The Many Faces of FKBP51


Abstract
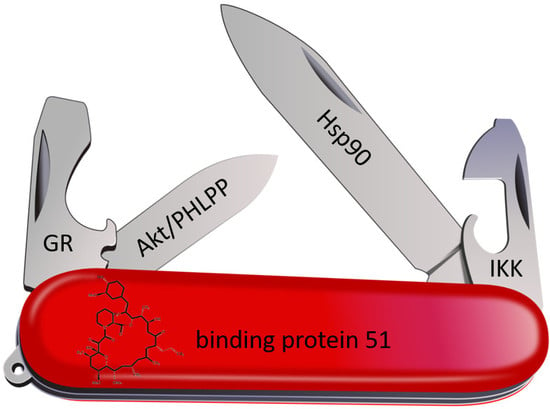
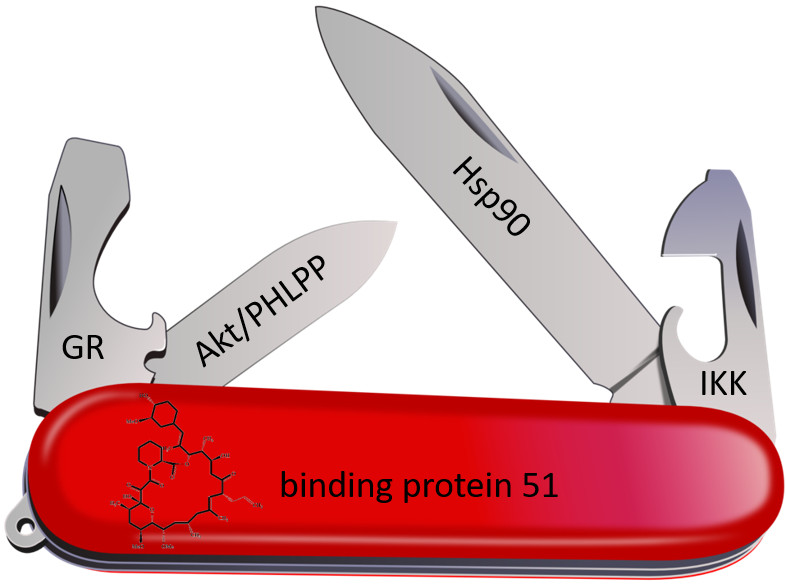
1. Introduction
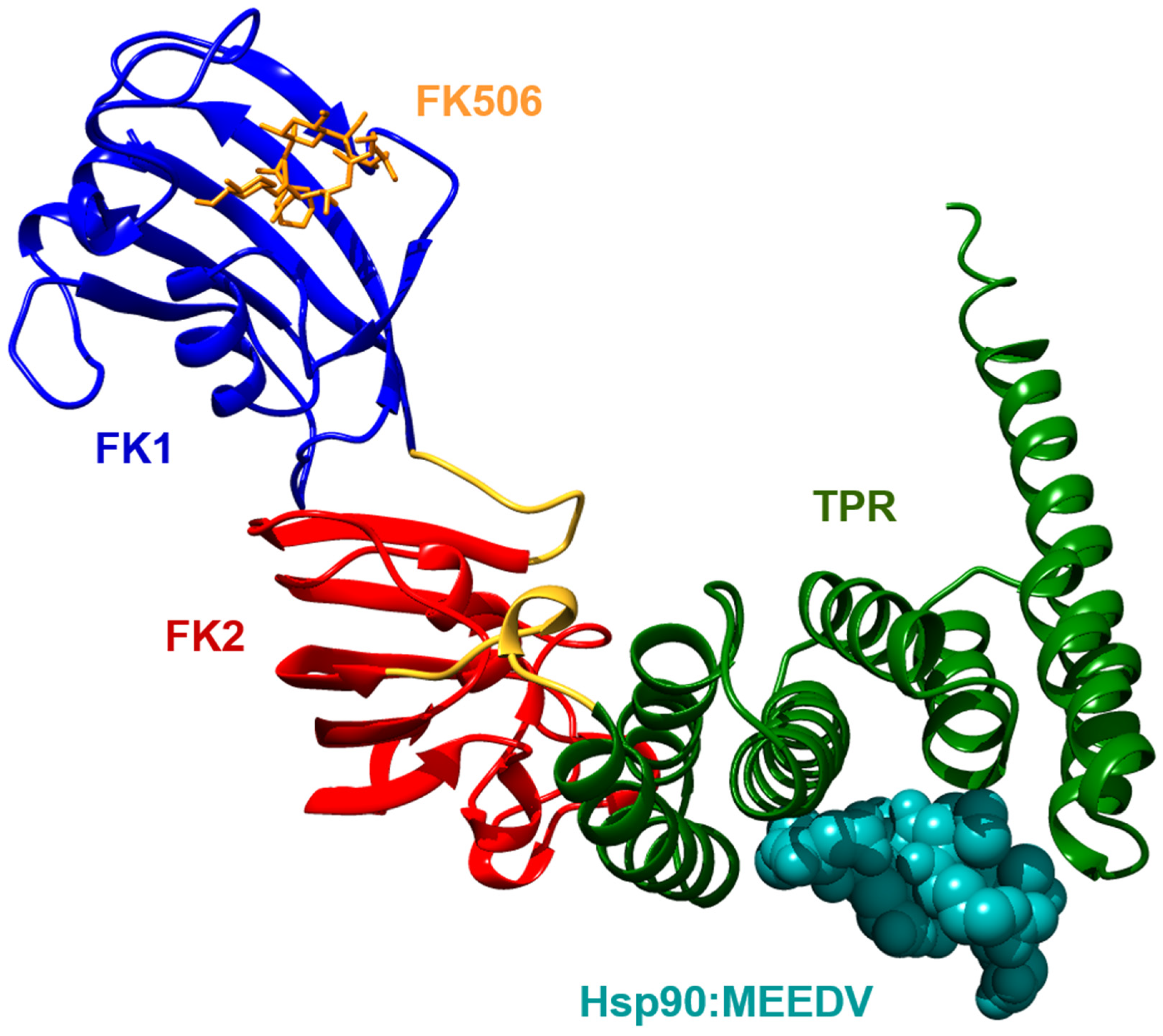
2. Structure of FKBP51
3. Human Genetics of FKBP51
4. Physiological Role of FKBP51 in Mammals
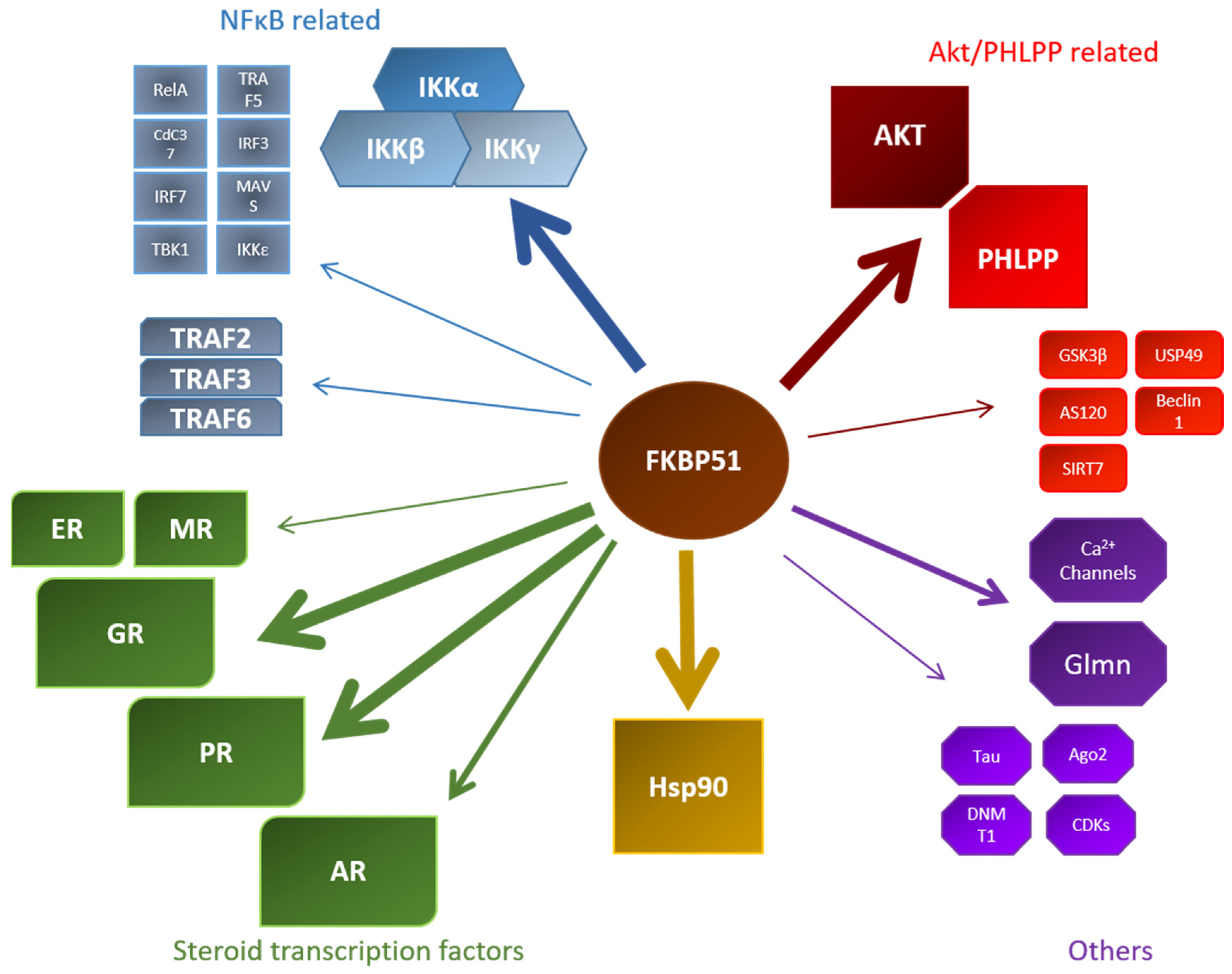
5. FKBP Interaction Partners
5.1. Hsp90
5.2. Steroid Hormone Receptors
5.3. Akt and PHLPP
5.4. Nuclear Factor ‘Kappa-Light-Chain-Enhancer’ of Activated B-Cells (NF-κB)
5.5. Other Interaction Partners
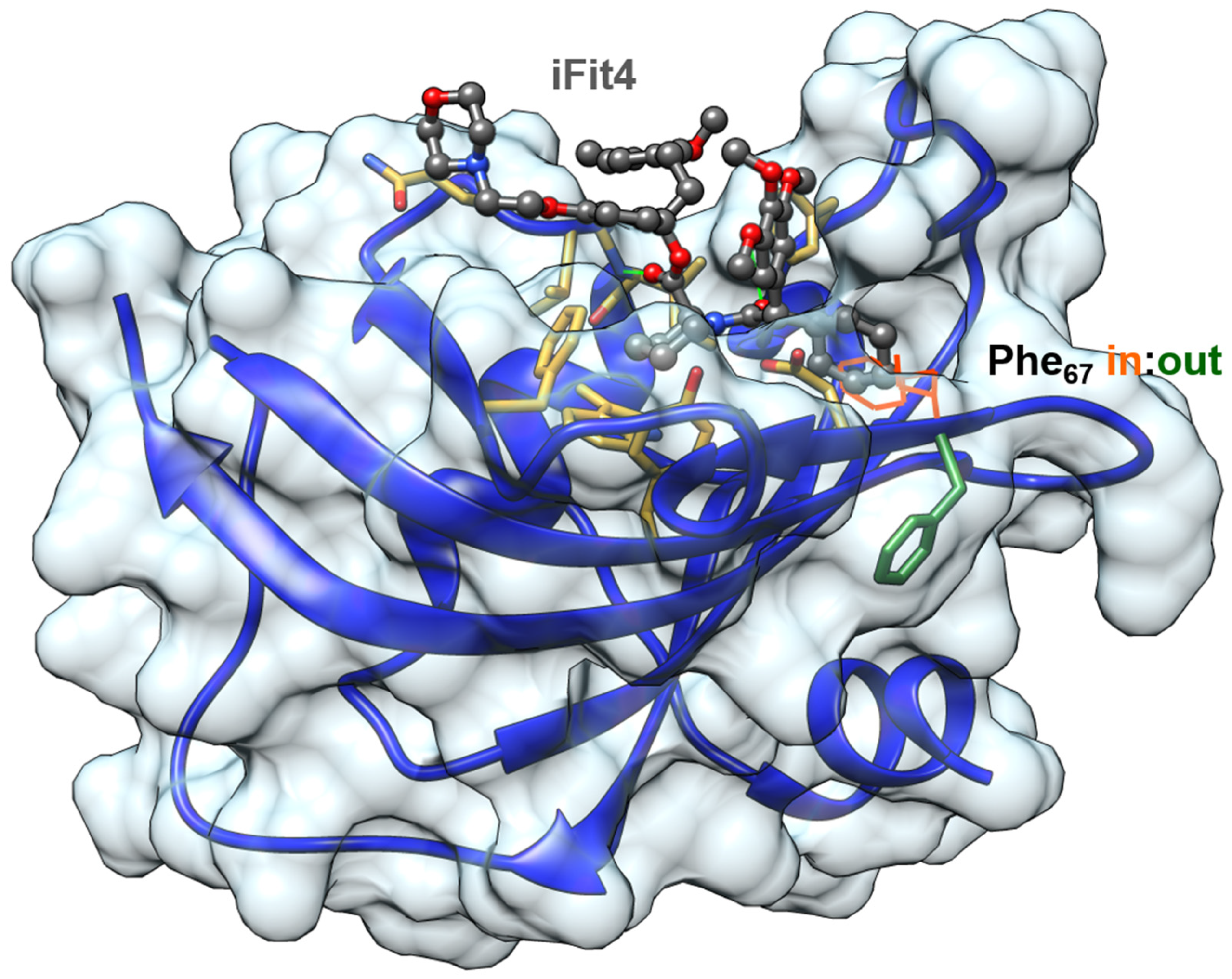
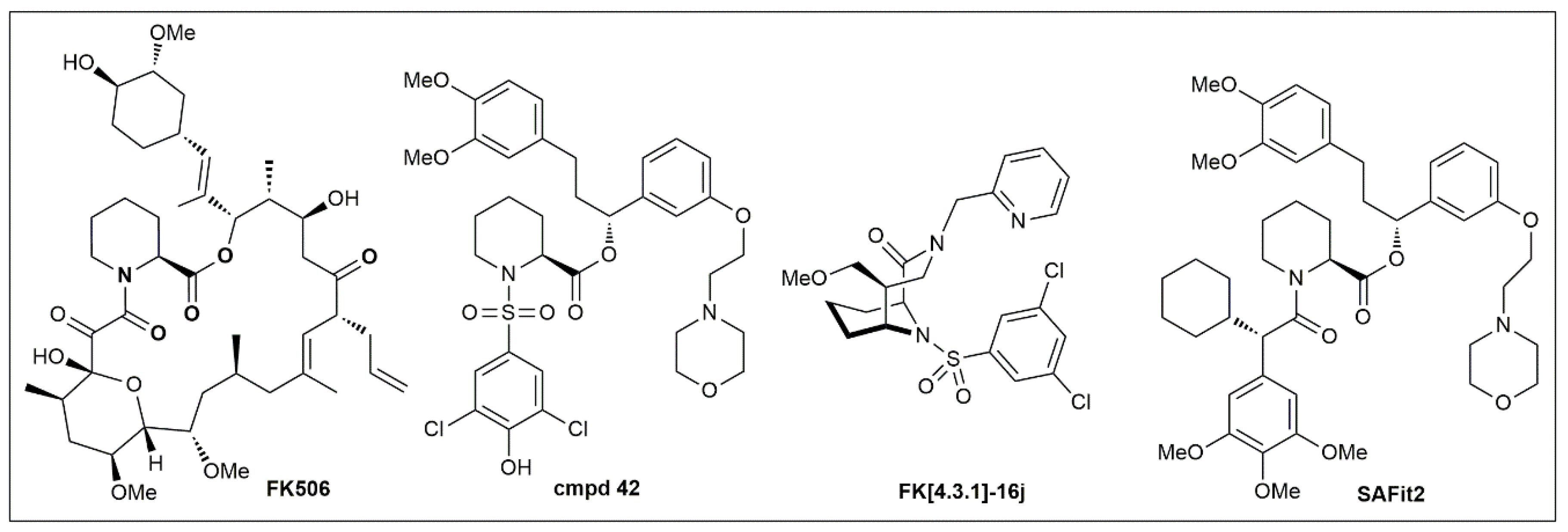
6. Implications for Drug Discovery
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sinars, C.R.; Cheung-Flynn, J.; Rimerman, R.A.; Scammell, J.G.; Smith, D.F.; Clardy, J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Li, P.; Liu, Y.; Lou, Z.; Ding, Y.; Shu, C.; Ye, S.; Bartlam, M.; Shen, B.; Rao, Z. 3D structure of human FK506-binding protein 52: Implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc. Natl. Acad. Sci. USA 2004, 101, 8348–8353. [Google Scholar] [CrossRef] [PubMed]
- Bracher, A.; Kozany, C.; Hahle, A.; Wild, P.; Zacharias, M.; Hausch, F. Crystal structures of the free and ligand-bound FK1-FK2 domain segment of FKBP52 reveal a flexible inter-domain hinge. J. Mol. Biol. 2013, 425, 4134–4144. [Google Scholar] [CrossRef]
- Marz, A.M.; Fabian, A.K.; Kozany, C.; Bracher, A.; Hausch, F. Large FK506-binding proteins shape the pharmacology of rapamycin. Mol. Cell. Biol. 2013, 33, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Kozany, C.; Marz, A.; Kress, C.; Hausch, F. Fluorescent probes to characterise FK506-binding proteins. Chembiochem 2009, 10, 1402–1410. [Google Scholar] [CrossRef]
- Wilson, K.P.; Yamashita, M.M.; Sintchak, M.D.; Rotstein, S.H.; Murcko, M.A.; Boger, J.; Thomson, J.A.; Fitzgibbon, M.J.; Black, J.R.; Navia, M.A. Comparative X-ray structures of the major binding protein for the immunosuppressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta Crystallogr. D Biol. Crystallogr. 1995, 51, 511–521. [Google Scholar] [CrossRef]
- Bracher, A.; Kozany, C.; Thost, A.K.; Hausch, F. Structural characterization of the PPIase domain of FKBP51, a cochaperone of human Hsp90. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 549–559. [Google Scholar] [CrossRef]
- Gopalakrishnan, R.; Kozany, C.; Gaali, S.; Kress, C.; Hoogeland, B.; Bracher, A.; Hausch, F. Evaluation of synthetic FK506 analogues as ligands for the FK506-binding proteins 51 and 52. J. Med. Chem. 2012, 55, 4114–4122. [Google Scholar] [CrossRef]
- Wang, Y.; Kirschner, A.; Fabian, A.K.; Gopalakrishnan, R.; Kress, C.; Hoogeland, B.; Koch, U.; Kozany, C.; Bracher, A.; Hausch, F. Increasing the efficiency of ligands for FK506-binding protein 51 by conformational control. J. Med. Chem. 2013, 56, 3922–3935. [Google Scholar] [CrossRef]
- Pomplun, S.; Sippel, C.; Hahle, A.; Tay, D.; Shima, K.; Klages, A.; Unal, C.M.; Riess, B.; Toh, H.T.; Hansen, G.; et al. Chemogenomic Profiling of Human and Microbial FK506-Binding Proteins. J. Med. Chem. 2018, 61, 3660–3673. [Google Scholar] [CrossRef]
- Gaali, S.; Kirschner, A.; Cuboni, S.; Hartmann, J.; Kozany, C.; Balsevich, G.; Namendorf, C.; Fernandez-Vizarra, P.; Sippel, C.; Zannas, A.S.; et al. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat. Chem. Biol. 2015, 11, 33–37. [Google Scholar] [CrossRef]
- LeMaster, D.M.; Mustafi, S.M.; Brecher, M.; Zhang, J.; Heroux, A.; Li, H.; Hernandez, G. Coupling of Conformational Transitions in the N-terminal Domain of the 51-kDa FK506-binding Protein (FKBP51) Near Its Site of Interaction with the Steroid Receptor Proteins. J. Biol. Chem. 2015, 290, 15746–15757. [Google Scholar] [CrossRef] [PubMed]
- Mustafi, S.M.; LeMaster, D.M.; Hernandez, G. Differential conformational dynamics in the closely homologous FK506-binding domains of FKBP51 and FKBP52. Biochem. J. 2014, 461, 115–123. [Google Scholar] [CrossRef] [PubMed]
