular ROS levels or whether the cellular dysfunction associated to oxidative stress also depends on alterations of BMP expression. Additionally, BMPs and ROS share a number of downstream signaling components, suggesting a putative signaling crosstalk between the pathways. For instance, BMP and ROS activation of p38 MAPK or PI3K/AKT are well reported. Such parallel behavior and close signaling connections suggest that BMPs, BMP antagonists and ROS might regulate in coordination cellular physiology. However, despite this compelling rationale, strong evidence that such mechanisms are relevant is still lacking. Elucidation of these molecular mechanisms will not only advance basic biology, it may also provide new strategies for the treatment of a number of pathologies.
Author Contributions
C.S.-d.-D., C.P.-L., J.A.V., J.L.R., and F.V. conceived, analyzed and discussed the manuscript. C.S.-d.-D. and F.V. wrote the manuscript and draw the figures.
Funding
This research was supported by grants from the M.E.C. (BFU2014-56313-P and BFU2017-8 2421-P).
Acknowledgments
Cristina Sánchez de Diego and Carolina Pimenta Costa are the recipient of a F.P.U. fellowship from the Spanish Ministry of Education.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Urist, M.R.; Rosen, V.; Celeste, A.; Mitsock, L.; Whitters, M.; Kriz, R.; Hewick, R.; Wang, E. Bone: Formation by Autoinduction; American Association for the Advancement of Science: Washington, DC, USA, 1965; Volume 150, pp. 893–899. [Google Scholar]
- Winnier, G.; Blessing, M.; Labosky, P.A.; Hogan, B.L. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995, 9, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Dehart, D.B.; Sulik, K.K.; Hogan, B.L.M. Distinct requirements for extra-embryonic and embryonic bone morphogenetic protein 4 in the formation of the node and primitive streak and coordination of left-right asymmetry in the mouse. Development 2002, 129, 4685–4696. [Google Scholar] [PubMed]
- Hébert, J.M.; Mishina, Y.; McConnell, S.K. BMP Signaling Is Required Locally to Pattern the Dorsal Telencephalic Midline. Neuron 2002, 35, 1029–1041. [Google Scholar] [CrossRef]
- Mehler, M.F.; Mabie, P.C.; Zhang, D.; Kessler, J.A. Bone morphogenetic proteins in the nervous system. Trends Neurosci. 1997, 20, 309–317. [Google Scholar] [CrossRef]
- Storm, E.E.; Huynh, T.V.; Copeland, N.G.; Jenkins, N.A.; Kingsley, D.M.; Lee, S.J. Limb alterations in brachypodism mice due to mutations in a new member of the TGFβ-superfamily. Nature 1994, 368, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Harris, S.E.; Horn, D.; Geng, Z.; Nishimura, R.; Mundy, G.R.; Chen, D. Bone morphogenetic protein receptor signaling is necessary for normal murine postnatal bone formation. J. Cell Biol. 2002, 157, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Pizette, S.; Niswander, L. BMPs Are Required at Two Steps of Limb Chondrogenesis: Formation of Prechondrogenic Condensations and Their Differentiation into Chondrocytes. Dev. Biol. 2000, 219, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Wozney, J.M. The bone morphogenetic protein family and osteogenesis. Mol. Reprod. Dev. 1992, 32, 160–167. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Oshima, K.; Fogo, A.; Hogan, B.L.; Ichikawa, I. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J. Clin. Investig. 2000, 105, 863–873. [Google Scholar] [CrossRef]
- Lowery, J.W.; De Caestecker, M.P. BMP signaling in vascular development and disease. Cytokine Growth Factor Rev. 2010, 21, 287–298. [Google Scholar] [CrossRef]
- De Vinuesa, A.G.; Abdelilah-Seyfried, S.; Knaus, P.; Zwijsen, A.; Bailly, S.; Information, P.E.K.F.C. BMP signaling in vascular biology and dysfunction. Cytokine Growth Factor Rev. 2016, 27, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Bloch, D.B.; Ten Dijke, P.; Goumans, M.J.T.H.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signaling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Q.; Liaw, L.; Hogan, B.L. Bone morphogenetic protein 8A plays a role in the maintenance of spermatogenesis and the integrity of the epididymis. Development 1998, 125, 1103–1112. [Google Scholar] [PubMed]
