Role of HIF-1α in Alcohol-Mediated Multiple Organ Dysfunction


Abstract
1. Introduction
2. Molecular Mechanisms Which Regulate HIF Activity
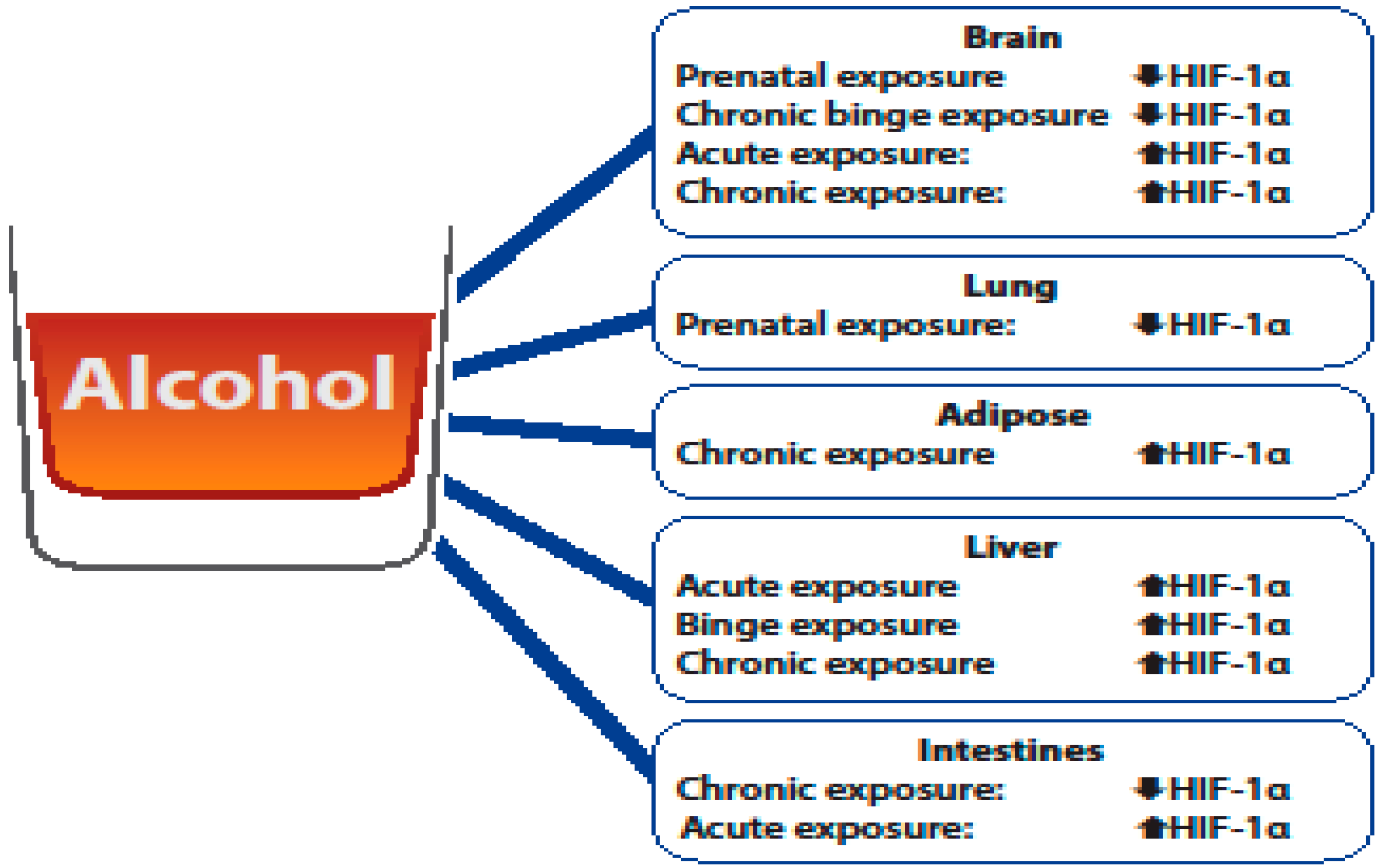
3. The Role of HIF-1α in Alcohol-Mediated Brain Damage
4. The Role of HIF-1α in Alcohol-Mediated Lung Damage
5. The Role of HIF-1α in Alcohol-Mediated Adipose Damage
6. The Role of HIF-1α in Alcohol-Mediated Liver Damage
7. The Role of HIF-1α in Alcohol-Mediated Intestinal Damage
8. Treatment Options for Alcohol-Mediated Tissue/Organ Damage
8.1. Antioxidants
8.2. microRNAs
8.3. Probiotics
8.4. Pharmacological Treatments
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Alcohol Facts and Statistics. NIAAA: Understanding the Impact of Alcohol on Human Health and Well-Being; NIAAA: Bethesda, MD, USA, 2017.