- LeMaster, D.M.; Hernandez, G. Conformational Dynamics in FKBP Domains: Relevance to Molecular Signaling and Drug Design. Curr. Mol. Pharmacol. 2015, 9, 5–26. [Google Scholar] [CrossRef] [PubMed]
- Riggs, D.L.; Cox, M.B.; Tardif, H.L.; Hessling, M.; Buchner, J.; Smith, D.F. Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol. Cell. Biol. 2007, 27, 8658–8669. [Google Scholar] [CrossRef]
- Kumar, R.; Moche, M.; Winblad, B.; Pavlov, P.F. Combined X-ray crystallography and computational modeling approach to investigate the Hsp90 C-terminal peptide binding to FKBP51. Sci. Rep. 2017, 7, 14288. [Google Scholar] [CrossRef] [PubMed]
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.U.; Moarefi, I. Structure of TPR domain-peptide complexes: Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 2000, 101, 199–210. [Google Scholar] [CrossRef]
- Yang, J.; Roe, S.M.; Cliff, M.J.; Williams, M.A.; Ladbury, J.E.; Cohen, P.T.; Barford, D. Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. EMBO J. 2005. [Google Scholar] [CrossRef] [PubMed]
- Blundell, K.L.; Pal, M.; Roe, S.M.; Pearl, L.H.; Prodromou, C. The structure of FKBP38 in complex with the MEEVD tetratricopeptide binding-motif of Hsp90. PLoS ONE 2017, 12, e0173543. [Google Scholar] [CrossRef]
- Ebong, I.O.; Beilsten-Edmands, V.; Patel, N.A.; Morgner, N.; Robinson, C.V. The interchange of immunophilins leads to parallel pathways and different intermediates in the assembly of Hsp90 glucocorticoid receptor complexes. Cell Discov. 2016, 2, 16002. [Google Scholar] [CrossRef]
- Assimon, V.A.; Southworth, D.R.; Gestwicki, J.E. Specific Binding of Tetratricopeptide Repeat Proteins to Heat Shock Protein 70 (Hsp70) and Heat Shock Protein 90 (Hsp90) Is Regulated by Affinity and Phosphorylation. Biochemistry 2015, 54, 7120–7131. [Google Scholar] [CrossRef] [PubMed]
- Antunica-Noguerol, M.; Budzinski, M.L.; Druker, J.; Gassen, N.C.; Sokn, M.C.; Senin, S.; Aprile-Garcia, F.; Holsboer, F.; Rein, T.; Liberman, A.C.; et al. The activity of the glucocorticoid receptor is regulated by SUMO conjugation to FKBP51. Cell Death Differ. 2016, 23, 1579–1591. [Google Scholar] [CrossRef] [PubMed]
- Cheung-Flynn, J.; Roberts, P.J.; Riggs, D.L.; Smith, D.F. C-terminal sequences outside the tetratricopeptide repeat domain of FKBP51 and FKBP52 cause differential binding to Hsp90. J. Biol. Chem. 2003, 278, 17388–17394. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Chang, B.J.; Wysoczanski, P.; Lee, C.T.; Perez-Lara, A.; Chakraborty, P.; Hofele, R.V.; Baker, J.D.; Blair, L.J.; Biernat, J.; et al. Structure and pro-toxic mechanism of the human Hsp90/PPIase/Tau complex. Nat. Commun. 2018, 9, 4532. [Google Scholar] [CrossRef] [PubMed]
- Zannas, A.S.; Wiechmann, T.; Gassen, N.C.; Binder, E.B. Gene-Stress-Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacology 2016, 41, 261–274. [Google Scholar] [CrossRef]
- Matosin, N.; Halldorsdottir, T.; Binder, E.B. Understanding the Molecular Mechanisms Underpinning Gene by Environment Interactions in Psychiatric Disorders: The FKBP5 Model. Biol. Psychiatry 2018, 83, 821–830. [Google Scholar] [CrossRef]
- Klengel, T.; Mehta, D.; Anacker, C.; Rex-Haffner, M.; Pruessner, J.C.; Pariante, C.M.; Pace, T.W.; Mercer, K.B.; Mayberg, H.S.; Bradley, B.; et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 2013, 16, 33–41. [Google Scholar] [CrossRef]
- Pereira, M.J.; Palming, J.; Svensson, M.K.; Rizell, M.; Dalenback, J.; Hammar, M.; Fall, T.; Sidibeh, C.O.; Svensson, P.A.; Eriksson, J.W. FKBP5 expression in human adipose tissue increases following dexamethasone exposure and is associated with insulin resistance. Metabolism 2014, 63, 1198–1208. [Google Scholar] [CrossRef]
- Bortsov, A.V.; Smith, J.E.; Diatchenko, L.; Soward, A.C.; Ulirsch, J.C.; Rossi, C.; Swor, R.A.; Hauda, W.E.; Peak, D.A.; Jones, J.S.; et al. Polymorphisms in the glucocorticoid receptor co-chaperone FKBP5 predict persistent musculoskeletal pain after traumatic stress exposure. Pain 2013, 154, 1419–1426. [Google Scholar] [CrossRef]
