Pre-Ribosomal RNA Processing in Human Cells: From Mechanisms to Congenital Diseases


Abstract
1. A Renewed Perspective on Pre-rRNA Processing in Human Cells
2. Human Pre-rRNA Processing Is Both Hierarchical and Modular
3. Pre-rRNA Processing Is Coordinated with RNA Folding and Modification
4. The Role of Ribosomal Proteins and Ribosomal Assembly Factors
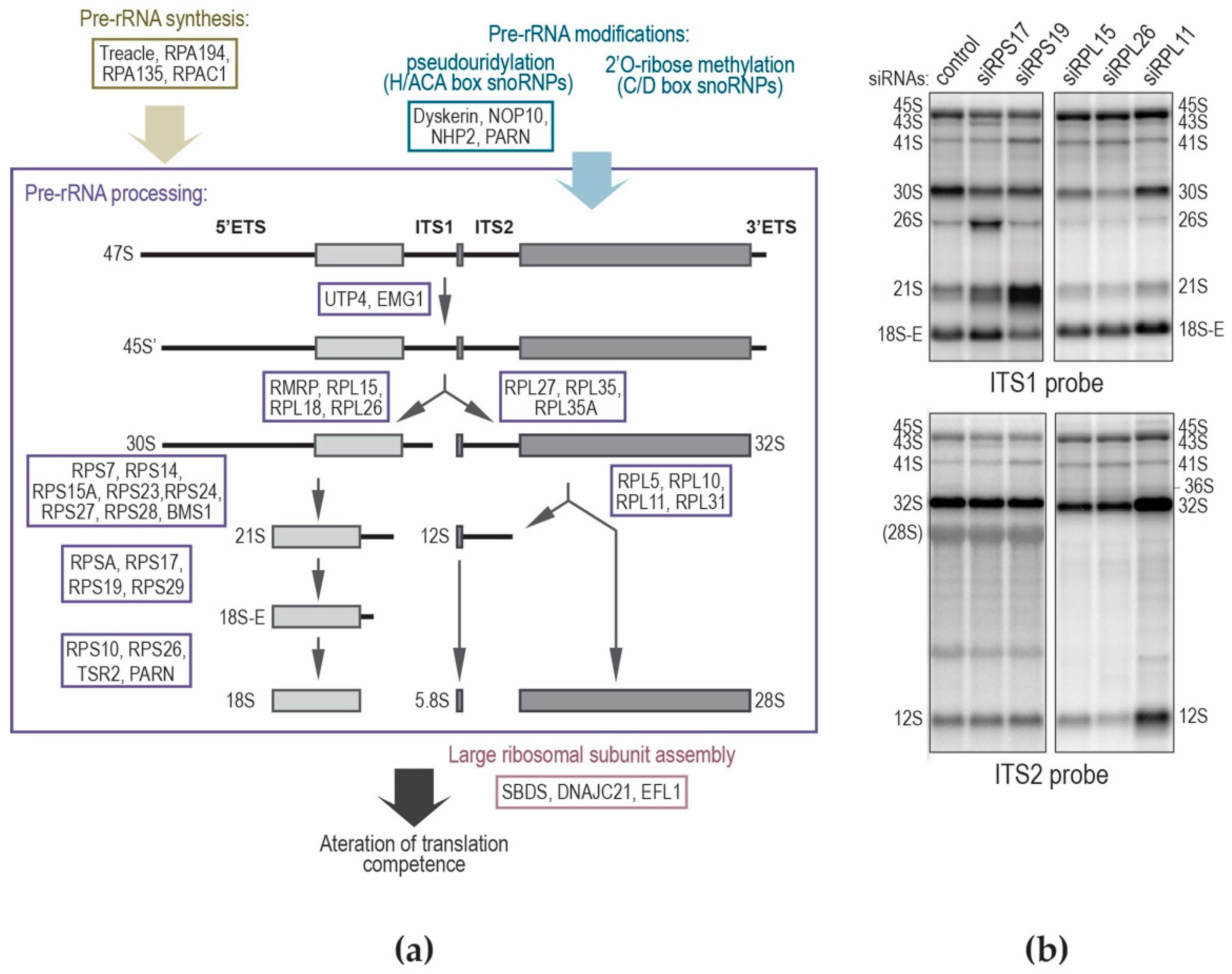
5. A Tour of the 18S rRNA Maturation Scheme
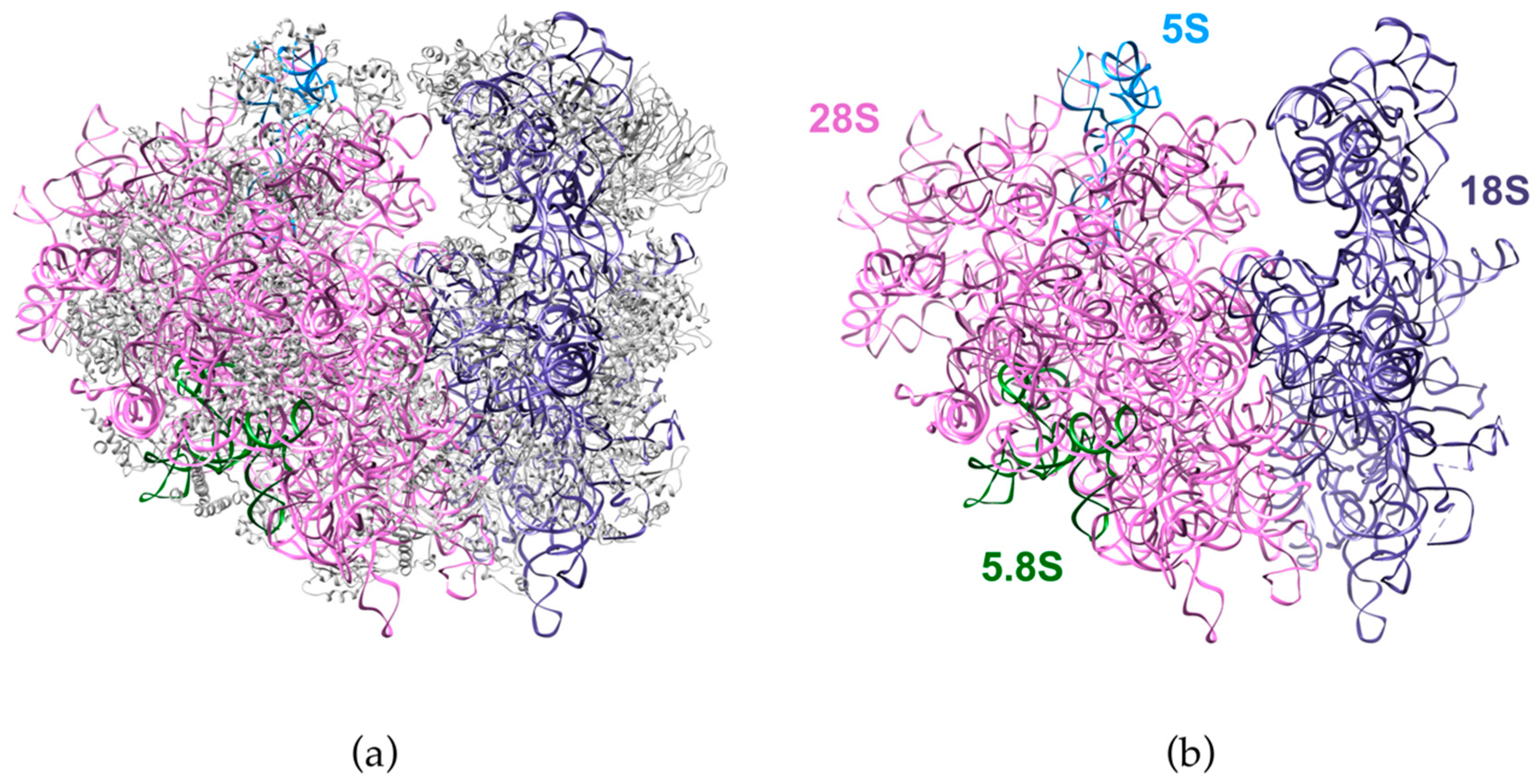
6. Processing of the Large Subunit rRNAs
7. 5S rRNA, the Fourth Musketeer
8. Defects of Pre-rRNA Processing in Congenital Diseases: The Case of Diamond–Blackfan Anemia
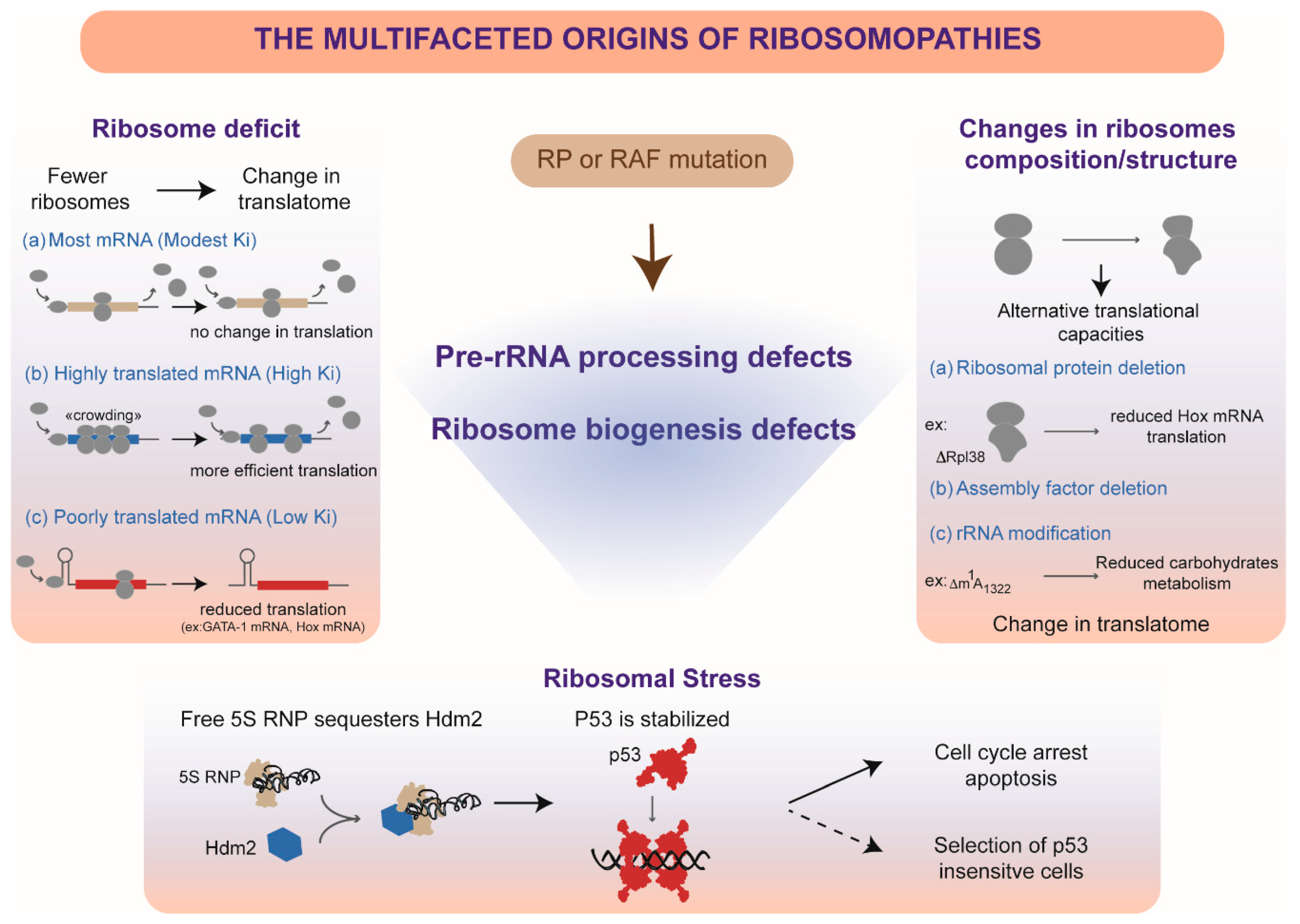
9. Do Pre-rRNA Processing Defects Contribute to Ribosomopathies?
10. Insufficient Production of Ribosomes
11. Alteration of Ribosome Quality by Pre-rRNA Processing Defects
12. Ribosomal Stress
13. Concluding Remarks on Cross-Talks between Pre-rRNA Processing and Other Gene Expression Processes
Funding
Acknowledgments
Conflicts of Interest
References
- Palade, G.E. A small particulate component of the cytoplasm. J. Biophys. Biochem. Cytol. 1955, 1, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Capel, M.; Moore, P.B.; Steitz, T.A. Placement of protein and RNA structures into a 5 Å-resolution map of the 50S ribosomal subunit. Nature 1999, 400, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Cate, J.H.; Yusupov, M.M.; Yusupova, G.Z.; Earnest, T.N.; Noller, H.F. X-ray crystal structures of 70S ribosome functional complexes. Science 1999, 285, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Clemons, W.M.; May, J.L.; Wimberly, B.T.; McCutcheon, J.P.; Capel, M.S.; Ramakrishnan, V. Structure of a bacterial 30S ribosomal subunit at 5.5 Å resolution. Nature 1999, 400, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Klinge, S.; Voigts-Hoffmann, F.; Leibundgut, M.; Arpagaus, S.; Ban, N. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science 2011, 334, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Rabl, J.; Leibundgut, M.; Ataide, S.F.; Haag, A.; Ban, N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 2011, 331, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Strunk, B.S.; Loucks, C.R.; Su, M.; Vashisth, H.; Cheng, S.; Schilling, J.; Brooks, C.L.; Karbstein, K.; Skiniotis, G. Ribosome assembly factors prevent premature translation initiation by 40S assembly intermediates. Science 2011, 333, 1449–1453. [Google Scholar] [CrossRef] [PubMed]
- Greber, B.J.; Boehringer, D.; Montellese, C.; Ban, N. Cryo-EM structures of Arx1 and maturation factors Rei1 and Jjj1 bound to the 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2012, 19, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Leidig, C.; Thoms, M.; Holdermann, I.; Bradatsch, B.; Berninghausen, O.; Bange, G.; Sinning, I.; Hurt, E.; Beckmann, R. 60S ribosome biogenesis requires rotation of the 5S ribonucleoprotein particle. Nat. Commun. 2014, 5, 3491. [Google Scholar] [CrossRef] [PubMed]
- Weis, F.; Giudice, E.; Churcher, M.; Jin, L.; Hilcenko, C.; Wong, C.C.; Traynor, D.; Kay, R.R.; Warren, A.J. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2015, 22, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Kornprobst, M.; Turk, M.; Kellner, N.; Cheng, J.; Flemming, D.; Koš-Braun, I.; Koš, M.; Thoms, M.; Berninghausen, O.; Beckmann, R.; et al. Architecture of the 90S Pre-ribosome: A Structural View on the Birth of the Eukaryotic Ribosome. Cell 2016, 166, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tutuncuoglu, B.; Yan, K.; Brown, H.; Zhang, Y.; Tan, D.; Gamalinda, M.; Yuan, Y.; Li, Z.; Jakovljevic, J.; et al. Diverse roles of assembly factors revealed by structures of late nuclear pre-60S ribosomes. Nature 2016, 534, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhu, X.; Qi, J.; An, W.; Lan, P.; Tan, D.; Chen, R.; Wang, B.; Zheng, S.; Zhang, C.; et al. Molecular architecture of the 90S small subunit pre-ribosome. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Chaker-Margot, M.; Barandun, J.; Hunziker, M.; Klinge, S. Architecture of the yeast small subunit processome. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Heuer, A.; Thomson, E.; Schmidt, C.; Berninghausen, O.; Becker, T.; Hurt, E.; Beckmann, R. Cryo-EM structure of a late pre-40S ribosomal subunit from Saccharomyces cerevisiae. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Scaiola, A.; Peña, C.; Weisser, M.; Böhringer, D.; Leibundgut, M.; Klingauf-Nerurkar, P.; Gerhardy, S.; Panse, V.G.; Ban, N. Structure of a eukaryotic cytoplasmic pre-40S ribosomal subunit. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Larburu, N.; Montellese, C.; O’Donohue, M.-F.; Kutay, U.; Gleizes, P.-E.; Plisson-Chastang, C. Structure of a human pre-40S particle points to a role for RACK1 in the final steps of 18S rRNA processing. Nucleic Acids Res. 2016, 44, 8465–8478. [Google Scholar] [CrossRef] [PubMed]
- Khatter, H.; Myasnikov, A.G.; Natchiar, S.K.; Klaholz, B.P. Structure of the human 80S ribosome. Nature 2015, 520, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Ameismeier, M.; Cheng, J.; Berninghausen, O.; Beckmann, R. Visualizing late states of human 40S ribosomal subunit maturation. Nature 2018, 558, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Henras, A.K.; Soudet, J.; Gérus, M.; Lebaron, S.; Caizergues-Ferrer, M.; Mougin, A.; Henry, Y. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. CMLS 2008, 65, 2334–2359. [Google Scholar] [CrossRef] [PubMed]
- Woolford, J.L.; Baserga, S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013, 195, 643–681. [Google Scholar] [CrossRef] [PubMed]
- Tafforeau, L.; Zorbas, C.; Langhendries, J.L.; Mullineux, S.T.; Stamatopoulou, V.; Mullier, R.; Wacheul, L.; Lafontaine, D.L. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol. Cell 2013, 51, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.S.; Lam, Y.W.; Leung, A.K.; Ong, S.E.; Lyon, C.E.; Lamond, A.I.; Mann, M. Nucleolar proteome dynamics. Nature 2005, 433, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Coute, Y.; Burgess, J.A.; Diaz, J.J.; Chichester, C.; Lisacek, F.; Greco, A.; Sanchez, J.C. Deciphering the human nucleolar proteome. Mass Spectrom. Rev. 2006, 25, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Wild, T.; Horvath, P.; Wyler, E.; Widmann, B.; Badertscher, L.; Zemp, I.; Kozak, K.; Csucs, G.; Lund, E.; Kutay, U. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol. 2010, 8, e1000522. [Google Scholar] [CrossRef] [PubMed]
- Farley-Barnes, K.I.; McCann, K.L.; Ogawa, L.M.; Merkel, J.; Surovtseva, Y.V.; Baserga, S.J. Diverse Regulators of Human Ribosome Biogenesis Discovered by Changes in Nucleolar Number. Cell Rep. 2018, 22, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Gazda, H.T. Ribosomopathies: how a common root can cause a tree of pathologies. Dis. Model. Mech. 2015, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Yelick, P.C.; Trainor, P.A. Ribosomopathies: Global process, tissue specific defects. Rare Dis. 2015, 3, e1025185. [Google Scholar] [CrossRef] [PubMed]