- Mokdad, A.H.; Marks, J.S.; Stroup, D.F.; Gerberding, J.L. Actual causes of death in the United States, 2000. JAMA 2004, 291, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Bouchery, E.E.; Harwood, H.J.; Sacks, J.J.; Simon, C.J.; Brewer, R.D. Economic costs of excessive alcohol consumption in the U.S., 2006. Am. J. Prev. Med. 2011, 41, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Alcohol Facts and Statistics. Available online: https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/alcohol-facts-and-statistics (accessed on 12 November 2018).
- Sacks, J.J.; Gonzales, K.R.; Bouchery, E.E.; Tomedi, L.E.; Brewer, R.D. 2010 National and State Costs of Excessive Alcohol Consumption. Am. J. Prev. Med. 2015, 49, e73–e79. [Google Scholar] [CrossRef] [PubMed]
- Esper, A.; Burnham, E.L.; Moss, M. The effect of alcohol abuse on ARDS and multiple organ dysfunction. Minerva Anestesiol. 2006, 72, 375–381. [Google Scholar]
- Moss, M. Epidemiology of sepsis:+ Race, sex, and chronic alcohol abuse. Clin. Infect. Dis. 2005, 41 (Suppl. 7), S490–S497. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Overview: How Is Alcohol Metabolized by the Body? Alcohol Res. Health 2006, 29, 10. [Google Scholar]
- Curtis, B.J.; Zahs, A.; Kovacs, E.J. Epigenetic targets for reversing immune defects caused by alcohol exposure. Alcohol Res. 2013, 35, 97–113. [Google Scholar]
- Massey, V.L.; Beier, J.I.; Ritzenthaler, J.D.; Roman, J.; Arteel, G.E. Potential Role of the Gut/Liver/Lung Axis in Alcohol-Induced Tissue Pathology. Biomolecules 2015, 5, 2477–2503. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.G.; Dolin, C.E.; Arteel, G.E. Organ-Organ Crosstalk and Alcoholic Liver Disease. Biomolecules 2017, 7, 62. [Google Scholar] [CrossRef]
- Dguzeh, U.; Haddad, N.C.; Smith, K.T.S.; Johnson, J.O.; Doye, A.A.; Gwathmey, J.K.; Haddad, G.E. Alcoholism: A Multi-Systemic Cellular Insult to Organs. Int. J. Environ. Res. Public Health 2018, 15, 1083. [Google Scholar] [CrossRef]
- Mehta, A.J. Alcoholism and critical illness: A review. World J. Crit. Care Med. 2016, 5, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Zhang, A.Y.; Yi, F.X.; Li, P.L.; Zou, A.P. Redox regulation of HIF-1alpha levels and HO-1 expression in renal medullary interstitial cells. Am. J. Physiol. Renal Physiol. 2003, 284, F1207–F1215. [Google Scholar] [CrossRef] [PubMed]
- Page, E.L.; Chan, D.A.; Giaccia, A.J.; Levine, M.; Richard, D.E. Hypoxia-inducible factor-1alpha stabilization in nonhypoxic conditions: Role of oxidation and intracellular ascorbate depletion. Mol. Biol. Cell 2008, 19, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell 2010, 21, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Page, E.L.; Robitaille, G.A.; Pouyssegur, J.; Richard, D.E. Induction of hypoxia-inducible factor-1alpha by transcriptional and translational mechanisms. J. Biol. Chem. 2002, 277, 48403–48409. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.E.; Berra, E.; Pouyssegur, J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J. Biol. Chem. 2000, 275, 26765–26771. [Google Scholar] [CrossRef]
- Biswas, S.; Mukherjee, R.; Tapryal, N.; Singh, A.K.; Mukhopadhyay, C.K. Insulin regulates hypoxia-inducible factor-1alpha transcription by reactive oxygen species sensitive activation of Sp1 in 3T3-L1 preadipocyte. PLoS ONE 2013, 8, e62128. [Google Scholar] [CrossRef]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [PubMed]