- Linnstaedt, S.D.; Riker, K.D.; Rueckeis, C.A.; Kutchko, K.M.; Lackey, L.; McCarthy, K.R.; Tsai, Y.H.; Parker, J.S.; Kurz, M.C.; Hendry, P.L.; et al. A Functional riboSNitch in the 3’ Untranslated Region of FKBP5 Alters MicroRNA-320a Binding Efficiency and Mediates Vulnerability to Chronic Post-Traumatic Pain. J. Neurosci. 2018, 38, 8407–8420. [Google Scholar] [CrossRef]
- Menke, A.; Arloth, J.; Putz, B.; Weber, P.; Klengel, T.; Mehta, D.; Gonik, M.; Rex-Haffner, M.; Rubel, J.; Uhr, M.; et al. Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology 2012, 37, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Baida, G.; Bhalla, P.; Yemelyanov, A.; Stechschulte, L.A.; Shou, W.; Readhead, B.; Dudley, J.T.; Sanchez, E.R.; Budunova, I. Deletion of the glucocorticoid receptor chaperone FKBP51 prevents glucocorticoid-induced skin atrophy. Oncotarget 2018, 9, 34772–34783. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Wagner, K.V.; Gaali, S.; Kirschner, A.; Kozany, C.; Ruhter, G.; Dedic, N.; Hausl, A.S.; Hoeijmakers, L.; Westerholz, S.; et al. Pharmacological Inhibition of the Psychiatric Risk Factor FKBP51 Has Anxiolytic Properties. J. Neurosci. 2015, 35, 9007–9016. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.H.; Liebl, C.; Binder, E.B.; Schmidt, M.V.; Muller, M.B. Expression and regulation of the FKBP5 gene in the adult mouse brain. PLoS ONE 2011, 6, e16883. [Google Scholar] [CrossRef] [PubMed]
- Maiaru, M.; Morgan, O.B.; Mao, T.; Breitsamer, M.; Bamber, H.; Pohlmann, M.; Schmidt, M.V.; Winter, G.; Hausch, F.; Geranton, S.M. The stress regulator FKBP51: A novel and promising druggable target for the treatment of persistent pain states across sexes. Pain 2018, 159, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, J.C., 3rd; Dharia, S.; Blair, L.J.; Brady, S.; Johnson, A.G.; Peters, M.; Cheung-Flynn, J.; Cox, M.B.; de Erausquin, G.; Weeber, E.J.; et al. A new anti-depressive strategy for the elderly: Ablation of FKBP5/FKBP51. PLoS ONE 2011, 6, e24840. [Google Scholar] [CrossRef]
- Touma, C.; Gassen, N.C.; Herrmann, L.; Cheung-Flynn, J.; Bull, D.R.; Ionescu, I.A.; Heinzmann, J.M.; Knapman, A.; Siebertz, A.; Depping, A.M.; et al. FK506 binding protein 5 shapes stress responsiveness: Modulation of neuroendocrine reactivity and coping behavior. Biol. Psychiatry 2011, 70, 928–936. [Google Scholar] [CrossRef]
- Hartmann, J.; Wagner, K.V.; Liebl, C.; Scharf, S.H.; Wang, X.D.; Wolf, M.; Hausch, F.; Rein, T.; Schmidt, U.; Touma, C.; et al. The involvement of FK506-binding protein 51 (FKBP5) in the behavioral and neuroendocrine effects of chronic social defeat stress. Neuropharmacology 2012, 62, 332–339. [Google Scholar] [CrossRef]
- Albu, S.; Romanowski, C.P.; Letizia Curzi, M.; Jakubcakova, V.; Flachskamm, C.; Gassen, N.C.; Hartmann, J.; Schmidt, M.V.; Schmidt, U.; Rein, T.; et al. Deficiency of FK506-binding protein (FKBP) 51 alters sleep architecture and recovery sleep responses to stress in mice. J. Sleep Res. 2014, 23, 176–185. [Google Scholar] [CrossRef]
- Stechschulte, L.A.; Qiu, B.; Warrier, M.; Hinds, T.D., Jr.; Zhang, M.; Gu, H.; Xu, Y.; Khuder, S.S.; Russo, L.; Najjar, S.M.; et al. FKBP51 Null Mice Are Resistant to Diet-Induced Obesity and the PPARγ Agonist Rosiglitazone. Endocrinology 2016, 157, 3888–3900. [Google Scholar] [CrossRef]
- Balsevich, G.; Hausl, A.S.; Meyer, C.W.; Karamihalev, S.; Feng, X.; Pohlmann, M.L.; Dournes, C.; Uribe-Marino, A.; Santarelli, S.; Labermaier, C.; et al. Stress-responsive FKBP51 regulates AKT2-AS160 signaling and metabolic function. Nat. Commun. 2017, 8, 1725. [Google Scholar] [CrossRef] [PubMed]
- Maiaru, M.; Tochiki, K.K.; Cox, M.B.; Annan, L.V.; Bell, C.G.; Feng, X.; Hausch, F.; Geranton, S.M. The stress regulator FKBP51 drives chronic pain by modul[ating spinal glucocorticoid signaling. Sci. Transl. Med. 2016, 8, 325ra319. [Google Scholar] [CrossRef] [PubMed]
