Nicotinamide Mononucleotide: Exploration of Diverse Therapeutic Applications of a Potential Molecule

 ,
,
Abstract
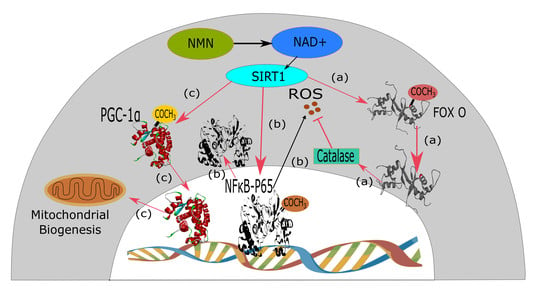
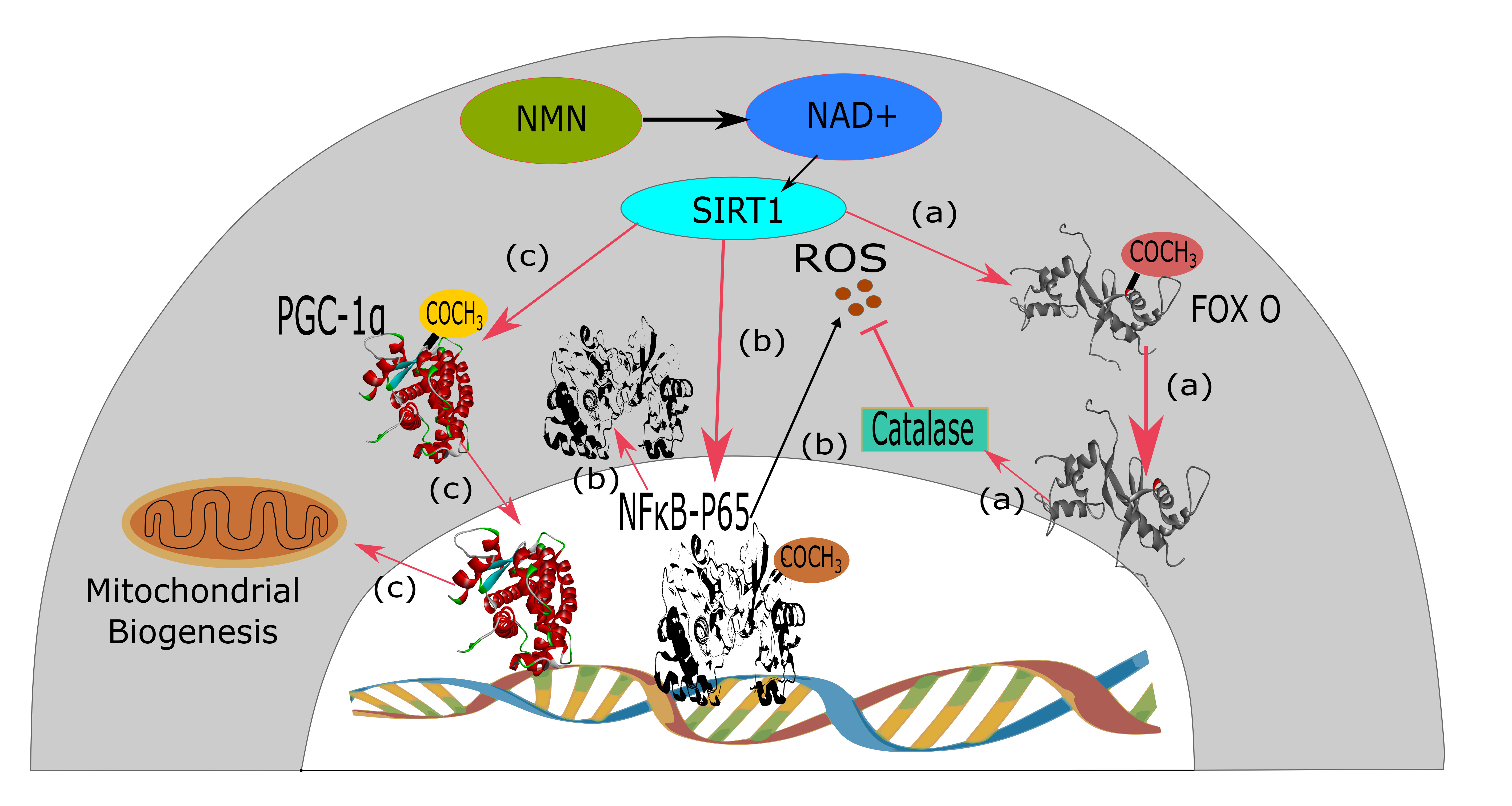{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Biosynthesis and Mechanism of Absorption
3. Pharmacological Activities
3.1. Ischemia-Reperfusion Injury
3.2. Neurological Disorders: Alzheimer’s Disease and Intracerebral Hemorrhage
3.3. Diabetes
3.4. Obesity and Its Related Complications
3.5. Ageing
4. Nicotinamide Mononucleotide or Nicotinamide Riboside: Which One is Better
5. Future Prospects
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef]
- Pubchem Nicotinamide Mononucleotide|C11H15N2O8P—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/nicotinamide_mononucleotide (accessed on 1 January 2019).