- Wyszynski, R.W.; Gibbs, B.F.; Varani, L.; Iannotta, D.; Sumbayev, V.V. Interleukin-1 beta induces the expression and production of stem cell factor by epithelial cells: Crucial involvement of the PI-3K/mTOR pathway and HIF-1 transcription complex. Cell. Mol. Immunol. 2016, 13, 47–56. [Google Scholar] [CrossRef]
- Kuo, H.P.; Lee, D.F.; Xia, W.; Wei, Y.; Hung, M.C. TNFalpha induces HIF-1alpha expression through activation of IKKbeta. Biochem. Biophys. Res. Commun. 2009, 389, 640–644. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G.Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar] [PubMed]
- Fratantonio, D.; Cimino, F.; Speciale, A.; Virgili, F. Need (more than) two to Tango: Multiple tools to adapt to changes in oxygen availability. Biofactors 2018, 44, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef]
- Wang, J.; Fan, Y.; Dong, Y.; Ma, M.; Ma, Y.; Dong, Y.; Niu, Y.; Jiang, Y.; Wang, H.; Wang, Z.; et al. Alterations in Brain Structure and Functional Connectivity in Alcohol Dependent Patients and Possible Association with Impulsivity. PLoS ONE 2016, 11, e0161956. [Google Scholar] [CrossRef]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef]
- Yon, J.M.; Lin, C.; Oh, K.W.; Baek, H.S.; Lee, B.J.; Yun, Y.W.; Nam, S.Y. Emodin prevents ethanol-induced developmental anomalies in cultured mouse fetus through multiple activities. Birth Defects Res. B Dev. Reprod. Toxicol. 2013, 98, 268–275. [Google Scholar] [CrossRef]
- Tong, M.; Ziplow, J.; Chen, W.C.; Nguyen, Q.G.; Kim, C.; de la Monte, S.M. Motor Function Deficits Following Chronic Prenatal Ethanol Exposure are Linked to Impairments in Insulin/IGF, Notch and Wnt Signaling in the Cerebellum. J. Diabetes Metab. 2013, 4, 238. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Sokolik, E.P.; Bukhtiarova, N.V.; Levich, S.V. Pharmacological Modulation of Heat Shock Protein 70 (HSP70)—Dependent Mechanisms of Endogenous Neuroprotection in Conditions of Prenatal Chronic Alcoholism by Cerebrocurin and Tiocetam. Bull. Clin. Psychopharmacol. 2016, 26, 103–108. [Google Scholar] [CrossRef]
- Treins, C.; Giorgetti-Peraldi, S.; Murdaca, J.; Monthouel-Kartmann, M.N.; Van Obberghen, E. Regulation of hypoxia-inducible factor (HIF)-1 activity and expression of HIF hydroxylases in response to insulin-like growth factor I. Mol. Endocrinol. 2005, 19, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Gonzalez-Navarrete, H.; Kirchberg, T.; Gotama, B.; Yalcin, E.B.; Kay, J.; de la Monte, S.M. Ethanol-Induced White Matter Atrophy Is Associated with Impaired Expression of Aspartyl-Asparaginyl-beta-Hydroxylase (ASPH) and Notch Signaling in an Experimental Rat Model. J. Drug Alcohol Res. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Y.; Geng, X.; Asmaro, K.; Peng, C.; Sullivan, J.M.; Ding, J.Y.; Ji, X.; Ding, Y. Neuroprotective effect of acute ethanol administration in a rat with transient cerebral ischemia. Stroke 2012, 43, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.D.; Padmavathi, P.; Kavitha, G.; Saradamma, B.; Varadacharyulu, N. Alcohol-induced oxidative/nitrosative stress alters brain mitochondrial membrane properties. Mol. Cell. Biochem. 