- Kastle, M.; Kistler, B.; Lamla, T.; Bretschneider, T.; Lamb, D.; Nicklin, P.; Wyatt, D. FKBP51 modulates steroid sensitivity and NFκB signalling: A novel anti-inflammatory drug target. Eur. J. Immunol. 2018, 48, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, J.J.; O’Leary, J.C., 3rd; Blair, L.J.; Klengel, T.; Nordhues, B.A.; Fontaine, S.N.; Binder, E.B.; Dickey, C.A. Age-associated epigenetic upregulation of the FKBP5 gene selectively impairs stress resiliency. PLoS ONE 2014, 9, e107241. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, M.L.; Hausl, A.S.; Harbich, D.; Balsevich, G.; Engelhardt, C.; Feng, X.; Breitsamer, M.; Hausch, F.; Winter, G.; Schmidt, M.V. Pharmacological Modulation of the Psychiatric Risk Factor FKBP51 Alters Efficiency of Common Antidepressant Drugs. Front. Behav. Neurosci. 2018, 12, 262. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.F.; Faber, L.E.; Toft, D.O. Purification of unactivated progesterone receptor and identification of novel receptor-associated proteins. J. Biol. Chem. 1990, 265, 3996–4003. [Google Scholar]
- Smith, D.F.; Albers, M.W.; Schreiber, S.L.; Leach, K.L.; Deibel, M.R., Jr. FKBP54, a novel FK506-binding protein in avian progesterone receptor complexes and HeLa extracts. J. Biol. Chem. 1993, 268, 24270–24273. [Google Scholar]
- Chen, S.; Sullivan, W.P.; Toft, D.O.; Smith, D.F. Differential interactions of p23 and the TPR-containing proteins Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaperones 1998, 3, 118–129. [Google Scholar] [CrossRef]
- Smith, D.F.; Whitesell, L.; Nair, S.C.; Chen, S.; Prapapanich, V.; Rimerman, R.A. Progesterone receptor structure and function altered by geldanamycin, an hsp90-binding agent. Mol. Cell. Biol. 1995, 15, 6804–6812. [Google Scholar] [CrossRef]
- Mayer, M.P.; Le Breton, L. Hsp90: Breaking the symmetry. Mol. Cell 2015, 58, 8–20. [Google Scholar] [CrossRef]
- Rohl, A.; Rohrberg, J.; Buchner, J. The chaperone Hsp90: Changing partners for demanding clients. Trends Biochem. Sci. 2013, 38, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Schülke, J.P.; Wochnik, G.M.; Lang-Rollin, I.; Gassen, N.C.; Knapp, R.T.; Berning, B.; Yassouridis, A.; Rein, T. Differential impact of tetratricopeptide repeat proteins on the steroid hormone receptors. PLoS ONE 2010, 5, e11717. [Google Scholar] [CrossRef] [PubMed]
- Pirkl, F.; Buchner, J. Functional analysis of the Hsp90-associated human peptidyl prolyl cis/trans isomerases FKBP51, FKBP52 and Cyp40. J. Mol. Biol. 2001, 308, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.Y.; Larsen, B.; Choi, H.; Berger, B.; Gingras, A.C.; Lindquist, S. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Richter, K.; Buchner, J. Mixed Hsp90-cochaperone complexes are important for the progression of the reaction cycle. Nat. Struct. Mol. Biol. 2011, 18, 61–66. [Google Scholar] [CrossRef]
- Echeverria, P.C.; Picard, D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim. Biophys. Acta 2010, 1803, 641–649. [Google Scholar] [CrossRef]
- Barent, R.L.; Nair, S.C.; Carr, D.C.; Ruan, Y.; Rimerman, R.A.; Fulton, J.; Zhang, Y.; Smith, D.F. Analysis of FKBP51/FKBP52 chimeras and mutants for Hsp90 binding and association with progesterone receptor complexes. Mol. Endocrinol. 1998, 12, 342–354. [Google Scholar] [CrossRef]
- Baughman, G.; Wiederrecht, G.J.; Chang, F.; Martin, M.M.; Bourgeois, S. Tissue distribution and abundance of human FKBP51, and FK506-binding protein that can mediate calcineurin inhibition. Biochem. Biophys. Res. Commun. 1997, 232, 437–443. [Google Scholar] [CrossRef]
- Reynolds, P.D.; Roveda, K.P.; Tucker, J.A.; Moore, C.M.; Valentine, D.L.; Scammell, J.G. Glucocorticoid-resistant B-lymphoblast cell line derived from the Bolivian squirrel monkey (Saimiri boliviensis boliviensis). Lab. Anim. Sci. 1998, 48, 364–370. [Google Scholar]