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Sorci, L.; Zhang, X.; Brautigam, C.A.; Li, X.; Raffaelli, N.; Magni, G.; Grishin, N.V.; Osterman, A.L.; Zhang, H. Bifunctional NMN Adenylyltransferase/ADP-Ribose Pyrophosphatase: Structure and Function in Bacterial NAD Metabolism. Structure 2008, 16, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular Compartmentation and Differential Catalytic Properties of the Three Human Nicotinamide Mononucleotide Adenylyltransferase Isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Byun, J.; Zhai, P.; Ikeda, Y.; Oka, S.; Sadoshima, J. Nicotinamide Mononucleotide, an Intermediate of NAD+ Synthesis, Protects the Heart from Ischemia and Reperfusion. PLoS ONE 2014, 9, e98972. [Google Scholar] [CrossRef] [PubMed]
- Long, A.N.; Owens, K.; Schlappal, A.E.; Kristian, T.; Fishman, P.S.; Schuh, R.A. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015, 15, 19. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through a SIRT3-dependent mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Schultz, M.B.; Sinclair, D.A. Why NAD+ Declines during Aging: It’s Destroyed. Cell Metab. 2016, 23, 965–966. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Gan, L.; Swanson, R.A. Poly(ADP-ribose) polymerase-1-induced NAD+ depletion promotes nuclear factor-κB transcriptional activity by preventing p65 de-acetylation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 1985–1991. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.; Auwerx, J. NAD+ metabolism and the control of energy homeostasis—A balancing act between mitochondria and the nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A. NAD+ and vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Douek, I.F.; Moore, W.P.T.; Gillmor, H.A.; McLean, A.E.; Bingley, P.J.; Gale, E.A.; European Nicotinamide Diabetes Intervention Trial Group. Safety of high-dose nicotinamide: A review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Mori, N.; Shibata, K. β-Nicotinamide Mononucleotide, an Anti-Aging Candidate Compound, Is Retained in the Body for Longer than Nicotinamide in Rats. J. Nutr. Sci. Vitaminol. 2016, 62, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Pieper, J.A. Overview of niacin formulations: Differences in pharmacokinetics, efficacy, and safety. Am. J. Health Syst. Pharm. 2003, 60, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Racette, F.G.; Bogan, K.L.; McClure, J.M.; Smith, J.S.; Brenner, C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 2007, 129, 473–484. [Google Scholar] [CrossRef]
- Wang, P.; Miao, C.-Y. NAMPT as a Therapeutic Target against Stroke. Trends Pharmacol. Sci. 2015, 36, 891–905. [Google Scholar] [CrossRef]
- Venter, G.; Oerlemans, F.T.J.J.; Willemse, M.; Wijers, M.; Fransen, J.A.M.; Wieringa, B. NAMPT-Mediated Salvage Synthesis of NAD+ Controls Morphofunctional Changes of Macrophages. PLoS ONE 2014, 9, e97378. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD+ Precursor Vitamins in Human Nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Eppig, J.T.; Bult, C.J.; Kadin, J.A.; Richardson, J.E.; Blake, J.A.; Anagnostopoulos, A.; Baldarelli, R.M.; Baya, M.; Beal, J.S.; Bello, S.M.; et al. The Mouse Genome Database (MGD): From genes to mice—A community resource for mouse biology. Nucleic Acids Res. 2005, 33, D471–D475. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marinescu, G.C.; Popescu, R.-G.; Dinischiotu, A. Size Exclusion Chromatography Method for Purification of Nicotinamide Mononucleotide (NMN) from Bacterial Cells. Sci. Rep. 2018, 8, 4433. [Google Scholar] [CrossRef] [PubMed]