- Otsuka, F.; Yao, Z.; Lee, T.; Yamamoto, S.; Erickson, G.F.; Shimasaki, S. Bone morphogenetic protein-15. Identification of target cells and biological functions. J. Biol. Chem. 2000, 275, 39523–39528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zou, M.H. Roles of Reactive Oxygen Species in Physiology and Pathology. In Atherosclerosis: Risks, Mechanisms, and Therapies; Wiley: Hoboken, NJ, USA, 2015; pp. 379–392. [Google Scholar]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Galano, J.M.; Durand, T.; Le Guennec, J.Y.; Lee, J.C.Y. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef]
- Reid, M.B.; Khawli, F.A.; Moody, M.R. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. 1993, 75, 1081–1087. [Google Scholar]
- Kraaij, M.D.; Savage, N.D.L.; Van Der Kooij, S.W.; Koekkoek, K.; Wang, J.; Berg, J.M.V.D.; Ottenhoff, T.H.M.; Kuijpers, T.W.; Holmdahl, R.; Van Kooten, C.; et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 17686–17691. [Google Scholar] [CrossRef]
- Sellak, H.; Franzini, E.; Hakim, J.; Pasquier, C. Reactive oxygen species rapidly increase endothelial ICAM-1 ability to bind neutrophils without detectable upregulation. Blood 1994, 83, 2669–2677. [Google Scholar]
- Kawabata, M. Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev. 1998, 9, 49–61. [Google Scholar] [CrossRef]
- Nohe, A. Signal transduction of bone morphogenetic protein receptors. Cell. Signal. 2004, 16, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P. Structural studies of the TGF-βs and their receptors—Insights into evolution of the TGF-β superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGF-β Signal Transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Gámez, B.; Rodríguez-Carballo, E.; Ventura, F. BMP signaling in telencephalic neural cell specification and maturation. Front. Cell. Neurosci. 2013, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef]
- Brazil, D.P.; Church, R.H.; Surae, S.; Godson, C.; Martin, F. BMP signalling: Agony and antagony in the family. Trends Cell Biol. 2015, 25, 249–264. [Google Scholar] [CrossRef]
- Rider, C.C.; Mulloy, B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem. J. 2010, 429, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone Morphogenetic Proteins: A critical review. Cell. Signal. 2011, 23, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Schoonbroodt, S.; Ferreira, V.; Best-Belpomme, M.; Boelaert, J.R.; Legrand-Poels, S.; Korner, M.; Piette, J. Crucial Role of the Amino-Terminal Tyrosine Residue 42 and the Carboxyl-Terminal PEST Domain of I B in NF- B Activation by an Oxidative Stress. J. Immunol. 2000, 164, 4292–4300. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [PubMed]
- León-Buitimea, A.; Rodríguez-Fragoso, L.; Lauer, F.T.; Bowles, H.; Thompson, T.A.; Burchiel, S.W. Ethanol-induced oxidative stress is associated with EGF receptor phosphorylation in MCF-10A cells overexpressing CYP2E1. Toxicol. Lett. 2012, 209, 161–165. [Google Scholar] [CrossRef]
- Wentworth, C.C.; Alam, A.; Jones, R.M.; Nusrat, A.; Neish, A.S. Enteric Commensal Bacteria Induce Extracellular Signal-regulated Kinase Pathway Signaling via Formyl Peptide Receptor-dependent Redox Modulation of Dual Specific Phosphatase 3. J. Biol. Chem. 2011, 286, 38448–38455. [Google Scholar] [CrossRef]
- Banan, A.; Fields, J.Z.; Zhang, Y.; Keshavarzian, A. Phospholipase C-γ inhibition prevents EGF protection of intestinal cytoskeleton and barrier against oxidants. Am. J. Physiol. Liver Physiol. 2001, 281, G412–G423. [Google Scholar] [CrossRef]
- Matsukawa, J.; Matsuzawa, A.; Takeda, K.; Ichijo, H. The ASK1-MAP Kinase Cascades in Mammalian Stress Response. J. Biochem. 2004, 136, 261–265. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Yue, P.; Tao, H.; Ramalingam, S.S.; Owonikoko, T.K.; Deng, X.; Wang, Y.; Fu, H.; Khuri, F.R.; et al. Protein Phosphatase 2A and DNA-dependent Protein Kinase Are Involved in Mediating Rapamycin-induced Akt Phosphorylation. J. Biol. Chem. 2013, 288, 13215–13224. [Google Scholar] [CrossRef]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Jeong, W.; Rhee, S.G.; Lee, C. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Shimura, T.; Sasatani, M.; Kamiya, K.; Kawai, H.; Inaba, Y.; Kunugita, N. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget 2016, 7, 3559–3570. [Google Scholar] [CrossRef]