- Wan, Y.; Nordeen, S.K. Identification of genes differentially regulated by glucocorticoids and progestins using a Cre/loxP-mediated retroviral promoter-trapping strategy. J. Mol. Endocrinol. 2002, 28, 177–192. [Google Scholar] [CrossRef][Green Version]
- Vermeer, H.; Hendriks-Stegeman, B.I.; van der Burg, B.; van Buul-Offers, S.C.; Jansen, M. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: A potential marker for glucocorticoid sensitivity, potency, and bioavailability. J. Clin. Endocrinol. Metab. 2003, 88, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Kester, H.A.; van der Leede, B.M.; van der Saag, P.T.; van der Burg, B. Novel progesterone target genes identified by an improved differential display technique suggest that progestin-induced growth inhibition of breast cancer cells coincides with enhancement of differentiation. J. Biol. Chem. 1997, 272, 16637–16643. [Google Scholar] [CrossRef] [PubMed]
- Hubler, T.R.; Denny, W.B.; Valentine, D.L.; Cheung-Flynn, J.; Smith, D.F.; Scammell, J.G. The FK506-binding immunophilin FKBP51 is transcriptionally regulated by progestin and attenuates progestin responsiveness. Endocrinology 2003, 144, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Hubler, T.R.; Scammell, J.G. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones 2004, 9, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Amler, L.C.; Agus, D.B.; LeDuc, C.; Sapinoso, M.L.; Fox, W.D.; Kern, S.; Lee, D.; Wang, V.; Leysens, M.; Higgins, B.; et al. Dysregulated expression of androgen-responsive and nonresponsive genes in the androgen-independent prostate cancer xenograft model CWR22-R1. Cancer Res. 2000, 60, 6134–6141. [Google Scholar] [PubMed]
- Mousses, S.; Wagner, U.; Chen, Y.; Kim, J.W.; Bubendorf, L.; Bittner, M.; Pretlow, T.; Elkahloun, A.G.; Trepel, J.B.; Kallioniemi, O.P. Failure of hormone therapy in prostate cancer involves systematic restoration of androgen responsive genes and activation of rapamycin sensitive signaling. Oncogene 2001, 20, 6718–6723. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, J.S.; Young, C.Y. Silymarin inhibits function of the androgen receptor by reducing nuclear localization of the receptor in the human prostate cancer cell line LNCaP. Carcinogenesis 2001, 22, 1399–1403. [Google Scholar] [CrossRef]
- Paakinaho, V.; Makkonen, H.; Jaaskelainen, T.; Palvimo, J.J. Glucocorticoid receptor activates poised FKBP51 locus through long-distance interactions. Mol. Endocrinol. 2010, 24, 511–525. [Google Scholar] [CrossRef]
- Riggs, D.L.; Roberts, P.J.; Chirillo, S.C.; Cheung-Flynn, J.; Prapapanich, V.; Ratajczak, T.; Gaber, R.; Picard, D.; Smith, D.F. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003, 22, 1158–1167. [Google Scholar] [CrossRef]
- Wochnik, G.M.; Ruegg, J.; Abel, G.A.; Schmidt, U.; Holsboer, F.; Rein, T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem. 2005, 280, 4609–4616. [Google Scholar] [CrossRef] [PubMed]
- Stechschulte, L.A.; Hinds, T.D., Jr.; Khuder, S.S.; Shou, W.; Najjar, S.M.; Sanchez, E.R. FKBP51 controls cellular adipogenesis through p38 kinase-mediated phosphorylation of GRα and PPARγ. Mol. Endocrinol. 2014, 28, 1265–1275. [Google Scholar] [CrossRef]
- Sabbagh, J.J.; Cordova, R.A.; Zheng, D.; Criado-Marrero, M.; Lemus, A.; Li, P.; Baker, J.D.; Nordhues, B.A.; Darling, A.L.; Martinez-Licha, C.; et al. Targeting the FKBP51/GR/Hsp90 Complex to Identify Functionally Relevant Treatments for Depression and PTSD. ACS Chem. Biol. 2018, 13, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Yang, C.S.; Gioeli, D.; Frierson, H.; Toft, D.O.; Paschal, B.M. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Mol. Cell. Biol. 2010, 30, 1243–1253. [Google Scholar] [CrossRef]
- Periyasamy, S.; Hinds, T., Jr.; Shemshedini, L.; Shou, W.; Sanchez, E.R. FKBP51 and Cyp40 are positive regulators of androgen-dependent prostate cancer cell growth and the targets of FK506 and cyclosporin A. Oncogene 2010, 29, 1691–1701. [Google Scholar] [CrossRef]