- Mullineux, S.T.; Lafontaine, D.L. Mapping the cleavage sites on mammalian pre-rRNAs: Where do we stand? Biochimie 2012, 94, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Henras, A.K.; Plisson-Chastang, C.; O’Donohue, M.F.; Chakraborty, A.; Gleizes, P.E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA 2015, 6, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Tomecki, R.; Sikorski, P.J.; Zakrzewska-Placzek, M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells–focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 2017, 591, 1801–1850. [Google Scholar] [CrossRef] [PubMed]
- Michot, B.; Bachellerie, J.P. Secondary structure of the 5′ external transcribed spacer of vertebrate pre-rRNA. Presence of phylogenetically conserved features. Eur. J. Biochem. FEBS 1991, 195, 601–609. [Google Scholar] [CrossRef]
- Ciganda, M.; Williams, N. Eukaryotic 5S rRNA biogenesis: Eukaryotic 5S rRNA biogenesis. Wiley Interdiscip. Rev. RNA 2011, 2, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Meyer, C.; Benjamin-Hong, S.; Rodriguez, J.; Briskin, D.; Bognanni, C.; Bogardus, K.; Steller, H.; Tuschl, T. The Conserved RNA Exonuclease Rexo5 Is Required for 3′ End Maturation of 28S rRNA, 5S rRNA, and snoRNAs. Cell Rep. 2017, 21, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Carron, C.; O’Donohue, M.F.; Choesmel, V.; Faubladier, M.; Gleizes, P.E. Analysis of two human pre-ribosomal factors, bystin and hTsr1, highlights differences in evolution of ribosome biogenesis between yeast and mammals. Nucleic Acids Res. 2011, 39, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Preti, M.; O’Donohue, M.F.; Montel-Lehry, N.; Bortolin-Cavaille, M.L.; Choesmel, V.; Gleizes, P.E. Gradual processing of the ITS1 from the nucleolus to the cytoplasm during synthesis of the human 18S rRNA. Nucleic Acids Res. 2013, 41, 4709–4723. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Mattijssen, S.; Lebaron, S.; Tollervey, D.; Pruijn, G.J.; Watkins, N.J. Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J. Cell Biol. 2013, 200, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Montellese, C.; Montel-Lehry, N.; Henras, A.K.; Kutay, U.; Gleizes, P.-E.; O’Donohue, M.-F. Poly(A)-specific ribonuclease is a nuclear ribosome biogenesis factor involved in human 18S rRNA maturation. Nucleic Acids Res. 2017, 45, 6822–6836. [Google Scholar] [CrossRef] [PubMed]
- O’Donohue, M.F.; Choesmel, V.; Faubladier, M.; Fichant, G.; Gleizes, P.E. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J. Cell Biol. 2010, 190, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Choesmel, V.; Bacqueville, D.; Rouquette, J.; Noaillac-Depeyre, J.; Fribourg, S.; Cretien, A.; Leblanc, T.; Tchernia, G.; Da Costa, L.; Gleizes, P.E. Impaired ribosome biogenesis in Diamond–Blackfan anemia. Blood 2007, 109, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.; Aspesi, A.; Bailey, J.C.; Miyake, K.; Caffrey, J.M.; Karlsson, S.; Ellis, S.R. Human RPS19, the gene mutated in Diamond–Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood 2007, 109, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Belin, S.; Beghin, A.; Solano-Gonzalez, E.; Bezin, L.; Brunet-Manquat, S.; Textoris, J.; Prats, A.C.; Mertani, H.C.; Dumontet, C.; Diaz, J.J. Dysregulation of ribosome biogenesis and translational capacity is associated with tumor progression of human breast cancer cells. PLoS ONE 2009, 4, e7147. [Google Scholar] [CrossRef] [PubMed]
- Henras, A.K.; Plisson-Chastang, C.; Humbert, O.; Romeo, Y.; Henry, Y. Synthesis, Function, and Heterogeneity of snoRNA-Guided Posttranscriptional Nucleoside Modifications in Eukaryotic Ribosomal RNAs. Enzymes 2017, 41, 169–213. [Google Scholar] [CrossRef] [PubMed]
- Watkins, N.J.; Bohnsack, M.T. The box C/D and H/ACA snoRNPs: Key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip. Rev. RNA 2012, 3, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Warda, A.S.; Sharma, S.; Entian, K.-D.; Lafontaine, D.L.J.; Bohnsack, M.T. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017, 14, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Taoka, M.; Nobe, Y.; Yamaki, Y.; Sato, K.; Ishikawa, H.; Izumikawa, K.; Yamauchi, Y.; Hirota, K.; Nakayama, H.; Takahashi, N.; et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Higa-Nakamine, S.; Suzuki, T.; Uechi, T.; Chakraborty, A.; Nakajima, Y.; Nakamura, M.; Hirano, N.; Kenmochi, N. Loss of ribosomal RNA modification causes developmental defects in zebrafish. Nucleic Acids Res. 2012, 40, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Gamalinda, M.; Ohmayer, U.; Jakovljevic, J.; Kumcuoglu, B.; Woolford, J.; Mbom, B.; Lin, L.; Woolford, J.L. A hierarchical model for assembly of eukaryotic 60S ribosomal subunit domains. Genes Dev. 2014, 28, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Kater, L.; Thoms, M.; Barrio-Garcia, C.; Cheng, J.; Ismail, S.; Ahmed, Y.L.; Bange, G.; Kressler, D.; Berninghausen, O.; Sinning, I.; et al. Visualizing the Assembly Pathway of Nucleolar Pre-60S Ribosomes. Cell 2017, 171, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Pertschy, B.; Schneider, C.; Gnadig, M.; Schafer, T.; Tollervey, D.; Hurt, E. RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1. J. Biol. Chem. 2009, 284, 35079–35091. [Google Scholar] [CrossRef] [PubMed]
- Lebaron, S.; Schneider, C.; van Nues, R.W.; Swiatkowska, A.; Walsh, D.; Böttcher, B.; Granneman, S.; Watkins, N.J.; Tollervey, D. Proofreading of pre-40S ribosome maturation by a translation initiation factor and 60S subunits. Nat. Struct. Mol. Biol. 2012, 19, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Espinar-Marchena, F.J.; Babiano, R.; Cruz, J. Placeholder factors in ribosome biogenesis: please, pave my way. Microb. Cell 2017, 4, 144–168. [Google Scholar] [CrossRef] [PubMed]