- Srinivasan, S.; Spear, J.; Chandran, K.; Joseph, J.; Kalyanaraman, B.; Avadhani, N.G. Oxidative Stress Induced Mitochondrial Protein Kinase A Mediates Cytochrome C Oxidase Dysfunction. PLoS ONE 2013, 8, e77129. [Google Scholar] [CrossRef]
- Thompson, J.W.; Narayanan, S.V.; Perez-Pinzon, M.A. Redox Signaling Pathways Involved in Neuronal Ischemic Preconditioning. Curr. Neuropharmacol. 2012, 10, 354–369. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Kimchi, A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012, 19, 788–797. [Google Scholar] [CrossRef]
- Kruk, J.S.; Vasefi, M.S.; Heikkila, J.J.; Beazely, M.A. Reactive Oxygen Species Are Required for 5-HT-Induced Transactivation of Neuronal Platelet-Derived Growth Factor and TrkB Receptors, but Not for ERK1/2 Activation. PLoS ONE 2013, 8, e77027. [Google Scholar] [CrossRef]
- Luczak, E.D.; Anderson, M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell. Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schröder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-induced Activation of Type I Protein Kinase A Is Mediated by RI Subunit Interprotein Disulfide Bond Formation. J. Biol. Chem. 2006, 281, 21827–21836. [Google Scholar] [CrossRef]
- Newton, A.C. Protein Kinase C: Structure, Function, and Regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free. Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell Signaling through Protein Kinase C Oxidation and Activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef]
- Cowell, C.F.; Döppler, H.; Yan, I.K.; Hausser, A.; Umezawa, Y.; Storz, P. Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J. Cell Sci. 2009, 122, 919–928. [Google Scholar] [CrossRef]
- Wang, Q.J. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol. Sci. 2006, 27, 317–323. [Google Scholar] [CrossRef]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by Protein Kinase C Regulates Antioxidant Response Element-mediated Transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef]
- Nakaso, K.; Yano, H.; Fukuhara, Y.; Takeshima, T.; Wada-Isoe, K.; Nakashima, K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003, 546, 181–184. [Google Scholar] [CrossRef]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef]
- Jain, A.; Jaiswal, A.K. GSK-3beta Acts Upstream of Fyn Kinase in Regulation of Nuclear Export and Degradation of NF-E2 Related Factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants 2017, 6, 90. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free. Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Hoyal, C.R.; Gutierrez, A.; Young, B.M.; Catz, S.D.; Lin, J.H.; Tsichlis, P.N.; Babior, B.M. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 5130–5135. [Google Scholar] [CrossRef]
- Kanai, F.; Liu, H.; Field, S.J.; Akbary, H.; Matsuo, T.; Brown, G.E.; Cantley, L.C.; Yaffe, M.B. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nature 2001, 3, 675–678. [Google Scholar] [CrossRef]
- Zhan, Y.; Virbasius, J.V.; Song, X.; Pomerleau, D.P.; Zhou, G.W. The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J. Biol. Chem. 2002, 277, 4512–4518. [Google Scholar] [CrossRef]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Wingler, K.; Wünsch, S.; Kreutz, R.; Rothermund, L.; Paul, M.; Schmidt, H.H. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic. Biol. Med. 2001, 31, 1456–1464. [Google Scholar] [CrossRef]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H Oxidase Homolog Nox4 Modulates Insulin-Stimulated Generation of H2O2 and Plays an Integral Role in Insulin Signal Transduction. Mol. Cell. Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef]
- Simone, S.; Cosola, C.; Loverre, A.; Cariello, M.; Sallustio, F.; Rascio, F.; Gesualdo, L.; Schena, F.P.; Grandaliano, G.; Pertosa, G. BMP-2 induces a profibrotic phenotype in adult renal progenitor cells through Nox4 activation. Am. J. Physiol. Physiol. 2012, 303, F23–F34. [Google Scholar] [CrossRef]