- Sorci, L.; Martynowski, D.; Rodionov, D.A.; Eyobo, Y.; Zogaj, X.; Klose, K.E.; Nikolaev, E.V.; Magni, G.; Zhang, H.; Osterman, A.L. Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. Proc. Natl. Acad. Sci. USA 2009, 106, 3083–3088. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, S.Y.; Kurnasov, O.V.; Shatalin, K.; Polanuyer, B.; Sloutsky, R.; Vonstein, V.; Overbeek, R.; Osterman, A.L. Comparative Genomics of NAD Biosynthesis in Cyanobacteria. J. Bacteriol. 2006, 188, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.J.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef]
- Tsubota, K. The first human clinical study for NMN has started in Japan. NPJ Aging Mech. Dis. 2016, 2, 16021. [Google Scholar] [CrossRef] [PubMed]
- Pittelli, M.; Felici, R.; Pitozzi, V.; Giovannelli, L.; Bigagli, E.; Cialdai, F.; Romano, G.; Moroni, F.; Chiarugi, A. Pharmacological Effects of Exogenous NAD on Mitochondrial Bioenergetics, DNA Repair, and Apoptosis. Mol. Pharmacol. 2011, 80, 1136–1146. [Google Scholar] [CrossRef]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Bolli, R. Preconditioning: A paradigm shift in the biology of myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H19–H27. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Perez-Pinzon, M.A. Sirt1 in cerebral ischemia. Brain Circ. 2015, 1, 69. [Google Scholar] [PubMed]
- Hsu, C.-P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.; Wang, Y.T.; Nehrke, K.; Munger, J.; Brookes, P.S. Cardioprotection by nicotinamide mononucleotide (NMN): Involvement of glycolysis and acidic pH. J. Mol. Cell. Cardiol. 2018, 121, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fu, L.; Sun, X.; Peng, W.; Chen, Z.; Li, Y. Remote ischemic conditioning improves myocardial parameters and clinical outcomes during primary percutaneous coronary intervention: A meta-analysis of randomized controlled trials. Oncotarget 2017, 9, 8653–8664. [Google Scholar] [CrossRef]
- Pantazi, E.; Bejaoui, M.; Folch-Puy, E.; Adam, R.; Roselló-Catafau, J. Advances in treatment strategies for ischemia reperfusion injury. Expert Opin. Pharmacother. 2016, 17, 169–179. [Google Scholar] [CrossRef]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar]
- De Graef, M.R.; Alexeeva, S.; Snoep, J.L.; Teixeira de Mattos, M.J. The Steady-State Internal Redox State (NADH/NAD) Reflects the External Redox State and Is Correlated with Catabolic Adaptation in Escherichia coli. J. Bacteriol. 1999, 181, 2351–2357. [Google Scholar]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimers Dis. 2014, 40, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Henriques, M.; Anton, F. Mechanistic perspective of mitochondrial fusion: Tubulation vs. fragmentation. Biochim. Biophys. Acta 2013, 1833, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, X.; Yang, Y.; Takata, T.; Sakurai, T. Nicotinamide mononucleotide protects against β-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 2016, 1643, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; MacTavish, D.; Yang, J.; Westaway, D.; Jhamandas, J.H. Beta Amyloid-Induced Depression of Hippocampal Long-Term Potentiation Is Mediated through the Amylin Receptor. J. Neurosci. 2012, 32, 17401–17406. [Google Scholar] [CrossRef] [PubMed]