- Schafer, T.; Strauss, D.; Petfalski, E.; Tollervey, D.; Hurt, E. The path from nucleolar 90S to cytoplasmic 40S pre-ribosomes. EMBO J. 2003, 22, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Dragon, F.; Gallagher, J.E.; Compagnone-Post, P.A.; Mitchell, B.M.; Porwancher, K.A.; Wehner, K.A.; Wormsley, S.; Settlage, R.E.; Shabanowitz, J.; Osheim, Y.; et al. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 2002, 417, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.J.; Knox, A.A.; Prieto, J.-L.; McStay, B.; Watkins, N.J. A novel small-subunit processome assembly intermediate that contains the U3 snoRNP, nucleolin, RRP5, and DBP4. Mol. Cell. Biol. 2009, 29, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Bohnsack, M.T.; Schneider, C.; Watkins, N.J. The roles of SSU processome components and surveillance factors in the initial processing of human ribosomal RNA. RNA 2014, 20, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Fayet-Lebaron, E.; Atzorn, V.; Henry, Y.; Kiss, T. 18S rRNA processing requires base pairings of snR30 H/ACA snoRNA to eukaryote-specific 18S sequences. EMBO J. 2009, 28, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.R.; Weichmann, F.; Sloan, K.E.; Colvin, D.; Watkins, N.J.; Schneider, C. The ribosome biogenesis factor yUtp23/hUTP23 coordinates key interactions in the yeast and human pre-40S particle and hUTP23 contains an essential PIN domain. Nucleic Acids Res. 2017, 45, 4796–4809. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, K.C.; Cech, T.R. Targeted CRISPR disruption reveals a role for RNase MRP RNA in human preribosomal RNA processing. Genes Dev. 2017, 31, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Anikin, L.; Pestov, D.G. Two orthogonal cleavages separate subunit RNAs in mouse ribosome biogenesis. Nucleic Acids Res. 2014, 42, 11180–11191. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.D.; Trahan, C.; Zindy, P.J.; Aguilar, L.C.; Delubac, M.Y.; Van Nostrand, E.L.; Adivarahan, S.; Wei, K.E.; Yeo, G.W.; Zenklusen, D.; et al. Nol12 is a multifunctional RNA binding protein at the nexus of RNA and DNA metabolism. Nucleic Acids Res. 2017, 45, 12509–12528. [Google Scholar] [CrossRef] [PubMed]
- Borovjagin, A.V.; Gerbi, S.A. Xenopus U3 snoRNA docks on pre-rRNA through a novel base-pairing interaction. RNA 2004, 10, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.R.; Weichmann, F.; Colvin, D.; Sloan, K.E.; Kudla, G.; Tollervey, D.; Watkins, N.J.; Schneider, C. The PIN domain endonuclease Utp24 cleaves pre-ribosomal RNA at two coupled sites in yeast and humans. Nucleic Acids Res. 2016, 44, 5399–5409. [Google Scholar] [CrossRef] [PubMed]
- Tomecki, R.; Labno, A.; Drazkowska, K.; Cysewski, D.; Dziembowski, A. hUTP24 is essential for processing of the human rRNA precursor at site A1, but not at site A0. RNA Biol. 2015, 12, 1010–1029. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Yoshikawa, H.; Izumikawa, K.; Miura, Y.; Taoka, M.; Nobe, Y.; Yamauchi, Y.; Nakayama, H.; Simpson, R.J.; Isobe, T. Poly(A)-specific ribonuclease regulates the processing of small-subunit rRNAs in human cells. Nucleic Acids Res. 2016, gkw1047. [Google Scholar] [CrossRef] [PubMed]
- Son, A.; Park, J.-E.; Kim, V.N. PARN and TOE1 Constitute a 3′ End Maturation Module for Nuclear Noncoding RNAs. Cell Rep. 2018, 23, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Park, J.; Ha, M.; Lim, J.; Chang, H.; Kim, V.N. PABP Cooperates with the CCR4-NOT Complex to Promote mRNA Deadenylation and Block Precocious Decay. Mol. Cell 2018, 70, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Berndt, H.; Harnisch, C.; Rammelt, C.; Stohr, N.; Zirkel, A.; Dohm, J.C.; Himmelbauer, H.; Tavanez, J.P.; Huttelmaier, S.; Wahle, E. Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA 2012, 18, 958–972. [Google Scholar] [CrossRef] [PubMed]
- Yoda, M.; Cifuentes, D.; Izumi, N.; Sakaguchi, Y.; Suzuki, T.; Giraldez, A.J.; Tomari, Y. Poly(A)-specific ribonuclease mediates 3′-end trimming of Argonaute2-cleaved precursor microRNAs. Cell Rep. 2013, 5, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Katoh, T.; Hojo, H.; Suzuki, T. Destabilization of microRNAs in human cells by 3′ deadenylation mediated by PARN and CUGBP1. Nucleic Acids Res. 2015, 43, 7521–7534. [Google Scholar] [CrossRef] [PubMed]
- Izumi, N.; Shoji, K.; Sakaguchi, Y.; Honda, S.; Kirino, Y.; Suzuki, T.; Katsuma, S.; Tomari, Y. Identification and Functional Analysis of the Pre-piRNA 3′ Trimmer in Silkworms. Cell 2016, 164, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.H.; Segal, M.; Boyraz, B.; Guinan, E.; Hofmann, I.; Cahan, P.; Tai, A.K.; Agarwal, S. Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat. Genet. 2015, 47, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.K.; Wang, H.F.; Burns, A.M.; Schroeder, M.R.; Gaspari, M.; Baumann, P. Human Telomerase RNA Processing and Quality Control. Cell Rep. 2015, 13, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Grenier St-Sauveur, V.; Bergeron, D.; Dupuis-Sandoval, F.; Scott, M.S.; Bachand, F. A Polyadenylation-Dependent 3′ End Maturation Pathway Is Required for the Synthesis of the Human Telomerase RNA. Cell Rep. 2015, 13, 2244–2257. [Google Scholar] [CrossRef] [PubMed]
- Rouquette, J.; Choesmel, V.; Gleizes, P.E. Nuclear export and cytoplasmic processing of precursors to the 40S ribosomal subunits in mammalian cells. EMBO J. 2005, 24, 2862–2872. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Zhang, J.; Li, T.; Hang, R.; Liu, Y.; Tian, Y.; Huang, D.; Qu, L.; Cao, X.; Ji, J.; et al. The ATPase hCINAP regulates 18S rRNA processing and is essential for embryogenesis and tumour growth. Nat. Commun. 2016, 7, 12310. [Google Scholar] [CrossRef] [PubMed]
- Gallouzi, I.E.; Wilusz, J. A DIStinctively novel exoribonuclease that really likes U. EMBO J. 2013, 32, 1799–1801. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Pestov, D.G. 5′-end surveillance by Xrn2 acts as a shared mechanism for mammalian pre-rRNA maturation and decay. Nucleic Acids Res. 2011, 39, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Schillewaert, S.; Wacheul, L.; Lhomme, F.; Lafontaine, D.L. The evolutionarily conserved protein Las1 is required for pre-rRNA processing at both ends of ITS2. Mol. Cell. Biol. 2012, 32, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Schilders, G.; van Dijk, E.; Pruijn, G.J. C1D and hMtr4p associate with the human exosome subunit PM/Scl-100 and are involved in pre-rRNA processing. Nucleic Acids Res. 2007, 35, 2564–2572. [Google Scholar] [CrossRef] [PubMed]
- Coute, Y.; Kindbeiter, K.; Belin, S.; Dieckmann, R.; Duret, L.; Bezin, L.; Sanchez, J.C.; Diaz, J.J. ISG20L2, a novel vertebrate nucleolar exoribonuclease involved in ribosome biogenesis. Mol. Cell. Proteomics 2008, 7, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.; Rothblum, L.I.; Subrahmanyam, C.S.; Liu, M.H.; Henning, D.; Cassidy, B.; Busch, H. The nucleotide sequence of 8 S RNA bound to preribosomal RNA of Novikoff hepatoma. The 5′-end of 8 S RNA is 5.8 S RNA. J. Biol. Chem. 1983, 258, 584–589. [Google Scholar] [PubMed]
- Michot, B.; Joseph, N.; Mazan, S.; Bachellerie, J.P. Evolutionarily conserved structural features in the ITS2 of mammalian pre-rRNAs and potential interactions with the snoRNA U8 detected by comparative analysis of new mouse sequences. Nucleic Acids Res. 1999, 27, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Ansel, K.M.; Pastor, W.A.; Rath, N.; Lapan, A.D.; Glasmacher, E.; Wolf, C.; Smith, L.C.; Papadopoulou, N.; Lamperti, E.D.; Tahiliani, M.; et al. Mouse Eri1 interacts with the ribosome and catalyzes 5.8S rRNA processing. Nat. Struct. Mol. Biol. 2008, 15, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Fedoriw, A.M.; Starmer, J.; Yee, D.; Magnuson, T. Nucleolar association and transcriptional inhibition through 5S rDNA in mammals. PLoS Genet. 2012, 8, e1002468. [Google Scholar] [CrossRef] [PubMed]