- Mandal, C.C.; Ganapathy, S.; Gorin, Y.; Mahadev, K.; Block, K.; Abboud, H.E.; Harris, S.E.; Ghosh-Choudhury, G.; Ghosh-Choudhury, N. Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem. J. 2011, 433, 393–402. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2016, 10, 301. [Google Scholar] [CrossRef]
- Chandrasekaran, V.; Lea, C.; Sosa, J.C.; Higgins, D.; Lein, P.J. Reactive oxygen species are involved in BMP-induced dendritic growth in cultured rat sympathetic neurons. Mol. Cell. Neurosci. 2015, 67, 116–125. [Google Scholar] [CrossRef][Green Version]
- Sorescu, G.P.; Song, H.; Tressel, S.L.; Hwang, J.; Dikalov, S.; Smith, D.A.; Boyd, N.L.; Platt, M.O.; Lassègue, B.; Griendling, K.K.; et al. Bone Morphogenic Protein 4 Produced in Endothelial Cells by Oscillatory Shear Stress Induces Monocyte Adhesion by Stimulating Reactive Oxygen Species Production From a Nox1-Based NADPH Oxidase. Circ. Res. 2004, 95, 773–779. [Google Scholar] [CrossRef]
- Pache, G.; Schäfer, C.; Wiesemann, S.; Springer, E.; Liebau, M.; Reinhardt, H.C.; August, C.; Pavenstädt, H.; Bek, M.J. Upregulation of Id-1 via BMP-2 receptors induces reactive oxygen species in podocytes. Am. J. Physiol. Physiol. 2006, 291, F654–F662. [Google Scholar] [CrossRef]
- Susperregui, A.R.G.; Gamell, C.; Rodríguez-Carballo, E.; Ortuño, M.J.; Bartrons, R.; Rosa, J.L.; Ventura, F. Noncanonical BMP Signaling Regulates Cyclooxygenase-2 Transcription. Mol. Endocrinol. 2011, 25, 1006–1017. [Google Scholar] [CrossRef]
- Hernanz, R.; Briones, A.M.; Salaices, M.; Alonso, M.J. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin. Sci. 2014, 126, 111–121. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef]
- Sampson, N.; Berger, P.; Zenzmaier, C. Redox Signaling as a Therapeutic Target to Inhibit Myofibroblast Activation in Degenerative Fibrotic Disease. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Csiszar, A.; Smith, K.E.; Koller, A.; Kaley, G.; Edwards, J.G.; Ungvari, Z. Regulation of Bone Morphogenetic Protein-2 Expression in Endothelial Cells. Circulation 2005, 111, 2364–2372. [Google Scholar] [CrossRef]
- Moriguchi, T.; Kuroyanagi, N.; Yamaguchi, K.; Gotoh, Y.; Irie, K.; Kano, T.; Shirakabe, K.; Muro, Y.; Shibuya, H.; Matsumoto, K. A Novel Kinase Cascade Mediated by Mitogen-activated Protein Kinase Kinase 6and MKK3. J. Biol. Chem. 1996, 271, 13675–13679. [Google Scholar] [CrossRef]
- Chen, X.; Liao, J.; Lu, Y.; Duan, X.; Sun, W. Activation of the PI3K/Akt Pathway Mediates Bone Morphogenetic Protein 2-Induced Invasion of Pancreatic Cancer Cells Panc-1. Pathol. Oncol. Res. 2011, 17, 257–261. [Google Scholar] [CrossRef]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin Exerts an Antiapoptotic Effect by Regulating the Redox State of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef]
- Hu, C.; Zhao, L.; Peng, C.; Li, L. Regulation of the mitochondrial reactive oxygen species: Strategies to control mesenchymal stem cell fates ex vivo and in vivo. J. Cell. Mol. Med. 2018, 22, 5196–5207. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, S. The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem Cells Int. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Kang, I.S.; Kim, C. NADPH oxidase gp91phox contributes to RANKL-induced osteoclast differentiation by upregulating NFATc1. Sci. Rep. 2016, 6, 38014. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Kwak, H.B.; Lee, S.W.; Jin, H.M.; Kim, H.M.; Kim, H.H.; Lee, Z.H. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp. Cell Res. 2004, 301, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B.; Bettiol, E.; Charnay, Y.; Steger, K.; Krause, K.H.; Jaconi, M.E. The NADPH Oxidase NOX4 Drives Cardiac Differentiation: Role in Regulating Cardiac Transcription Factors and MAP Kinase Activation. Mol. Biol. Cell 2006, 17, 3978–3988. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Gong, F. BMP-2 and icariin synergistically promote p38MAPK-mediated cardiomyocyte differentiation of mesenchymal stem cells via enhanced NOX4-driven ROS generation. Med. Chem. Res. 2017, 26, 2547–2556. [Google Scholar] [CrossRef]