- Keep, R.F.; Hua, Y.; Xi, G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurol. 2012, 11, 720–731. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y.; Wang, J.; Anne Stetler, R.; Yang, Q.-W. Inflammation in intracerebral hemorrhage: From mechanisms to clinical translation. Prog. Neurobiol. 2014, 115, 25–44. [Google Scholar] [CrossRef]
- Wei, C.-C.; Kong, Y.-Y.; Li, G.-Q.; Guan, Y.-F.; Wang, P.; Miao, C.-Y. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 2017, 7, 717. [Google Scholar] [CrossRef]
- Mattson, M.P. Roles of the Lipid Peroxidation Product 4-Hydroxynonenal in Obesity, the Metabolic Syndrome, and Associated Vascular and Neurodegenerative Disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef]
- Nov, O.; Kohl, A.; Lewis, E.C.; Bashan, N.; Dvir, I.; Ben-Shlomo, S.; Fishman, S.; Wueest, S.; Konrad, D.; Rudich, A. Interleukin-1beta may mediate insulin resistance in liver-derived cells in response to adipocyte inflammation. Endocrinology 2010, 151, 4247–4256. [Google Scholar] [CrossRef]
- Liu, T.F.; McCall, C.E. Deacetylation by SIRT1 Reprograms Inflammation and Cancer. Genes Cancer 2013, 4, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Kieswich, J.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia 2011, 54, 3083–3092. [Google Scholar] [CrossRef] [PubMed]
- Ofei, F. Obesity—A Preventable Disease. Ghana Med. J. 2005, 39, 98–101. [Google Scholar] [PubMed]
- Agil, A.; El-Hammadi, M.; Jiménez-Aranda, A.; Tassi, M.; Abdo, W.; Fernández-Vázquez, G.; Reiter, R.J. Melatonin reduces hepatic mitochondrial dysfunction in diabetic obese rats. J. Pineal Res. 2015, 59, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial Dysfunction and Oxidative Stress in the Pathogenesis of Alcohol and Obesity Induced Fatty Liver Diseases. Free Radic. Biol. Med. 2008, 44, 1259–1272. [Google Scholar] [CrossRef] [PubMed]
- Huskisson, E.; Maggini, S.; Ruf, M. The role of vitamins and minerals in energy metabolism and well-being. J. Int. Med. Res. 2007, 35, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, H.; Ding, S. The effects of NAD+ on apoptotic neuronal death and mitochondrial biogenesis and function after glutamate excitotoxicity. Int. J. Mol. Sci. 2014, 15, 20449–20468. [Google Scholar] [CrossRef]
- Uddin, G.M.; Youngson, N.A.; Sinclair, D.A.; Morris, M.J. Head to Head Comparison of Short-Term Treatment with the NAD(+) Precursor Nicotinamide Mononucleotide (NMN) and 6 Weeks of Exercise in Obese Female Mice. Front. Pharmacol. 2016, 7, 258. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-L.; Guo, S.-J. Sirtuins Function as the Modulators in Aging-related Diseases in Common or Respectively. Chin. Med. J. (Engl.) 2015, 128, 1671–1678. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Li, J.; Bonkowski, M.S.; Moniot, S.; Zhang, D.; Hubbard, B.P.; Ling, A.J.Y.; Rajman, L.A.; Qin, B.; Lou, Z.; Gorbunova, V.; et al. A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science 2017, 355, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Zoukhri, D. Effect of inflammation on lacrimal gland function. Exp. Eye Res. 2006, 82, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Aredo, B.; Zhang, K.; Chen, X.; Wang, C.; Li, T.; Ufret-Vincenty, R.L. Differences in the distribution, phenotype and gene expression of subretinal microglia/macrophages in C57BL/6N (Crb1rd8/rd8) versus C57BL6/J (Crb1wt/wt) mice. J. Neuroinflamm. 2015, 12, 6. [Google Scholar] [CrossRef]