- Engelke, D.R.; Ng, S.Y.; Shastry, B.S.; Roeder, R.G. Specific interaction of a purified transcription factor with an internal control region of 5S RNA genes. Cell 1980, 19, 717–728. [Google Scholar] [CrossRef]
- Moorefield, B.; Roeder, R.G. Purification and characterization of human transcription factor IIIA. J. Biol. Chem. 1994, 269, 20857–20865. [Google Scholar] [PubMed]
- Dieci, G.; Fiorino, G.; Castelnuovo, M.; Teichmann, M.; Pagano, A. The expanding RNA polymerase III transcriptome. Trends Genet. 2007, 23, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Dieci, G.; Conti, A.; Pagano, A.; Carnevali, D. Identification of RNA polymerase III-transcribed genes in eukaryotic genomes. Biochim. Biophys. Acta BBA–Gene Regul. Mech. 2013, 1829, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Rinke, J.; Steitz, J.A. Precursor molecules of both human 5S ribosomal RNA and transfer RNAs are bound by a cellular protein reactive with anti-La Lupus antibodies. Cell 1982, 29, 149–159. [Google Scholar] [CrossRef]
- Silva, S.; Homolka, D.; Pillai, R.S. Characterization of the mammalian RNA exonuclease 5/NEF-sp as a testis-specific nuclear 3′ → 5′ exoribonuclease. RNA 2017, 23, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Steitz, J.A.; Berg, C.; Hendrick, J.P.; Branche-Chabot, H.L.; Metspalu, A.; Rinke, J.; Yario, T. A 5S rRNA/L5 complex is a precursor to ribosome assembly in mammalian cells. J. Cell Biol. 1988, 106, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Harnpicharnchai, P.; Jakovljevic, J.; Tang, L.; Guo, Y.; Oeffinger, M.; Rout, M.P.; Hiley, S.L.; Hughes, T.; Woolford, J.L. Assembly factors Rpf2 and Rrs1 recruit 5S rRNA and ribosomal proteins rpL5 and rpL11 into nascent ribosomes. Genes Dev. 2007, 21, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Bange, G.; Ogawa, Y.; Stjepanovic, G.; Bradatsch, B.; Pratte, D.; Amlacher, S.; Strauss, D.; Yoneda, Y.; Katahira, J.; et al. Synchronizing Nuclear Import of Ribosomal Proteins with Ribosome Assembly. Science 2012, 338, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Madru, C.; Lebaron, S.; Blaud, M.; Delbos, L.; Pipoli, J.; Pasmant, E.; Réty, S.; Leulliot, N. Chaperoning 5S RNA assembly. Genes Dev. 2015, 29, 1432–1446. [Google Scholar] [CrossRef] [PubMed]
- Calviño, F.R.; Kharde, S.; Ori, A.; Hendricks, A.; Wild, K.; Kressler, D.; Bange, G.; Hurt, E.; Beck, M.; Sinning, I. Symportin 1 chaperones 5S RNP assembly during ribosome biogenesis by occupying an essential rRNA-binding site. Nat. Commun. 2015, 6, 6510. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Bohnsack, M.T.; Watkins, N.J. The 5S RNP Couples p53 Homeostasis to Ribosome Biogenesis and Nucleolar Stress. Cell Rep. 2013, 5, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Parisot, P.; Pinto-Monteiro, C.; de Walque, R.; Vleeschouwer, C.D.; Lafontaine, D.L.J. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 2016, 7, 11390. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, R.D.; Weinberg, R.A.; Penman, S. Unusual metabolism of 5 S RNA in HeLa cells. J. Mol. Biol. 1973, 73, 139–144. [Google Scholar] [CrossRef]
- Donati, G.; Peddigari, S.; Mercer, C.A.; Thomas, G. 5S Ribosomal RNA Is an Essential Component of a Nascent Ribosomal Precursor Complex that Regulates the Hdm2-p53 Checkpoint. Cell Rep. 2013, 4, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.; Narla, A.; Mohandas, N. An update on the pathogenesis and diagnosis of Diamond–Blackfan anemia. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.R.; Gleizes, P.-E. Diamond Blackfan anemia: Ribosomal proteins going rogue. Semin. Hematol. 2011, 48, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The Genetic Landscape of Diamond–Blackfan Anemia. bioRxiv 2018, 365890. [Google Scholar] [CrossRef]
- Gregory, L.A.; Aguissa-Touré, A.-H.; Pinaud, N.; Legrand, P.; Gleizes, P.-E.; Fribourg, S. Molecular basis of Diamond–Blackfan anemia: Structure and function analysis of RPS19. Nucleic Acids Res. 2007, 35, 5913–5921. [Google Scholar] [CrossRef] [PubMed]
- Angelini, M.; Cannata, S.; Mercaldo, V.; Gibello, L.; Santoro, C.; Dianzani, I.; Loreni, F. Missense mutations associated with Diamond–Blackfan anemia affect the assembly of ribosomal protein S19 into the ribosome. Hum. Mol. Genet. 2007, 16, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Cerca, S.; Poll, G.; Gleizes, P.E.; Tschochner, H.; Milkereit, P. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol. Cell 2005, 20, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Gazda, H.T.; Sheen, M.R.; Vlachos, A.; Choesmel, V.; O’Donohue, M.F.; Schneider, H.; Darras, N.; Hasman, C.; Sieff, C.A.; Newburger, P.E.; et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond–Blackfan anemia patients. Am. J. Hum. Genet. 2008, 83, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Choesmel, V.; Fribourg, S.; Aguissa-Toure, A.H.; Pinaud, N.; Legrand, P.; Gazda, H.T.; Gleizes, P.E. Mutation of ribosomal protein RPS24 in Diamond–Blackfan anemia results in a ribosome biogenesis disorder. Hum. Mol. Genet. 2008, 17, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Quarello, P.; Garelli, E.; Carando, A.; Mancini, C.; Foglia, L.; Botto, C.; Farruggia, P.; De Keersmaecker, K.; Aspesi, A.; Ellis, S.R.; et al. Ribosomal RNA analysis in the diagnosis of Diamond–Blackfan Anaemia. Br. J. Haematol. 2016, 172, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.; O’Donohue, M.-F.; van Dooijeweert, B.; Albrecht, K.; Unal, S.; Ramenghi, U.; Leblanc, T.; Dianzani, I.; Tamary, H.; Bartels, M.; et al. Molecular approaches to diagnose Diamond–Blackfan anemia: The EuroDBA experience. Eur. J. Med. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.E.; Quarello, P.; Fisher, R.; O’Brien, K.A.; Aspesi, A.; Parrella, S.; Henson, A.L.; Seidel, N.E.; Atsidaftos, E.; Prakash, S.; et al. Exploiting pre-rRNA processing in Diamond Blackfan anemia gene discovery and diagnosis. Am. J. Hematol. 2014, 89, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Curry, C.; Olney, A.H.; Sandoval, C.; Fisher, J.; Chong, J.X.L.; Pilchman, L.; Sahraoui, R.; Stabley, D.L.; Sol-Church, K. Diamond–Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am. J. Med. Genet. A 2014, 164, 2240–2249. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Ghazvinian, R.; Do, R.; Thiru, P.; Vergilio, J.-A.; Beggs, A.H.; Sieff, C.A.; Orkin, S.H.; Nathan, D.G.; Lander, E.S.; et al. Exome sequencing identifies GATA1 mutations resulting in Diamond–Blackfan anemia. J. Clin. Investig. 2012, 122, 2439–2443. [Google Scholar] [CrossRef] [PubMed]