- Caratti, G.; Matthews, L.; Poolman, T.; Kershaw, S.; Baxter, M.; Ray, D. Glucocorticoid receptor function in health and disease. Clin. Endocrinol. 2015, 83, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Li, L.; Fridley, B.L.; Jenkins, G.D.; Kalari, K.R.; Lingle, W.; Petersen, G.; Lou, Z.; Wang, L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009, 16, 259–266. [Google Scholar] [CrossRef]
- Luo, K.; Li, Y.; Yin, Y.; Li, L.; Wu, C.; Chen, Y.; Nowsheen, S.; Hu, Q.; Zhang, L.; Lou, Z.; et al. USP49 negatively regulates tumorigenesis and chemoresistance through FKBP51-AKT signaling. EMBO J. 2017, 36, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Hartmann, J.; Zschocke, J.; Stepan, J.; Hafner, K.; Zellner, A.; Kirmeier, T.; Kollmannsberger, L.; Wagner, K.V.; Dedic, N.; et al. Association of FKBP51 with priming of autophagy pathways and mediation of antidepressant treatment response: Evidence in cells, mice, and humans. PLoS Med. 2014, 11, e1001755. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Qin, B.; Wu, F.; Qin, S.; Nowsheen, S.; Shan, S.; Zayas, J.; Pei, H.; Lou, Z.; Wang, L. Regulation of Serine-Threonine Kinase Akt Activation by NAD+-Dependent Deacetylase SIRT7. Cell Rep. 2017, 18, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Fabian, A.K.; Marz, A.; Neimanis, S.; Biondi, R.M.; Kozany, C.; Hausch, F. InterAKTions with FKBPs–mutational and pharmacological exploration. PLoS ONE 2013, 8, e57508. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Hartmann, J.; Zannas, A.S.; Kretzschmar, A.; Zschocke, J.; Maccarrone, G.; Hafner, K.; Zellner, A.; Kollmannsberger, L.K.; Wagner, K.V.; et al. FKBP51 inhibits GSK3β and augments the effects of distinct psychotropic medications. Mol. Psychiatry 2016, 21, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Cazacu, S.; Xiang, C.; Zenklusen, J.C.; Fine, H.A.; Berens, M.; Armstrong, B.; Brodie, C.; Mikkelsen, T. FK506 binding protein mediates glioma cell growth and sensitivity to rapamycin treatment by regulating NF-κB signaling pathway. Neoplasia 2008, 10, 235–243. [Google Scholar] [CrossRef]
- Romano, S.; D’Angelillo, A.; Pacelli, R.; Staibano, S.; De Luna, E.; Bisogni, R.; Eskelinen, E.L.; Mascolo, M.; Cali, G.; Arra, C.; et al. Role of FK506-binding protein 51 in the control of apoptosis of irradiated melanoma cells. Cell Death Differ. 2010, 17, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Xiao, Y.; Nakaya, M.; D’Angelillo, A.; Chang, M.; Jin, J.; Hausch, F.; Masullo, M.; Feng, X.; Romano, M.F.; et al. FKBP51 employs both scaffold and isomerase functions to promote NF-κB activation in melanoma. Nucleic Acids Res. 2015, 43, 6983–6993. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Bhardwaj, A.; Arora, S.; Tyagi, N.; Singh, A.P.; Carter, J.E.; Scammell, J.G.; Fodstad, O.; Singh, S. Interleukin-8 is a key mediator of FKBP51-induced melanoma growth, angiogenesis and metastasis. Br. J. Cancer 2015, 112, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.F.; Avellino, R.; Petrella, A.; Bisogni, R.; Romano, S.; Venuta, S. Rapamycin inhibits doxorubicin-induced NF-κB/Rel nuclear activity and enhances the apoptosis of melanoma cells. Eur. J. Cancer 2004, 40, 2829–2836. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Akiyama, T.; Shiraishi, T.; Qin, J.; Konno, H.; Akiyama, N.; Shinzawa, M.; Miyauchi, M.; Takizawa, N.; Yanai, H.; Ohashi, H.; et al. Mitochondria-nucleus shuttling FK506-binding protein 51 interacts with TRAF proteins and facilitates the RIG-I-like receptor-mediated expression of type I IFN. PLoS ONE 2014, 9, e95992. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, T.; Bauch, A.; Ruffner, H.; Angrand, P.O.; Bergamini, G.; Croughton, K.; Cruciat, C.; Eberhard, D.; Gagneur, J.; Ghidelli, S.; et al. A physical and functional map of the human TNF-α/NF-κ B signal transduction pathway. Nat. Cell Biol. 2004, 6, 97–105. [Google Scholar] [CrossRef]