2013, 375, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Katada, R.; Nishitani, Y.; Honmou, O.; Mizuo, K.; Okazaki, S.; Tateda, K.; Watanabe, S.; Matsumoto, H. Expression of aquaporin-4 augments cytotoxic brain edema after traumatic brain injury during acute ethanol exposure. Am. J. Pathol. 2012, 180, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, R.; Hao, S.; Xu, F.; Mao, X.; Liu, B. A pre-injury high ethanol intake in rats promotes brain edema following traumatic brain injury. Br. J. Neurosurg. 2014, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Yan, E.B.; Satgunaseelan, L.; Paul, E.; Bye, N.; Nguyen, P.; Agyapomaa, D.; Kossmann, T.; Rosenfeld, J.V.; Morganti-Kossmann, M.C. Post-traumatic hypoxia is associated with prolonged cerebral cytokine production, higher serum biomarker levels, and poor outcome in patients with severe traumatic brain injury. J. Neurotrauma 2014, 31, 618–629. [Google Scholar] [CrossRef]
- Lazic, T.; Sow, F.B.; Van Geelen, A.; Meyerholz, D.K.; Gallup, J.M.; Ackermann, M.R. Exposure to ethanol during the last trimester of pregnancy alters the maturation and immunity of the fetal lung. Alcohol (Fayetteville, N.Y.) 2011, 45, 673–680. [Google Scholar] [CrossRef]
- Mehta, A.J.; Yeligar, S.M.; Elon, L.; Brown, L.A.; Guidot, D.M. Alcoholism causes alveolar macrophage zinc deficiency and immune dysfunction. Am. J. Respir. Crit. Care Med. 2013, 188, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.M.; Mehta, A.J.; Harris, F.L.; Brown, L.A.; Hart, C.M. Peroxisome Proliferator-Activated Receptor gamma Regulates Chronic Alcohol-Induced Alveolar Macrophage Dysfunction. Am. J. Respir. Cell Mol. Biol. 2016, 55, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.J.; Guidot, D.M. Alcohol abuse, the alveolar macrophage and pneumonia. Am. J. Med Sci. 2012, 343, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.M.; Harris, F.L.; Hart, C.M.; Brown, L.A. Glutathione attenuates ethanol-induced alveolar macrophage oxidative stress and dysfunction by downregulating NADPH oxidases. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L429–L441. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Harris, F.L.; Ping, X.D.; Gauthier, T.W. Chronic ethanol ingestion and the risk of acute lung injury: A role for glutathione availability? Alcohol 2004, 33, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.I.; Bijli, K.M.; Murphy, T.C.; Kleinhenz, J.M.; Hart, C.M. Time-dependent PPARgamma Modulation of HIF-1alpha Signaling in Hypoxic Pulmonary Artery Smooth Muscle Cells. Am. J. Med. Sci. 2016, 352, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.M.; Harris, F.L.; Hart, C.M.; Brown, L.A. Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J. Immunol. 2012, 188, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Addolorato, G.; Capristo, E.; Marini, M.; Santini, P.; Scognamiglio, U.; Attilia, M.L.; Messineo, D.; Sasso, G.F.; Gasbarrini, G.; Ceccanti, M. Body composition changes induced by chronic ethanol abuse: Evaluation by dual energy X-ray absorptiometry. Am. J. Gastroenterol. 2000, 95, 2323–2327. [Google Scholar] [CrossRef]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Sun, X.; Kim, S.; et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: Role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef]