- Klar, J.; Khalfallah, A.; Arzoo, P.S.; Gazda, H.T.; Dahl, N. Recurrent GATA1 mutations in Diamond–Blackfan anaemia. Br. J. Haematol. 2014, 166, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Parrella, S.; Aspesi, A.; Quarello, P.; Garelli, E.; Pavesi, E.; Carando, A.; Nardi, M.; Ellis, S.R.; Ramenghi, U.; Dianzani, I. Loss of GATA-1 full length as a cause of Diamond–Blackfan anemia phenotype. Pediatr. Blood Cancer 2014, 61, 1319–1321. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, L.S.; Gazda, H.T.; Eng, J.C.; Eichhorn, S.W.; Thiru, P.; Ghazvinian, R.; George, T.I.; Gotlib, J.R.; Beggs, A.H.; Sieff, C.A.; et al. Altered translation of GATA1 in Diamond–Blackfan anemia. Nat. Med. 2014, 20, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yoshida, K.; Toki, T.; Sawada, T.; Uechi, T.; Okuno, Y.; Sato-Otsubo, A.; Kudo, K.; Kamimaki, I.; Kanezaki, R.; et al. Loss of function mutations in RPL27 and RPS27 identified by whole-exome sequencing in Diamond–Blackfan anaemia. Br. J. Haematol. 2015, 168, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Smetanina, N.S.; Mersiyanova, I.V.; Kurnikova, M.A.; Ovsyannikova, G.S.; Hachatryan, L.A.; Bobrynina, V.O.; Maschan, M.A.; Novichkova, G.A.; Lipton, J.M.; Maschan, A.A. Clinical and genomic heterogeneity of Diamond Blackfan anemia in the Russian Federation. Pediatr. Blood Cancer 2015, 62, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Yoshida, K.; Okuno, Y.; Yujiri, T.; Nagai, K.; Nishi, M.; Shiraishi, Y.; Ueno, H.; Toki, T.; Chiba, K.; et al. Diagnostic challenge of Diamond–Blackfan anemia in mothers and children by whole-exome sequencing. Int. J. Hematol. 2017, 105, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Doherty, L.; Sheen, M.R.; Vlachos, A.; Choesmel, V.; O’Donohue, M.F.; Clinton, C.; Schneider, H.E.; Sieff, C.A.; Newburger, P.E.; Ball, S.E.; et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond–Blackfan anemia. Am. J. Hum. Genet. 2010, 86, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Yoshida, K.; Toki, T.; Uechi, T.; Ishida, S.; Nakajima, Y.; Sasahara, Y.; Okuno, Y.; Kanezaki, R.; Terui, K.; et al. Exome sequencing identified RPS15A as a novel causative gene for Diamond–Blackfan anemia. Haematologica 2017, 102, e93–e96. [Google Scholar] [CrossRef] [PubMed]
- Cmejla, R.; Cmejlova, J.; Handrkova, H.; Petrak, J.; Pospisilova, D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond–Blackfan anemia. Hum. Mutat. 2007, 28, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-J.; Yoo, E.-H.; Lee, K.-O.; Kim, G.-N.; Kim, H.-J.; Kim, S.-Y.; Kim, S.-H. A novel initiation codon mutation in the ribosomal protein S17 gene (RPS17) in a patient with Diamond–Blackfan anemia. Pediatr. Blood Cancer 2009, 54. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Toki, T.; Tandai, S.; Xu, G.; Wang, R.N.; Terui, K.; Ohga, S.; Hara, T.; Hama, A.; Kojima, S.; et al. Mutations in the ribosomal protein genes in Japanese patients with Diamond–Blackfan anemia. Haematologica 2010, 95, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.E.; Vlachos, A.; Atsidaftos, E.; Carlson-Donohoe, H.; Markello, T.C.; Arceci, R.J.; Ellis, S.R.; Lipton, J.M.; Bodine, D.M. Ribosomal protein gene deletions in Diamond–Blackfan anemia. Blood 2011, 118, 6943–6951. [Google Scholar] [CrossRef] [PubMed]
- Draptchinskaia, N.; Gustavsson, P.; Andersson, B.; Pettersson, M.; Willig, T.N.; Dianzani, I.; Ball, S.; Tchernia, G.; Klar, J.; Matsson, H.; et al. The gene encoding ribosomal protein S19 is mutated in Diamond- Blackfan anaemia. Nat. Genet. 1999, 21, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Gazda, H.T.; Grabowska, A.; Merida-Long, L.B.; Latawiec, E.; Schneider, H.E.; Lipton, J.M.; Vlachos, A.; Atsidaftos, E.; Ball, S.E.; Orfali, K.A.; et al. Ribosomal protein S24 gene is mutated in Diamond–Blackfan anemia. Am. J. Hum. Genet. 2006, 79, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Macari, E.R.; Jessop, L.; Ellis, S.R.; Myers, T.; Giri, N.; Alison, M.; Mcgrath, K.E.; Humphries, J.M.; Ballew, B.J.; et al. Whole-exome sequencing and functional studies identifyRPS29 as a novel gene mutated in multi-case Diamond–Blackfan anemia families. Blood 2014, 124, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Landowski, M.; O’Donohue, M.F.; Buros, C.; Ghazvinian, R.; Montel-Lehry, N.; Vlachos, A.; Sieff, C.A.; Newburger, P.E.; Niewiadomska, E.; Matysiak, M.; et al. Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond–Blackfan anemia. Hum. Genet. 2013, 132, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Khincha, P.P.; Ellis, S.R.; Giri, N.; Brodie, S.; Chandrasekharappa, S.C.; Donovan, F.X.; Zhou, W.; Hicks, B.D.; Boland, J.F.; et al. Novel and known ribosomal causes of Diamond–Blackfan anaemia identified through comprehensive genomic characterisation. J. Med. Genet. 2017, 54, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Gazda, H.T.; Preti, M.; Sheen, M.R.; O’Donohue, M.F.; Vlachos, A.; Davies, S.M.; Kattamis, A.; Doherty, L.; Landowski, M.; Buros, C.; et al. Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in Diamond–Blackfan anemia. Hum. Mutat. 2012, 33, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.E.; Nater, M.; Caywood, E.; McDevitt, M.A.; Kowalski, J.; Takemoto, C.M.; Talbot, C.C.; Meltzer, P.; Esposito, D.; Beggs, A.H.; et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond–Blackfan anemia. Blood 2008, 112, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q-syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Bolze, A.; Mahlaoui, N.; Byun, M.; Turner, B.; Trede, N.; Ellis, S.R.; Abhyankar, A.; Itan, Y.; Patin, E.; Brebner, S.; et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science 2013, 340, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Paolini, N.A.; Attwood, M.; Sondalle, S.B.; dos Santos Vieira, C.M.; van Adrichem, A.M.; di Summa, F.M.; O’Donohue, M.-F.; Gleizes, P.-E.; Rachuri, S.; Briggs, J.W.; et al. A Ribosomopathy Reveals Decoding Defective Ribosomes Driving Human Dysmorphism. Am. J. Hum. Genet. 2017, 100, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.S.; Wall, A.L.; Golzio, C.; Reid, D.W.; Kondyles, A.; Willer, J.R.; Botti, C.; Nicchitta, C.V.; Katsanis, N.; Davis, E.E. A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X-linked microcephaly in humans. Genetics 2014, 198, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Thevenon, J.; Michot, C.; Bole, C.; Nitschke, P.; Nizon, M.; Faivre, L.; Munnich, A.; Lyonnet, S.; Bonnefont, J.P.; Portes, V.D.; et al. RPL10 mutation segregating in a family with X-linked syndromic Intellectual Disability. Am. J. Med. Genet. A 2015, 167, 1908–1912. [Google Scholar] [CrossRef] [PubMed]