- Erlejman, A.G.; De Leo, S.A.; Mazaira, G.I.; Molinari, A.M.; Camisay, M.F.; Fontana, V.; Cox, M.B.; Piwien-Pilipuk, G.; Galigniana, M.D. NF-κB transcriptional activity is modulated by FK506-binding proteins FKBP51 and FKBP52: A role for peptidyl-prolyl isomerase activity. J. Biol. Chem. 2014, 289, 26263–26276. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Broemer, M.; Arslan, S.C.; Otto, A.; Mueller, E.C.; Dettmer, R.; Scheidereit, C. Signal responsiveness of IκB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 2007, 282, 32311–32319. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Fries, G.R.; Zannas, A.S.; Hartmann, J.; Zschocke, J.; Hafner, K.; Carrillo-Roa, T.; Steinbacher, J.; Preissinger, S.N.; Hoeijmakers, L.; et al. Chaperoning epigenetics: FKBP51 decreases the activity of DNMT1 and mediates epigenetic effects of the antidepressant paroxetine. Sci. Signal. 2015, 8, ra119. [Google Scholar] [CrossRef] [PubMed]
- Jirawatnotai, S.; Sharma, S.; Michowski, W.; Suktitipat, B.; Geng, Y.; Quackenbush, J.; Elias, J.E.; Gygi, S.P.; Wang, Y.E.; Sicinski, P. The cyclin D1-CDK4 oncogenic interactome enables identification of potential novel oncogenes and clinical prognosis. Cell Cycle 2014, 13, 2889–2900. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Koren, J., 3rd; Borysov, S.I.; Schmid, A.B.; Abisambra, J.F.; Blair, L.J.; Johnson, A.G.; Jones, J.R.; Shults, C.L.; O’Leary, J.C., 3rd; et al. The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J. Neurosci. 2010, 30, 591–599. [Google Scholar] [CrossRef]
- Quinta, H.R.; Maschi, D.; Gomez-Sanchez, C.; Piwien-Pilipuk, G.; Galigniana, M.D. Subcellular rearrangement of hsp90-binding immunophilins accompanies neuronal differentiation and neurite outgrowth. J. Neurochem. 2010, 115, 716–734. [Google Scholar] [CrossRef]
- Gaali, S.; Feng, X.; Hahle, A.; Sippel, C.; Bracher, A.; Hausch, F. Rapid, Structure-Based Exploration of Pipecolic Acid Amides as Novel Selective Antagonists of the FK506-Binding Protein 51. J. Med. Chem. 2016, 59, 2410–2422. [Google Scholar] [CrossRef]
- Pomplun, S.; Wang, Y.; Kirschner, A.; Kozany, C.; Bracher, A.; Hausch, F. Rational design and asymmetric synthesis of potent and neurotrophic ligands for FK506-binding proteins (FKBPs). Angew. Chem. Int. Ed. 2015, 54, 345–348. [Google Scholar] [CrossRef]
- Hamilton, C.L.; Abney, K.A.; Vasauskas, A.A.; Alexeyev, M.; Li, N.; Honkanen, R.E.; Scammell, J.G.; Cioffi, D.L. Serine/threonine phosphatase 5 (PP5C/PPP5C) regulates the ISOC channel through a PP5C-FKBP51 axis. Pulm. Circ. 2018, 8, 2045893217753156. [Google Scholar] [CrossRef] [PubMed]
- Kadeba, P.I.; Vasauskas, A.A.; Chen, H.; Wu, S.; Scammell, J.G.; Cioffi, D.L. Regulation of store-operated calcium entry by FK506-binding immunophilins. Cell Calcium 2013, 53, 275–285. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sinkins, W.G.; Goel, M.; Estacion, M.; Schilling, W.P. Association of immunophilins with mammalian TRPC channels. J. Biol. Chem. 2004, 279, 34521–34529. [Google Scholar] [CrossRef] [PubMed]
- Lopez, E.; Berna-Erro, A.; Hernandez-Cruz, J.M.; Salido, G.M.; Redondo, P.C.; Rosado, J.A. Immunophilins are involved in the altered platelet aggregation observed in patients with type 2 diabetes mellitus. Curr. Med. Chem. 2013, 20, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Lopez, E.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. FKBP52 is involved in the regulation of SOCE channels in the human platelets and MEG 01 cells. Biochim. Biophys. Acta 2013, 1833, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Lopez, E.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. FKBP25 and FKBP38 regulate non-capacitative calcium entry through TRPC6. Biochim. Biophys. Acta 2015, 1853, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Chambraud, B.; Radanyi, C.; Camonis, J.H.; Shazand, K.; Rajkowski, K.; Baulieu, E.E. FAP48, a new protein that forms specific complexes with both immunophilins FKBP59 and FKBP12. Prevention by the immunosuppressant drugs FK506 and rapamycin. J. Biol. Chem. 1996, 271, 32923–32929. [Google Scholar] [CrossRef][Green Version]