- He, Z.; Li, M.; Zheng, D.; Chen, Q.; Liu, W.; Feng, L. Adipose tissue hypoxia and low-grade inflammation: A possible mechanism for ethanol-related glucose intolerance? Br. J. Nutr. 2015, 113, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.K.; Drager, L.F.; Yao, Q.; Bevans-Fonti, S.; Yoo, D.Y.; Jun, J.C.; Aja, S.; Bhanot, S.; Polotsky, V.Y. Metabolic consequences of high-fat diet are attenuated by suppression of HIF-1alpha. PLoS ONE 2012, 7, e46562. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.D.; Liu, Y.H.; Zhang, Q.L.; Zhang, B.G.; Zhao, N.; Wang, Q.L.; Wang, X.D. Pre-endurance training prevents acute alcoholic liver injury in rats through the regulation of damaged mitochondria accumulation and mitophagy balance. Hepatol. Int. 2014, 8, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Sidhu, A.; Ma, Z.; McClain, C.; Feng, W. Lactobacillus rhamnosus GG culture supernatant ameliorates acute alcohol-induced intestinal permeability and liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G32–G41. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kirpich, I.; Liu, Y.; Ma, Z.; Barve, S.; McClain, C.J.; Feng, W. Lactobacillus rhamnosus GG treatment potentiates intestinal hypoxia-inducible factor, promotes intestinal integrity and ameliorates alcohol-induced liver injury. Am. J. Pathol. 2011, 179, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.W.; Son, M.J.; Abdelmegeed, M.A.; Banerjee, A.; Morgan, T.R.; Yoo, S.H.; Song, B.J. Binge alcohol promotes hypoxic liver injury through a CYP2E1-HIF-1alpha-dependent apoptosis pathway in mice and humans. Free Radic. Biol. Med. 2014, 77, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xu, W.; Shao, J.; Zhang, F.; Chen, A.; Zheng, S. Nrf2 Activation Is Required for Ligustrazine to Inhibit Hepatic Steatosis in Alcohol-Preferring Mice and Hepatocytes. Toxicol. Sci. 2017, 155, 432–443. [Google Scholar] [CrossRef]
- Ambade, A.; Satishchandran, A.; Szabo, G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stemness and miR-122-mediated HIF-1alpha activation. Sci. Rep. 2016, 6, 21340. [Google Scholar] [CrossRef] [PubMed]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, Y.; Shen, Y.; Gao, Y.; Chang, Y.; Duan, X. Gene expression profiles of sodium-dependent vitamin C transporters in mice after alcohol consumption. Acta Biochim. Biophys. Sin. 2013, 45, 912–920. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Jiang, Z.A.; Zhao, C.Y.; Zhen, Z.; Wang, W.; Nanji, A.A. Long-term binge and escalating ethanol exposure causes necroinflammation and fibrosis in rat liver. Alcohol. Clin. Exp. Res. 2013, 37, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, Y.; Li, Q.; Xu, M.; Bai, J.; Wu, S. Resveratrol improves alcoholic fatty liver disease by downregulating HIF-1alpha expression and mitochondrial ROS production. PLoS ONE 2017, 12, e0183426. [Google Scholar] [CrossRef]
- Machado, M.V.; Janeiro, A.; Miltenberger-Miltenyi, G.; Cortez-Pinto, H. Genetic polymorphisms of proangiogenic factors seem to favor hepatocellular carcinoma development in alcoholic cirrhosis. Eur. J. Gastroenterol. Hepatol. 2014, 26, 438–443. [Google Scholar] [CrossRef]