- Zanni, G.; Kalscheuer, V.M.; Friedrich, A.; Barresi, S.; Alfieri, P.; Di Capua, M.; Haas, S.A.; Piccini, G.; Karl, T.; Klauck, S.M.; et al. A Novel Mutation in RPL10 (Ribosomal Protein L10) Causes X-Linked Intellectual Disability, Cerebellar Hypoplasia, and Spondylo-Epiphyseal Dysplasia. Hum. Mutat. 2015, 36, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Klauck, S.M.; Felder, B.; Kolb-Kokocinski, A.; Schuster, C.; Chiocchetti, A.; Schupp, I.; Wellenreuther, R.; Schmötzer, G.; Poustka, F.; Breitenbach-Koller, L.; et al. Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol. Psychiatry 2006, 11, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Chiocchetti, A.; Pakalapati, G.; Duketis, E.; Wiemann, S.; Poustka, A.; Poustka, F.; Klauck, S.M. Mutation and expression analyses of the ribosomal protein gene RPL10 in an extended German sample of patients with autism spectrum disorder. Am. J. Med. Genet. A 2011, 155, 1472–1475. [Google Scholar] [CrossRef] [PubMed]
- Bourque, D.K.; Hartley, T.; Nikkel, S.M.; Pohl, D.; Tétreault, M.; Kernohan, K.D.; Dyment, D.A. A de novo mutation in RPL10 causes a rare X-linked ribosomopathy characterized by syndromic intellectual disability and epilepsy: A new case and review of the literature. Eur. J. Med. Genet. 2018, 61, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Boocock, G.R.B.; Morrison, J.A.; Popovic, M.; Richards, N.; Ellis, L.; Durie, P.R.; Rommens, J.M. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 2003, 33, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Traynor, D.; Basse, N.; Kay, R.R.; Warren, A.J. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood 2011, 118, 4305–4312. [Google Scholar] [CrossRef] [PubMed]
- Tummala, H.; Walne, A.J.; Williams, M.; Bockett, N.; Collopy, L.; Cardoso, S.; Ellison, A.; Wynn, R.; Leblanc, T.; Fitzgibbon, J.; et al. DNAJC21 Mutations Link a Cancer-Prone Bone Marrow Failure Syndrome to Corruption in 60S Ribosome Subunit Maturation. Am. J. Hum. Genet. 2016, 99, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dhanraj, S.; Matveev, A.; Li, H.; Lauhasurayotin, S.; Jardine, L.; Cada, M.; Zlateska, B.; Tailor, C.S.; Zhou, J.; Mendoza-Londono, R.; et al. Biallelic mutations in DNAJC21 cause Shwachman-Diamond syndrome. Blood 2017, 129, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Morini, J.; Nacci, L.; Babini, G.; Cesaro, S.; Valli, R.; Ottolenghi, A.; Nicolis, E.; Pintani, E.; Maserati, E.; Cipolli, M.; et al. Whole exome sequencing discloses heterozygous variants in the DNAJC21 and EFL1 genes but not in SRP54 in 6 out of 16 patients with Shwachman-Diamond Syndrome carrying biallelic SBDS mutations. Br. J. Haematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stepensky, P.; Chacón-Flores, M.; Kim, K.H.; Abuzaitoun, O.; Bautista-Santos, A.; Simanovsky, N.; Siliqi, D.; Altamura, D.; Méndez-Godoy, A.; Gijsbers, A.; et al. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in aShwachman-Diamond like syndrome. J. Med. Genet. 2017, 54, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Armistead, J.; Khatkar, S.; Meyer, B.; Mark, B.L.; Patel, N.; Coghlan, G.; Lamont, R.E.; Liu, S.; Wiechert, J.; Cattini, P.A.; et al. Mutation of a Gene Essential for Ribosome Biogenesis, EMG1, Causes Bowen-Conradi Syndrome. Am. J. Hum. Genet. 2009, 84, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Wurm, J.P.; Kötter, P.; Leisegang, M.S.; Schilling, V.; Buchhaupt, M.; Held, M.; Bahr, U.; Karas, M.; Heckel, A.; et al. The Bowen-Conradi syndrome protein Nep1 (Emg1) has a dual role in eukaryotic ribosome biogenesis, as an essential assembly factor and in the methylation of Ψ1191 in yeast 18S rRNA. Nucleic Acids Res. 2011, 39, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Warda, A.S.; Freytag, B.; Haag, S.; Sloan, K.E.; Görlich, D.; Bohnsack, M.T. Effects of the Bowen-Conradi syndrome mutation in EMG1 on its nuclear import, stability and nucleolar recruitment. Hum. Mol. Genet. 2016, 25, 5353–5364. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.F.; Baserga, S.J. The C-terminus of Utp4, mutated in childhood cirrhosis, is essential for ribosome biogenesis. Nucleic Acids Res. 2010, 38, 4798–4806. [Google Scholar] [CrossRef] [PubMed]
- Chagnon, P.; Michaud, J.; Mitchell, G.; Mercier, J.; Marion, J.-F.; Drouin, E.; Rasquin-Weber, A.; Hudson, T.J.; Richter, A. A missense mutation (R565W) in cirhin (FLJ14728) in North American Indian childhood cirrhosis. Am. J. Hum. Genet. 2002, 71, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Marneros, A.G. BMS1 is mutated in aplasia cutis congenita. PLoS Genet. 2013, 9, e1003573. [Google Scholar] [CrossRef] [PubMed]
- Ridanpää, M.; Van Eenennaam, H.; Pelin, K.; Chadwick, R.; Johnson, C.; Yuan, B.; VanVenrooij, W.; Pruijn, G.; Salmela, R.; Rockas, S.; et al. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell 2001, 104, 195–203. [Google Scholar] [CrossRef]
- Heiss, N.S.; Knight, S.W.; Vulliamy, T.J.; Klauck, S.M.; Wiemann, S.; Mason, P.J.; Poustka, A.; Dokal, I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998, 19, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.W.; Heiss, N.S.; Vulliamy, T.J.; Greschner, S.; Stavrides, G.; Pai, G.S.; Lestringant, G.; Varma, N.; Mason, P.J.; Dokal, I.; et al. X-Linked Dyskeratosis Congenita Is Predominantly Caused by Missense Mutations in the DKC1 Gene. Am. J. Hum. Genet. 1999, 65, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Walne, A.J.; Vulliamy, T.; Marrone, A.; Beswick, R.; Kirwan, M.; Masunari, Y.; Al-Qurashi, F.H.; Aljurf, M.; Dokal, I. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum. Mol. Genet. 2007, 16, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Vulliamy, T.; Beswick, R.; Kirwan, M.; Marrone, A.; Digweed, M.; Walne, A.; Dokal, I. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc. Natl. Acad. Sci. USA 2008, 105, 8073–8078. [Google Scholar] [CrossRef] [PubMed]
- Dhanraj, S.; Gunja, S.M.; Deveau, A.P.; Nissbeck, M.; Boonyawat, B.; Coombs, A.J.; Renieri, A.; Mucciolo, M.; Marozza, A.; Buoni, S.; et al. Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN). J. Med. Genet. 2015, 52, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Tummala, H.; Walne, A.; Collopy, L.; Cardoso, S.; de la Fuente, J.; Lawson, S.; Powell, J.; Cooper, N.; Foster, A.; Mohammed, S.; et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J. Clin. Investig. 2015, 125, 2151–2160. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.; Oldridge, M.; Archer, C.; O’Rourke, A.; McParland, J.; Brekelmans, R.; Seller, A.; Lester, T. Gross deletions in TCOF1 are a cause of Treacher–Collins–Franceschetti syndrome. Eur. J. Hum. Genet. 2012, 20, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Dauwerse, J.G.; Dixon, J.; Seland, S.; Ruivenkamp, C.A.; van Haeringen, A.; Hoefsloot, L.H.; Peters, D.J.; Boers, A.C.; Daumer-Haas, C.; Maiwald, R.; et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 2011, 43, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Kadakia, S.; Helman, S.N.; Badhey, A.K.; Saman, M.; Ducic, Y. Treacher Collins Syndrome: The genetics of a craniofacial disease. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.; Collet, C.; Genevieve, D.; Vincent, M.; Lohmann, D.R.; Sanchez, E.; Bolender, C.; Eliot, M.M.; Nürnberg, G.; Passos-Bueno, M.R.; et al. Autosomal recessive POLR1D mutation with decrease of TCOF1 mRNA is responsible for Treacher Collins syndrome. Genet. Med. 2014, 16, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.N.; Watt, K.E.N.; Hufnagel, R.B.; Navajas Acedo, J.; Linscott, L.L.; Sund, K.L.; Bender, P.L.; König, R.; Lourenco, C.M.; Hehr, U.; et al. Acrofacial Dysostosis, Cincinnati Type, a Mandibulofacial Dysostosis Syndrome with Limb Anomalies, Is Caused by POLR1A Dysfunction. Am. J. Hum. Genet. 2015, 96, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Kara, B.; Köroğlu, Ç.; Peltonen, K.; Steinberg, R.C.; Maraş Genç, H.; Hölttä-Vuori, M.; Güven, A.; Kanerva, K.; Kotil, T.; Solakoğlu, S.; et al. Severe neurodegenerative disease in brothers with homozygous mutation in POLR1A. Eur. J. Hum. Genet. EJHG 2017, 25, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Sinturel, F.; Gachon, F. Diurnal liver mass is associated with ribosome biogenesis. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.; Owens, N.D.L.; Baserga, S.J.; Khokha, M.K.; Griffin, J.N. Expression of ribosomopathy genes during Xenopus tropicalis embryogenesis. BMC Dev. Biol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358, eaan2755. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.F.; Desalu, O. Regulation of synthesis of non-globin proteins in cell-free extracts of rabbit reticulocytes. J. Biol. Chem. 1973, 248, 3420–3427. [Google Scholar] [PubMed]
- Van de Waterbeemd, M.; Tamara, S.; Fort, K.L.; Damoc, E.; Franc, V.; Bieri, P.; Itten, M.; Makarov, A.; Ban, N.; Heck, A.J.R. Dissecting ribosomal particles throughout the kingdoms of life using advanced hybrid mass spectrometry methods. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.M.; Kurylo, C.M.; Dass, R.A.; Bojmar, L.; Lyden, D.; Vincent, C.T.; Blanchard, S.C. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv. 2018, 4, eaao0665. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, S.; Manakongtreecheep, K.; Söll, D. Revising the Structural Diversity of Ribosomal Proteins across the Three Domains of Life. Mol. Biol. Evol. 2018, 35, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Marchand, V.; Motorin, Y.; Lafontaine, D.L.J. Identification of sites of 2′-O-methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, N.; Pusic, A.; Stumpf, C.R.; Shimizu, K.; Hsieh, A.C.; Xue, S.; Ishijima, J.; Shiroishi, T.; Barna, M. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 2011, 145, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Genuth, N.R.; Barna, M. The Discovery of Ribosome Heterogeneity and Its Implications for Gene Regulation and Organismal Life. Mol. Cell 2018, 71, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Hurt, E.; Baßler, J. A Puzzle of Life: Crafting Ribosomal Subunits. Trends Biochem. Sci. 2017, 42, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Peña, C.; Hurt, E.; Panse, V.G. Eukaryotic ribosome assembly, transport and quality control. Nat. Struct. Mol. Biol. 2017, 24, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.L.J. A ‘garbage can’ for ribosomes: how eukaryotes degrade their ribosomes. Trends Biochem. Sci. 2010, 35, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Strunk, B.S.; Novak, M.N.; Young, C.L.; Karbstein, K. A Translation-Like Cycle Is a Quality Control Checkpoint for Maturing 40S Ribosome Subunits. Cell 2012, 150, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.E.; LaRiviere, F.J.; Merrikh, C.N.; Moore, M.J. A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol. Cell 2009, 34, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J.; Bennett, E.J. Protecting the proteome: Eukaryotic cotranslational quality control pathways. J. Cell Biol. 2014, 204, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Kitabatake, M.; Sakata, T.; Ohno, M. 40S subunit dissociation and proteasome-dependent RNA degradation in nonfunctional 25S rRNA decay. EMBO J. 2012, 31, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Thoms, M.; Barrio-Garcia, C.; Thomson, E.; Flemming, D.; Beckmann, R.; Hurt, E. Preribosomes escaping from the nucleus are caught during translation by cytoplasmic quality control. Nat. Struct. Mol. Biol. 2017, 24, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Soudet, J.; Gélugne, J.-P.; Belhabich-Baumas, K.; Caizergues-Ferrer, M.; Mougin, A. Immature small ribosomal subunits can engage in translation initiation in Saccharomyces cerevisiae. EMBO J. 2010, 29, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Galán, O.; García-Gómez, J.J.; Kressler, D.; de la Cruz, J. Immature large ribosomal subunits containing the 7S pre-rRNA can engage in translation in Saccharomyces cerevisiae. RNA Biol. 2015, 12, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Belhabich-Baumas, K.; Joret, C.; Jády, B.E.; Plisson-Chastang, C.; Shayan, R.; Klopp, C.; Henras, A.K.; Henry, Y.; Mougin, A. The Rio1p ATPase hinders premature entry into translation of late pre-40S pre-ribosomal particles. Nucleic Acids Res. 2017, 45, 10824–10836. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.B.; Ghalei, H.; Ward, E.A.; Potts, E.L.; Karbstein, K. Rps26 directs mRNA-specific translation by recognition of Kozak sequence elements. Nat. Struct. Mol. Biol. 2017, 24, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Jansson, M.D.; Häfner, S.J.; Tehler, D.; Birkedal, U.; Christensen-Dalsgaard, M.; Lund, A.H.; Nielsen, H. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016, 44, 7884–7895. [Google Scholar] [CrossRef] [PubMed]
- Erales, J.; Marchand, V.; Panthu, B.; Gillot, S.; Belin, S.; Ghayad, S.E.; Garcia, M.; Laforêts, F.; Marcel, V.; Baudin-Baillieu, A.; et al. Evidence for rRNA 2′-O-methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proc. Natl. Acad. Sci. USA 2017, 114, 12934–12939. [Google Scholar] [CrossRef] [PubMed]
- Bundred, J.R.; Hendrix, E.; Coleman, M.L. The emerging roles of ribosomal histidyl hydroxylases in cell biology, physiology and disease. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, T.; Singh, A.K.; Veland, N.; Vemulapalli, V.; Chen, J.; Hardikar, S.; Bao, J.; Fry, C.J.; Yang, V.; Lee, K.A.; et al. Identification of Rpl29 as a major substrate of the lysine methyltransferase Set7/9. J. Biol. Chem. 2018, 293, 12770–12780. [Google Scholar] [CrossRef] [PubMed]