- Lein, P.; Johnson, M.; Guo, X.; Rueger, D.; Higgins, D. Osteogenic protein-1 induces dendritic growth in rat sympathetic neurons. Neuron 1995, 15, 597–605. [Google Scholar] [CrossRef]
- Charette, M.; Banker, G.; Withers, G.S.; Higgins, D. Bone morphogenetic protein-7 enhances dendritic growth and receptivity to innervation in cultured hippocampal neurons. Eur. J. Neurosci. 2000, 12, 106–116. [Google Scholar]
- Le Roux, P.; Behar, S.; Higgins, D.; Charette, M. OP-1 Enhances Dendritic Growth from Cerebral Cortical Neurons in Vitro. Exp. Neurol. 1999, 160, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Hocking, J.C.; Hehr, C.L.; Chang, R.Y.; Johnston, J.; McFarlane, S. TGFβ ligands promote the initiation of retinal ganglion cell dendrites in vitro and in vivo. Mol. Cell. Neurosci. 2008, 37, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Thangnipon, W.; Puangmalai, N.; Suwanna, N.; Soi-Ampornkul, R.; Phonchai, R.; Kotchabhakdi, N.; Mukda, S.; Phermthai, T.; Julavijitphong, S.; Tuchinda, P.; et al. Potential role of N-benzylcinnamide in inducing neuronal differentiation from human amniotic fluid mesenchymal stem cells. Neurosci. Lett. 2016, 610, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Salmon, R.M.; Upton, P.D.; Morrell, N.W.; Li, W. Regulation of bone morphogenetic protein 9 (BMP9) by redox-dependent proteolysis. J. Biol. Chem. 2014, 289, 31150–31159. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Weissman, I.L.; Reya, T.; Clarke, M.F. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar]
- Wang, L.; Park, P.; Zhang, H.; La Marca, F.; Claeson, A.; Valdivia, J.; Lin, C.Y. BMP-2 inhibits the tumorigenicity of cancer stem cells in human osteosarcoma OS99-1 cell line. Cancer Biol. Ther. 2011, 11, 457–463. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lombardo, Y.; Scopelliti, A.; Cammareri, P.; Todaro, M.; Iovino, F.; Ricci–Vitiani, L.; Gulotta, G.; Dieli, F.; De Maria, R.; Stassi, G. Bone Morphogenetic Protein 4 Induces Differentiation of Colorectal Cancer Stem Cells and Increases Their Response to Chemotherapy in Mice. Gastroenterology 2011, 140, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Wu, Q.; Yan, D.H.; Lee, C.H.; Rahim, N.; Tritschler, I.; DeVecchio, J.; Kalady, M.F.; Hjelmeland, A.B.; Rich, J.N. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014, 28, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxidative Med. Cell. Longev. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Epstein, F.H.; Gibbons, G.H.; Dzau, V.J. The Emerging Concept of Vascular Remodeling. N. Engl. J. Med. 1994, 330, 1431–1438. [Google Scholar] [CrossRef]
- Mueller, C.F.H.; Laude, K.; McNally, J.S.; Harrison, D.G. Redox Mechanisms in Blood Vessels. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 274–278. [Google Scholar] [CrossRef]
- Kojda, G.; Harrison, D. Interactions between NO and reactive oxygen species: Pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc. Res. 1999, 43, 562–571. [Google Scholar] [PubMed]
- Csiszar, A.; Lehoux, S.; Ungvari, Z. Hemodynamic Forces, Vascular Oxidative Stress, and Regulation of BMP-2/4 Expression. Antioxid. Redox Signal. 2009, 11, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone Morphogenetic Proteins in Vascular Calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sara, J.D.; Wang, F.L.; Liu, L.P.; Su, L.X.; Zhe, J.; Wu, X.; Liu, J.H. Increased plasma BMP-2 levels are associated with atherosclerosis burden and coronary calcification in type 2 diabetic patients. Cardiovasc. Diabetol. 2015, 14, 64. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef]
- Liberman, M.; Johnson, R.C.; Handy, D.E.; Loscalzo, J.; Leopold, J.A. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem. Biophys. Res. Commun. 2011, 413, 436–441. [Google Scholar] [PubMed]
- Csiszar, A.; Ahmad, M.; Smith, K.E.; Labinskyy, N.; Gao, Q.; Kaley, G.; Edwards, J.G.; Wolin, M.S.; Ungvari, Z. Bone Morphogenetic Protein-2 Induces Proinflammatory Endothelial Phenotype. Am. J. Pathol. 2006, 168, 629–638. [Google Scholar] [CrossRef]