- Satishchandran, A.; Ambade, A.; Rao, S.; Hsueh, Y.C.; Iracheta-Vellve, A.; Tornai, D.; Lowe, P.; Gyongyosi, B.; Li, J.; Catalano, D.; et al. MicroRNA 122, Regulated by GRLH2, Protects Livers of Mice and Patients from Ethanol-Induced Liver Disease. Gastroenterology 2018, 154, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Lv, Y.; Lin, B.; Tipoe, G.L.; Youdim, M.B.; Xing, F.; Liu, Y. A novel antioxidant multitarget iron chelator M30 protects hepatocytes against ethanol-induced injury. Oxid. Med. Cell. Longev. 2015, 2015, 607271. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1alpha induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef]
- Wang, X.; Wu, D.; Yang, L.; Gan, L.; Cederbaum, A.I. Cytochrome P450 2E1 potentiates ethanol induction of hypoxia and HIF-1alpha in vivo. Free Radic. Biol. Med. 2013, 63, 175–186. [Google Scholar] [CrossRef]
- Chacko, B.K.; Srivastava, A.; Johnson, M.S.; Benavides, G.A.; Chang, M.J.; Ye, Y.; Jhala, N.; Murphy, M.P.; Kalyanaraman, B.; Darley-Usmar, V.M. Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology 2011, 54, 153–163. [Google Scholar] [CrossRef]
- Heritage, M.L.; Murphy, T.L.; Bridle, K.R.; Anderson, G.J.; Crawford, D.H.; Fletcher, L.M. Hepcidin regulation in wild-type and Hfe knockout mice in response to alcohol consumption: Evidence for an alcohol-induced hypoxic response. Alcohol. Clin. Exp. Res. 2009, 33, 1391–1400. [Google Scholar] [CrossRef]
- Li, L.; Chen, S.H.; Zhang, Y.; Yu, C.H.; Li, S.D.; Li, Y.M. Is the hypoxia-inducible factor-1 alpha mRNA expression activated by ethanol-induced injury, the mechanism underlying alcoholic liver disease? Hepatobiliary Pancreat Dis. Int. 2006, 5, 560–563. [Google Scholar]
- Yeligar, S.; Tsukamoto, H.; Kalra, V.K. Ethanol-induced expression of ET-1 and ET-BR in liver sinusoidal endothelial cells and human endothelial cells involves hypoxia-inducible factor-1alpha and microrNA-199. J. Immunol. 2009, 183, 5232–5243. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.M.; Machida, K.; Tsukamoto, H.; Kalra, V.K. Ethanol augments RANTES/CCL5 expression in rat liver sinusoidal endothelial cells and human endothelial cells via activation of NF-kappa B, HIF-1 alpha, and AP-1. J. Immunol. 2009, 183, 5964–5976. [Google Scholar] [CrossRef] [PubMed]
- Na, T.Y.; Han, Y.H.; Ka, N.L.; Park, H.S.; Kang, Y.P.; Kwon, S.W.; Lee, B.H.; Lee, M.O. 22-S-Hydroxycholesterol protects against ethanol-induced liver injury by blocking the auto/paracrine activation of MCP-1 mediated by LXRalpha. J. Pathol. 2015, 235, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Raskopf, E.; Gonzalez Carmona, M.A.; Van Cayzeele, C.J.; Strassburg, C.; Sauerbruch, T.; Schmitz, V. Toxic damage increases angiogenesis and metastasis in fibrotic livers via PECAM-1. Biomed. Res. Int. 2014, 2014, 712893. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.M.; Machida, K.; Kalra, V.K. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. J. Biol. Chem. 2010, 285, 35359–35373. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Zahs, A.; Brown, M.M.; Ramirez, L.; Turner, J.R.; Choudhry, M.A.; Kovacs, E.J. An alteration of the gut-liver axis drives pulmonary inflammation after intoxication and burn injury in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G711–G718. [Google Scholar] [CrossRef]
- Chen, M.M.; O’Halloran, E.B.; Shults, J.A.; Kovacs, E.J. Kupffer Cell p38 Mitogen-Activated Protein Kinase Signaling Drives Postburn Hepatic Damage and Pulmonary Inflammation When Alcohol Intoxication Precedes Burn Injury. Crit. Care Med. 2016, 44, e973–e979. [Google Scholar] [CrossRef]