- Loenarz, C.; Sekirnik, R.; Thalhammer, A.; Ge, W.; Spivakovsky, E.; Mackeen, M.M.; McDonough, M.; Cockman, M.E.; Kessler, B.; Ratcliffe, P.; et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc. Natl. Acad. Sci. USA 2014, 111. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.C.; Lynn, M.L.; Gaudenz, K.; Sakai, D.; Aoto, K.; Rey, J.-P.; Glynn, E.F.; Ellington, L.; Du, C.; Dixon, J.; et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 2008, 14, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Bursac, S.; Brdovcak, M.C.; Pfannkuchen, M.; Orsolic, I.; Golomb, L.; Zhu, Y.; Katz, C.; Daftuar, L.; Grabusic, K.; Vukelic, I.; et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc. Natl. Acad. Sci. USA 2012, 109, 20467–20472. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-S.; Arnold, H.; Sun, X.-X.; Sears, R.; Lu, H. Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 2007, 26, 3332–3345. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.J.; Kuiper, E.G.; Jones, S.K.; Leung, S.W.; Corbett, A.H.; Fasken, M.B. The RNA exosome and RNA exosome-linked disease. RNA 2018, 24, 127–142. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Manifestation/Clinical Features | Frequency | Inheritance | Genes Involved in Ribosome Biogenesis | Proteins | Functional Validation of Impact on Ribosome Biogenesis | % of Patients | References | |
|---|---|---|---|---|---|---|---|---|
| Patient Cells | Models * | |||||||
| Diamond-Blackfan anemia | ||||||||
| Aregenerative macrocytic anemia, high eADA levels, short stature, craniofacial and upper limb anomalies, heart or genitourinary malformations, predisposition to MDS, AML, and solid tumors | 1:150,000–200,000 births | Autosomal dominant | RPS7 | RPS7 | no | yes | <0.1% | [109,120,121,122] |
| RPS10 | RPS10 | yes | yes | 3% | [123] | |||
| RPS15A | RPS15A | yes | yes | <1% | [124] | |||
| RPS17 | RPS17 | yes | yes | 1% | [109,125,126,127,128] | |||
| RPS19 | RPS19 | yes | yes | 25% | [41,42,129] | |||
| RPS24 | RPS24 | yes | yes | 2.4% | [110,130] | |||
| RPS26 | RPS26 | yes | yes | 6.6% | [123] | |||
| RPS27 | RPS27 | no | yes | <0.1% | [120] | |||
| RPS28 | RPS28 | no | yes | <0.1% | [114] | |||
| RPS29 | RPS29 | yes | yes | <0.1% | [131] | |||
| RPL5 | RPL5 | yes | yes | 7% | [109] | |||
| RPL11 | RPL11 | yes | yes | 5% | [109] | |||
| RPL15 | RPL15 | yes | yes | <0.5% | [132] | |||
| RPL18 | RPL18 | yes | yes | <0.1% | [133] | |||
| RPL26 | RPL26 | yes | yes | <0.1% | [134] | |||
| RPL27 | RPL27 | yes | yes | <0.1% | [120] | |||
| RPL31 | RPL31 | yes | yes | <0.1% | [113] | |||
| RPL35 | RPL35 | yes | yes | <1% | [133] | |||
| RPL35A | RPL35A | yes | yes | 3% | [135] | |||
| One family | X-linked recessive | TSR2 | TSR2 | no | yes | <0.1% | [114] | |
| 5q-syndrome | ||||||||
| MDS, severe macrocytic anemia | 10–15% of patients with MDS or AML | Sporadic | RPS14 | RPS14 | yes | yes | 100% | [136] |
| Isolated congenital asplenia | ||||||||
| Absence of spleen, high susceptibility to infections | 1:60,000 births | Autosomal dominant | RPSA | RPSA | no | yes | 100% | [137] |
| Other syndromes caused by RP mutations | ||||||||
| Intellectual disability, autism, microcephaly, hearing loss | Two patients | Autosomal dominant | RPS23 | RPS23 | yes | yes | n.a. | [138] |
| Autism, microcephaly, mental retardation, growth retardation, seizures, skeletal malformations | Three families/Nine patients | RPL10 | RPL10 | no | yes | n.a. | [139,140,141] | |
| Autism, microcephaly | Two families/Four patients | RPL10 | RPL10 | no | n.a. | [142,143] | ||
| Intellectual disability, epilepsy | One patient | RPL10 | RPL10 | no | n.a. | [144] | ||
| Schwachman-Diamond anemia | ||||||||
| Neutropenia, exocrine pancreatic dysfunction, metaphyseal dysplasia, osteopenia, mild mental retardation, high predisposition to MDS and AML | 1:77,000 births | Autosomal recessive | SBDS | SBDS | yes | yes | >95% | [145,146] |
| DNAJC21 | DNAJC21 ** | yes | yes | 2% | [147,148,149] | |||
| EFL1 | EFL1 | no | yes | <0.5% | [149,150] | |||
| Bowen-Conradi Syndrome | ||||||||
| Growth retardation, psychomotor delay, microcephaly, micrognatia, joint contractures, rockerbottom feet | 1:355 in the Hutterite populations | Autosomal recessive | EMG1 | EMG1 | no | yes | 100% | [151,152,153] |
| North American Indian chilhood cirrhosis | ||||||||
| Cirrhosis | 1:250 in the Ojibway-Cree First Nations population | Autosomal recessive | CIRH1A | hUTP4 | no | yes | 100% | [154,155] |
| Familial Aplasia Cutis Congenita | ||||||||
| Scalp skin defect | 1 patient | Autosomal dominant | BMS1 | BMS1 | yes | yes | n.a. | [156] |
| Cartilage-hair hypoplasia | ||||||||
| Hypoplastic macrocytic anemia, neutropenia, defective T-cell, response, short limb dwarfism, fine, sparse hair, skeletal abnormalities, nail dysplasia, gastrointestinal malabsorption, abnormal dentition, predisposition to non-Hodgkin lymphomas and other cancers | 1–2:1000 in the Amish population, 1:23,000 in the Finnish population | Autosomal recessive | RMRP | - | yes | yes | 100% | [61,157] |
| Diskeratosis congenita and Hoyeraal-Hreidarsson syndrome | ||||||||
| Bone marrow failure, pancytopenia, aplastic anemia, mucocutaneous defects, nail dystrophy, developmental delay, pulmonary fibrosis, reduced telomere length, cancer predisposition, immunodeficiency | 1:1,000,000 births | X-linked recessive | DKC1 | Dyskerin | no | yes | 25% | [158,159] |
| Autosomal recessive | NOP10 | NOP10 | no | no | <1% | [160] | ||
| NHP2 | NHP2 | no | no | <1% | [161] | |||
| PARN | PARN | yes | yes | <1% | [162,163] | |||
| Treacher-Collins syndrome | ||||||||
| Severe craniofacial defects, mental retardation | 1:10,000–50,000 births | Autosomal dominant | TCOF1 | Treacle | no | yes | 78–93% | [164] |
| Autosomal dominant or recessive | POLR1D | RPA16 | no | yes | 8% *** | [165,166,167] | ||
| autosomal recessive | POLR1C | RPA39 | no | yes | [165] | |||
| Other RNA polymerase I-related diseases | ||||||||
| Acrofacial dysostosis | Three patients | Autosomal dominant | POLR1A | RPA194 | no | yes | n.a. | [168] |
| Severe neurodegenerative disease, psychomotor retardation, intellectual disability | Two patients | POLR1A | RPA194 | no | n.a. | [169] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aubert, M.; O’Donohue, M.-F.; Lebaron, S.; Gleizes, P.-E. Pre-Ribosomal RNA Processing in Human Cells: From Mechanisms to Congenital Diseases. Biomolecules 2018, 8, 123. https://doi.org/10.3390/biom8040123
Aubert M, O’Donohue M-F, Lebaron S, Gleizes P-E. Pre-Ribosomal RNA Processing in Human Cells: From Mechanisms to Congenital Diseases. Biomolecules. 2018; 8(4):123. https://doi.org/10.3390/biom8040123
Chicago/Turabian StyleAubert, Maxime, Marie-Françoise O’Donohue, Simon Lebaron, and Pierre-Emmanuel Gleizes. 2018. "Pre-Ribosomal RNA Processing in Human Cells: From Mechanisms to Congenital Diseases" Biomolecules 8, no. 4: 123. https://doi.org/10.3390/biom8040123
APA StyleAubert, M., O’Donohue, M.-F., Lebaron, S., & Gleizes, P.-E. (2018). Pre-Ribosomal RNA Processing in Human Cells: From Mechanisms to Congenital Diseases. Biomolecules, 8(4), 123. https://doi.org/10.3390/biom8040123