- Tian, X.Y.; Yung, L.H.; Wong, W.T.; Liu, J.; Leung, F.P.; Liu, L.; Chen, Y.; Kong, S.K.; Kwan, K.M.; Ng, S.M.; et al. Bone morphogenic protein-4 induces endothelial cell apoptosis through oxidative stress-dependent p38MAPK and JNK pathway. J. Mol. Cell. Cardiol. 2012, 52, 237–244. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Jo, H.; Ballabh, P.; Ungvari, Z. Differential proinflammatory and prooxidant effects of bone morphogenetic protein-4 in coronary and pulmonary arterial endothelial cells. Am. J. Physiol. Circ. Physiol. 2008, 295, H569–H577. [Google Scholar] [CrossRef]
- Wong, W.T.; Tian, X.Y.; Chen, Y.; Leung, F.P.; Liu, L.; Lee, H.K.; Ng, C.F.; Xu, A.; Yao, X.; Vanhoutte, P.M.; et al. Bone Morphogenic Protein-4 Impairs Endothelial Function Through Oxidative Stress–Dependent Cyclooxygenase-2 Upregulation. Circ. Res. 2010, 107, 984–991. [Google Scholar] [CrossRef]
- Luo, J.Y.; Zhang, Y.; Wang, L.; Huang, Y. Regulators and effectors of bone morphogenetic protein signalling in the cardiovascular system. J. Physiol. 2015, 593, 2995–3011. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Zhang, Y.; Huang, Y. Bone morphogenic protein-4-induced oxidant signaling via protein carbonylation for endothelial dysfunction. Free Radic. Biol. Med. 2014, 75, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Grgurevic, L.; Christensen, G.L.; Schulz, T.J.; Vukicevic, S.; Information, P.E. Bone morphogenetic proteins in inflammation, glucose homeostasis and adipose tissue energy metabolism. Cytokine Growth Factor Rev. 2016, 27, 105–118. [Google Scholar] [CrossRef]
- Yung, L.M.; Sánchez-Duffhues, G.; Dijke, P.T.; Yu, P.B. Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc. Res. 2015, 108, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Derwall, M.; Malhotra, R.; Lai, C.S.; Beppu, Y.; Aikawa, E.; Seehra, J.S.; Zapol, W.M.; Bloch, K.D.; Yu, P.B. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arter. Thromb. Vasc. Biol. 2012, 32, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Duffhues, G.; De Vinuesa, A.G.; Van De Pol, V.; Geerts, M.E.; De Vries, M.R.; Janson, S.G.T.; Van Dam, H.; Lindeman, J.H.; Goumans, M.J.; Dijke, P.T. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Huo, R.; Sheng, Y.; Li, Y.; Xie, X.; Chen, C.; Liu, H.B.; Li, N.; Li, C.B.; Guo, W.T.; et al. Bone Morphogenetic Protein-4 Mediates Cardiac Hypertrophy, Apoptosis, and Fibrosis in Experimentally Pathological Cardiac Hypertrophy. Hypertension 2013, 61, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Tao, T.; Wang, X.; Liu, M.; Song, D.; Liu, X.; Shi, D. Zedoarondiol Attenuates Endothelial Cells Injury Induced by Oxidized Low-Density Lipoprotein via Nrf2 Activation. Cell. Physiol. Biochem. 2018, 48, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.P.; Cui, H.J.; Yang, A.L.; Luo, J.K.; Tang, T. Astragaloside IV Improves Vasodilatation Function by Regulating the PI3K/Akt/eNOS Signaling Pathway in Rat Aorta Endothelial Cells. J. Vasc. Res. 2018, 55, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Rösen, P.; Nawroth, P.P.; King, G.; Möller, W.; Tritschler, H.J.; Packer, L. The role of oxidative stress in the onset and progression of diabetes and its complications: A summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes. Metab. Res. Rev. 2001, 17, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Asaba, K.; Tojo, A.; Onozato, M.L.; Goto, A.; Quinn, M.T.; Fujita, T.; Wilcox, C.S. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int. 2005, 67, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Martín, A.S.; Du, P.; Dikalova, A.; Lassegue, B.; Aleman, M.; Góngora, M.C.; Brown, K.; Joseph, G.; Harrison, D.G.; Taylor, W.R.; et al. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am. J. Physiol. Circ. Physiol. 2007, 292, H2073–H2082. [Google Scholar] [CrossRef] [PubMed]