- Hammer, A.M.; Morris, N.L.; Earley, Z.M.; Choudhry, M.A. The First Line of Defense the Effects of Alcohol on Post-Burn Intestinal Barrier, Immune Cells, and Microbiome. Alcohol Res. Curr. Rev. 2015, 37, 209–222. [Google Scholar]
- Hammer, A.M.; Morris, N.L.; Cannon, A.R.; Khan, O.M.; Gagnon, R.C.; Movtchan, N.V.; van Langeveld, I.; Li, X.; Gao, B.; Choudhry, M.A. Interleukin-22 Prevents Microbial Dysbiosis and Promotes Intestinal Barrier Regeneration Following Acute Injury. Shock (Augusta, Ga.) 2017, 48, 657–665. [Google Scholar] [CrossRef]
- Hammer, A.M.; Khan, O.M.; Morris, N.L.; Li, X.; Movtchan, N.V.; Cannon, A.R.; Choudhry, M.A. The Effects of Alcohol Intoxication and Burn Injury on the Expression of Claudins and Mucins in the Small and Large Intestines. Shock (Augusta, Ga.) 2015. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, L.; Forsyth, C.B.; Shaikh, M.; Song, S.; Keshavarzian, A. The Role of miR-212 and iNOS in Alcohol-Induced Intestinal Barrier Dysfunction and Steatohepatitis. Alcoholism. Clin. Exp. Res. 2015, 39, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Banan, A.; Forsyth, C.B.; Fields, J.Z.; Lau, C.K.; Zhang, L.J.; Keshavarzian, A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcoholism. Clin. Exp. Res. 2008, 32, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.Y.; Nguyen, D.; Bui, V.; Nguyen, H.; Hoa, N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999, 276, G965–G974. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, J.; Chang, B.; Wang, B.; Zhang, D.; Wang, B. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol. Med. Rep. 2014, 9, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Akhtar, S.; Choudhry, M.A. Alteration in intestine tight junction protein phosphorylation and apoptosis is associated with increase in IL-18 levels following alcohol intoxication and burn injury. Biochim. Biophys. Acta 2012, 1822, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Zahs, A.; Bird, M.D.; Ramirez, L.; Turner, J.R.; Choudhry, M.A.; Kovacs, E.J. Inhibition of long myosin light-chain kinase activation alleviates intestinal damage after binge ethanol exposure and burn injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G705–G712. [Google Scholar] [CrossRef] [PubMed]
- Zahs, A.; Bird, M.D.; Ramirez, L.; Choudhry, M.A.; Kovacs, E.J. Anti-IL-6 antibody treatment but not IL-6 knockout improves intestinal barrier function and reduces inflammation after binge ethanol exposure and burn injury. Shock (Augusta, Ga.) 2013, 39, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.; Zhao, C.; Li, F.; Gu, Z.; Liu, L.; Zhang, L.; Wang, Y.; He, L.; Liu, Y.; Liu, Q.; et al. Intestinal HIF-1alpha deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Morris, N.L.; Li, X.; Earley, Z.M.; Choudhry, M.A. Regional variation in expression of pro-inflammatory mediators in the intestine following a combined insult of alcohol and burn injury. Alcohol (Fayetteville, N.Y.) 2015, 49, 507–511. [Google Scholar] [CrossRef]
- Morris, N.L.; Hammer, A.M.; Cannon, A.R.; Gagnon, R.C.; Li, X.; Choudhry, M.A. Dysregulation of microRNA biogenesis in the small intestine after ethanol and burn injury. Biochim. Biophys. Acta 2017, 1863, 2645–2653. [Google Scholar] [CrossRef]