- Brouillard, P.; Boon, L.M.; Mulliken, J.B.; Enjolras, O.; Ghassibe, M.; Warman, M.L.; Tan, O.T.; Olsen, B.R.; Vikkula, M. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am. J. Hum. Genet. 2002, 70, 866–874. [Google Scholar] [CrossRef]
- Brouillard, P.; Boon, L.M.; Revencu, N.; Berg, J.; Dompmartin, A.; Dubois, J.; Garzon, M.; Holden, S.; Kangesu, L.; Labreze, C.; et al. Genotypes and phenotypes of 162 families with a glomulin mutation. Mol. Syndromol. 2013, 4, 157–164. [Google Scholar] [CrossRef]
- Brouillard, P.; Ghassibe, M.; Penington, A.; Boon, L.M.; Dompmartin, A.; Temple, I.K.; Cordisco, M.; Adams, D.; Piette, F.; Harper, J.I.; et al. Four common glomulin mutations cause two thirds of glomuvenous malformations (“familial glomangiomas”): Evidence for a founder effect. J. Med. Genet. 2005, 42, e13. [Google Scholar] [CrossRef]
- Neye, H. Mutation of FKBP associated protein 48 (FAP48) at proline 219 disrupts the interaction with FKBP12 and FKBP52. Regul. Pept. 2001, 97, 147–152. [Google Scholar] [CrossRef]
- Martinez, N.J.; Chang, H.M.; Borrajo Jde, R.; Gregory, R.I. The co-chaperones FKBP4/5 control Argonaute2 expression and facilitate RISC assembly. RNA 2013, 19, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, R.; Kozany, C.; Wang, Y.; Schneider, S.; Hoogeland, B.; Bracher, A.; Hausch, F. Exploration of pipecolate sulfonamides as binders of the FK506-binding proteins 51 and 52. J. Med. Chem. 2012, 55, 4123–4131. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, M.; Sippel, C.; Bracher, A.; Hausch, F. Stereoselective construction of the 5-hydroxy diazabicyclo[4.3.1]decane-2-one scaffold, a privileged motif for FK506-binding proteins. Org. Lett. 2014, 16, 5254–5257. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Sippel, C.; Bracher, A.; Hausch, F. Structure-Affinity Relationship Analysis of Selective FKBP51 Ligands. J. Med. Chem. 2015, 58, 7796–7806. [Google Scholar] [CrossRef]
- Sidibeh, C.O.; Pereira, M.J.; Abalo, X.M.; Gretha, J.B.; Skrtic, S.; Lundkvist, P.; Katsogiannos, P.; Hausch, F.; Castillejo-Lopez, C.; Eriksson, J.W. FKBP5 expression in human adipose tissue: Potential role in glucose and lipid metabolism, adipogenesis and type 2 diabetes. Endocrine 2018, 62, 116–128. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction Partner | FK1 Domain Dependency | TPR Domain Dependency | Remarks |
|---|---|---|---|
| Hsp90 | a [49] | d, e [49] | FKBP52 competitive [53] |
| GR | via Hsp90 [58] | SUMOylation dependent [22] FKBP52 competitive [70] | |
| PR | b [58] | d [58] | |
| AR | c [74,75] | [74] | |
| Akt | a [77] | e [81] | Deubiquitination [78] and acetylation dependent [80] |
| PHLPP | a [77] | d [77] | |
| AS160 | c [41] | ||
| GSK3β | a [82] | d [82] | |
| DNMT1 | b [95] | FKBP52 competitive [95] | |
| SIRT7 | [80] | ||
| IKKα | b,c [83,85] | e [85] | |
| IKKβ | b [85] | e [85] | |
| IKKγ | b [85] | e [85] | |
| TRAF2 | b [85] | d,e [85] | |
| Tau | b [97] | [97] | |
| Ago | c [112] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hähle, A.; Merz, S.; Meyners, C.; Hausch, F. The Many Faces of FKBP51. Biomolecules 2019, 9, 35. https://doi.org/10.3390/biom9010035
Hähle A, Merz S, Meyners C, Hausch F. The Many Faces of FKBP51. Biomolecules. 2019; 9(1):35. https://doi.org/10.3390/biom9010035
Chicago/Turabian StyleHähle, Andreas, Stephanie Merz, Christian Meyners, and Felix Hausch. 2019. "The Many Faces of FKBP51" Biomolecules 9, no. 1: 35. https://doi.org/10.3390/biom9010035
APA StyleHähle, A., Merz, S., Meyners, C., & Hausch, F. (2019). The Many Faces of FKBP51. Biomolecules, 9(1), 35. https://doi.org/10.3390/biom9010035