- Zhen, D.; Chen, Y.; Tang, X. Metformin reverses the deleterious effects of high glucose on osteoblast function. J. Diabetes Complicat. 2010, 24, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Diabetic Retinopathy: Mitochondrial Dysfunction and Retinal Capillary Cell Death. Antioxid. Redox Signal. 2005, 7, 1581. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef]
- Wang, H.T.; Liu, C.F.; Tsai, T.H.; Chen, Y.L.; Chang, H.W.; Tsai, C.Y.; Leu, S.; Zhen, Y.Y.; Chai, H.T.; Chung, S.Y.; et al. Effect of obesity reduction on preservation of heart function and attenuation of left ventricular remodeling, oxidative stress and inflammation in obese mice. J. Transl. Med. 2012, 10, 145. [Google Scholar] [CrossRef]
- Saeed, O.; Otsuka, F.; Polavarapu, R.; Karmali, V.; Weiss, D.; Davis, T.; Rostad, B.; Pachura, K.; Adams, L.; Elliott, J.; et al. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Svensson, P.A.; Grimsby, J.; Rondinone, C.; Smith, U. BMP4 and BMP Antagonists Regulate Human White and Beige Adipogenesis. Diabetes 2015, 64, 1670–1681. [Google Scholar] [CrossRef]
- De Villiers, D.; Potgieter, M.; Ambele, M.A.; Adam, L.; Durandt, C.; Pepper, M.S. The Role of Reactive Oxygen Species in Adipogenic Differentiation. In Genome Editing; Springer: Cham, Switzerland, 2017; Volume 1083, pp. 125–144. [Google Scholar]
- Youn, J.Y.; Gao, L.; Cai, H. The p47phox-and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetology 2012, 55, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Zhou, J.; Cai, H. Bone Morphogenic Protein 4 Mediates NOX1-Dependent eNOS Uncoupling, Endothelial Dysfunction, and COX2 Induction in Type 2 Diabetes Mellitus. Mol. Endocrinol. 2015, 29, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhong, H.; Liang, J.Y.; Fu, P.; Luo, Z.J.; Zhou, L.; Gou, R.; Huang, J. Effect of high glucose levels on the calcification of vascular smooth muscle cells by inducing osteoblastic differentiation and intracellular calcium deposition via BMP-2/Cbfα-1 pathway. J. Zhejiang Univ. Sci. B 2010, 11, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Song, H.; Kumar, S.; Nam, D.; Kwon, H.S.; Chang, K.H.; Son, D.J.; Kang, D.W.; Brodie, S.A.; Weiss, D.; et al. Anti-inflammatory and antiatherogenic role of BMP receptor II in endothelial cells. Arter. Thromb. Vasc. Biol. 2013, 33, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.S.; Aly, Z.A.; Lai, C.F.; Cheng, S.L.; Cai, J.; Huang, E.; Behrmann, A.; Towler, D.A. Vascular Bmp Msx2 Wnt Signaling and Oxidative Stress in Arterial Calcification. Ann. N. Y. Acad. Sci. 2007, 1117, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Boström, K.I.; Jumabay, M.; Matveyenko, A.; Nicholas, S.B.; Yao, Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ. Res. 2011, 108, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Kolset, S.O.; Reinholt, F.P.; Jenssen, T. Diabetic Nephropathy and Extracellular Matrix. J. Histochem. Cytochem. 2012, 60, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Chang, C.K.; Cheng, M.F.; Lin, H.J.; Cheng, J.T. The antioxidative effect of bone morphogenetic protein-7 against high glucose-induced oxidative stress in mesangial cells. Biochem. Biophys. Res. Commun. 2009, 382, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Díez, R.; Lavoz, C.; Carvajal, G.; Rayego-Mateos, S.; Diez, R.R.; Arduan, A.O.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Gremlin Is a Downstream Profibrotic Mediator of Transforming Growth Factor-Beta in Cultured Renal Cells. Nephron Exp. Nephrol. 2012, 122, 62–74. [Google Scholar] [CrossRef]
- Kane, R.; Stevenson, L.; Godson, C.; Stitt, A.W.; O’Brien, C. Gremlin gene expression in bovine retinal pericytes exposed to elevated glucose. Br. J. Ophthalmol 2005, 89, 1638–1642. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.S. Oxidative Stress and Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Q.; Simon, T.C.; Strebeck, F.; Chaudhary, L.; Morrissey, J.; Liapis, H.; Klahr, S.; Hruska, K.A. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy11Professor Robert Chevalier served as a guest editor for this paper. Kidney Int. 2003, 63, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.I.