- Cannon, A.R.; Kuprys, P.V.; Cobb, A.N.; Ding, X.; Kothari, A.N.; Kuo, P.C.; Eberhardt, J.M.; Hammer, A.M.; Morris, N.L.; Li, X.; et al. Alcohol enhances symptoms and propensity for infection in inflammatory bowel disease patients and a murine model of DSS-induced colitis. J. Leukoc. Biol. 2018, 104, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Son, Y.O.; Ding, S.; Wang, X.; Hitron, J.A.; Budhraja, A.; Lee, J.C.; Lin, Q.; Poyil, P.; Zhang, Z.; et al. Ethanol enhances tumor angiogenesis in vitro induced by low-dose arsenic in colon cancer cells through hypoxia-inducible factor 1 alpha pathway. Toxicol. Sci. Off. J. Soc. Toxicol. 2012, 130, 269–280. [Google Scholar] [CrossRef]
- Anderson, E.R.; Taylor, M.; Xue, X.; Martin, A.; Moons, D.S.; Omary, M.B.; Shah, Y.M. The hypoxia-inducible factor-C/EBPalpha axis controls ethanol-mediated hepcidin repression. Mol. Cell Biol. 2012, 32, 4068–4077. [Google Scholar] [CrossRef]
- Gerjevic, L.N.; Lu, S.; Chaky, J.P.; Harrison-Findik, D.D. Regulation of heme oxygenase expression by alcohol, hypoxia and oxidative stress. World J. Biol. Chem. 2011, 2, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Zamore, P.D. Beginning to understand microRNA function. Cell Res. 2007, 17, 661–663. [Google Scholar] [CrossRef]


{kind=link}
| Treatment Type | Effects on HIF-1α | Source |
|---|---|---|
| Antioxidants | ||
| M30 | Decreased HIF-1α | Xiao et al. Oxid Med Cell Longev, 2015. |
| Resveratrol | Decreased HIF-1α | Ma et al. PLoS One, 2017. |
| Vitamin C | Decreased HIF-1α | Guo et al. Acta Biochimica et Biophysica Sinic, 2013. |
| MitoQ | Decreased HIF-1α | Chacko et al. Hepatology, 2011. |
| Pre-endurance Training | Decreased HIF-1α | Ma et al. Hepatol Int, 2014. |
| microRNAs | ||
| miR-122 | Decreased HIF-1α | Satishchandran et al. Gastroenterology, 2018. |
| Probiotics | ||
| Lactobacillus rhamnosus GG | Increased HIF-1α | Wang et al. The American Journal of Pathology, 2012. |
| Pharmacological Treatments | ||
| Tiocetam | Increased HIF-1α | Belnichev et al. Klinik Psikofarmakoloji Bülteni-Bulletin of Clinical Psychopharmacology,2016. |
| Cerebrocurin | Increased HIF-1α | Belnichev et al. Klinik Psikofarmakoloji Bülteni-Bulletin of Clinical Psychopharmacology,2016. |
| Emodin | Increased HIF-1α | Yon et al. Birth Defects Res B Dev Reprod Toxicol, 2013. |
| Ligustrazine | Decreased HIF-1α | Lu et al. Toxicol Sci, 2017. |
| PX-478 | Decreased HIF-1α | Yun et al. Free radical biology & Medicine, 2014. |
| Methoxyestradiol | Decreased HIF-1α | Wang et al. Free Radic Biol Med, 2013. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morris, N.L.; Yeligar, S.M. Role of HIF-1α in Alcohol-Mediated Multiple Organ Dysfunction. Biomolecules 2018, 8, 170. https://doi.org/10.3390/biom8040170
Morris NL, Yeligar SM. Role of HIF-1α in Alcohol-Mediated Multiple Organ Dysfunction. Biomolecules. 2018; 8(4):170. https://doi.org/10.3390/biom8040170
Chicago/Turabian StyleMorris, Niya L., and Samantha M. Yeligar. 2018. "Role of HIF-1α in Alcohol-Mediated Multiple Organ Dysfunction" Biomolecules 8, no. 4: 170. https://doi.org/10.3390/biom8040170
APA StyleMorris, N. L., & Yeligar, S. M. (2018). Role of HIF-1α in Alcohol-Mediated Multiple Organ Dysfunction. Biomolecules, 8(4), 170. https://doi.org/10.3390/biom8040170