- De Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Ferrante, A.W. The Immune Cells in Adipose Tissue. Diabetes Obes. Metab. 2013, 15, 34–38. [Google Scholar] [CrossRef]
- Dollerup, O.L.; Christensen, B.; Svart, M.; Schmidt, M.S.; Sulek, K.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Møller, N.; Brenner, C.; Treebak, J.T.; et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: Safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 2018, 108, 343–353. [Google Scholar] [CrossRef]
- Martin, A.S.; Abraham, D.M.; Hershberger, K.A.; Bhatt, D.P.; Mao, L.; Cui, H.; Liu, J.; Liu, X.; Muehlbauer, M.J.; Grimsrud, P.A.; et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2017, 2, 93885. [Google Scholar] [CrossRef] [PubMed]
- Stram, A.R.; Pride, P.M.; Payne, R.M. NAD+ replacement therapy with nicotinamide riboside does not improve cardiac function in a model of mitochondrial heart disease. FASEB J. 2017, 31, 602.15. [Google Scholar]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yang, W.; Gao, Z.; Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 2017, 647, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Bürkle, A. Poly(ADP-ribose). The most elaborate metabolite of NAD+. FEBS J. 2005, 272, 4576–4589. [Google Scholar] [CrossRef]
- Cipriani, G.; Rapizzi, E.; Vannacci, A.; Rizzuto, R.; Moroni, F.; Chiarugi, A. Nuclear Poly(ADP-ribose) Polymerase-1 Rapidly Triggers Mitochondrial Dysfunction. J. Biol. Chem. 2005, 280, 17227–17234. [Google Scholar] [CrossRef]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Carcillo, J.A.; Szabó, C.; Clark, R.S.B. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003, 278, 18426–18433. [Google Scholar] [CrossRef]
- Andreone, T.; Meares, G.P.; Hughes, K.J.; Hansen, P.A.; Corbett, J.A. Cytokine-mediated β-cell damage in PARP-1-deficient islets. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E172–E179. [Google Scholar] [CrossRef]
- Yamamoto, H.; Uchigata, Y.; Okamoto, H. Streptozotocin and alloxan induce DNA strand breaks and poly(ADP-ribose) synthetase in pancreatic islets. Nature 1981, 294, 284–286. [Google Scholar] [CrossRef]
- Lu, L.-M.; Zhang, X.-B.; Kong, R.-M.; Yang, B.; Tan, W. A Ligation-Triggered DNAzyme Cascade for Amplified Fluorescence Detection of Biological Small Molecules with Zero-Background Signal. J. Am. Chem. Soc. 2011, 133, 11686–11691. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poddar, S.K.; Sifat, A.E.; Haque, S.; Nahid, N.A.; Chowdhury, S.; Mehedi, I. Nicotinamide Mononucleotide: Exploration of Diverse Therapeutic Applications of a Potential Molecule. Biomolecules 2019, 9, 34. https://doi.org/10.3390/biom9010034
Poddar SK, Sifat AE, Haque S, Nahid NA, Chowdhury S, Mehedi I. Nicotinamide Mononucleotide: Exploration of Diverse Therapeutic Applications of a Potential Molecule. Biomolecules. 2019; 9(1):34. https://doi.org/10.3390/biom9010034
Chicago/Turabian StylePoddar, Saikat Kumar, Ali Ehsan Sifat, Sanjana Haque, Noor Ahmed Nahid, Sabiha Chowdhury, and Imtias Mehedi. 2019. "Nicotinamide Mononucleotide: Exploration of Diverse Therapeutic Applications of a Potential Molecule" Biomolecules 9, no. 1: 34. https://doi.org/10.3390/biom9010034
APA StylePoddar, S. K., Sifat, A. E., Haque, S., Nahid, N. A., Chowdhury, S., & Mehedi, I. (2019). Nicotinamide Mononucleotide: Exploration of Diverse Therapeutic Applications of a Potential Molecule. Biomolecules, 9(1), 34. https://doi.org/10.3390/biom9010034