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-β1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Hsing, C.H.; Chou, W.; Wang, J.J.; Chen, H.W.; Yeh, C.H. Propofol increases bone morphogenetic protein-7 and decreases oxidative stress in sepsis-induced acute kidney injury. Nephrol. Dial. Transplant. 2011, 26, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Ha, H. Mechanisms of Epithelial-Mesenchymal Transition of Peritoneal Mesothelial Cells During Peritoneal Dialysis. J. Korean Med. Sci. 2007, 22, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Dolan, V.; Murphy, M.; Sadlier, D.; Lappin, D.; Doran, P.; Godson, C.; Martin, F.; O’Meara, Y.; Schmid, H.; Henger, A.; et al. Expression of gremlin, a bone morphogenetic protein antagonist, in human diabetic nephropathy. Am. J. Kidney Dis. 2005, 45, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q. Bone morphogenetic protein-7 and Gremlin: New emerging therapeutic targets for diabetic nephropathy. Biochem. Biophys. Res. Commun. 2009, 383, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF2 System to Prevent Kidney Disease Progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef]
- Choi, B.H.; Kang, K.S.; Kwak, M.K. Effect of Redox Modulating NRF2 Activators on Chronic Kidney Disease. Molecules 2014, 19, 12727–12759. [Google Scholar] [CrossRef]
- Verdijk, L.B.; Snijders, T.; Drost, M.; Delhaas, T.; Kadi, F.; van Loon, L.J.C. Satellite cells in human skeletal muscle; from birth to old age. Age (Omaha) 2014, 36, 545–557. [Google Scholar] [CrossRef]
- Frühbeck, G.; Sesma, P.; Burrell, M.A. PRDM16: The interconvertible adipo-myocyte switch. Trends Cell Biol. 2009, 19, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Morozzi, G.; Beccafico, S.; Bianchi, R.; Riuzzi, F.; Bellezza, I.; Giambanco, I.; Arcuri, C.; Minelli, A.; Donato, R. Oxidative stress-induced S100B accumulation converts myoblasts into brown adipocytes via an NF-κB/YY1/miR-133 axis and NF-κB/YY1/BMP-7 axis. Cell Death Differ. 2017, 24, 2077–2088. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Bian, H.; Aya-Ay, J.; Garces, A.; Morgan, E.F.; Gilbert, S.R. Hypoxia and HIF-1α expression in the epiphyseal cartilage following ischemic injury to the immature femoral head. Bone 2009, 45, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Nakase, T.; Miyaji, T.; Tomita, T.; Kaneko, M.; Kuriyama, K.; Myoui, A.; Sugamoto, K.; Ochi, T.; Yoshikawa, H. Localization of bone morphogenetic protein-2 in human osteoarthritic cartilage and osteophyte. Osteoarthr. Cartil. 2003, 11, 278–284. [Google Scholar] [CrossRef]
- Kamiya, N.; Shafer, S.; Oxendine, I.; Mortlock, D.P.; Chandler, R.L.; Oxburgh, L.; Kim, H.K. Acute BMP2 upregulation following induction of ischemic osteonecrosis in immature femoral head. Bone 2013, 53, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Ku, W.Y.; Zhou, Z.; Dellon, E.S.; Falk, G.W.; Nakagawa, H.; Wang, M.L.; Liu, K.; Wang, J.; Katzka, D.A.; et al. BMP-driven NRF2 activation in esophageal basal cell differentiation and eosinophilic esophagitis. J. Clin. Investig. 2015, 125, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Tardif, G.; Hum, D.; Pelletier, J.P.; Boileau, C.; Ranger, P.; Martel-Pelletier, J. Differential gene expression and regulation of the bone morphogenetic protein antagonists follistatin and gremlin in normal and osteoarthritic human chondrocytes and synovial fibroblasts. Arthritis Rheum. 2004, 50, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Tardif, G.; Pelletier, J.P.; Boileau, C.; Martel-Pelletier, J. The BMP antagonists follistatin and gremlin in normal and early osteoarthritic cartilage: An immunohistochemical study. Osteoarthr. Cartil. 2009, 17, 263–270. [Google Scholar] [CrossRef]
- Han, E.J.; Yoo, S.A.; Kim, G.M.; Hwang, D.; Cho, C.S.; You, S.; Kim, W.U. GREM1 Is a Key Regulator of Synoviocyte Hyperplasia and Invasiveness. J. Rheumatol. 2016, 43, 474–485. [Google Scholar] [CrossRef]
- Yi, J.; Jin, Q.; Zhang, B.; Wu, X.; Ge, D. Gremlin-1 Concentrations Are Correlated with the Severity of Knee Osteoarthritis. Med. Sci. Monit. 2016, 22, 4062–4065. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).