Hepatitis C Virus-Infected Apoptotic Hepatocytes Program Macrophages and Hepatic Stellate Cells for Liver Inflammation and Fibrosis Development: Role of Ethanol as a Second Hit

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Media
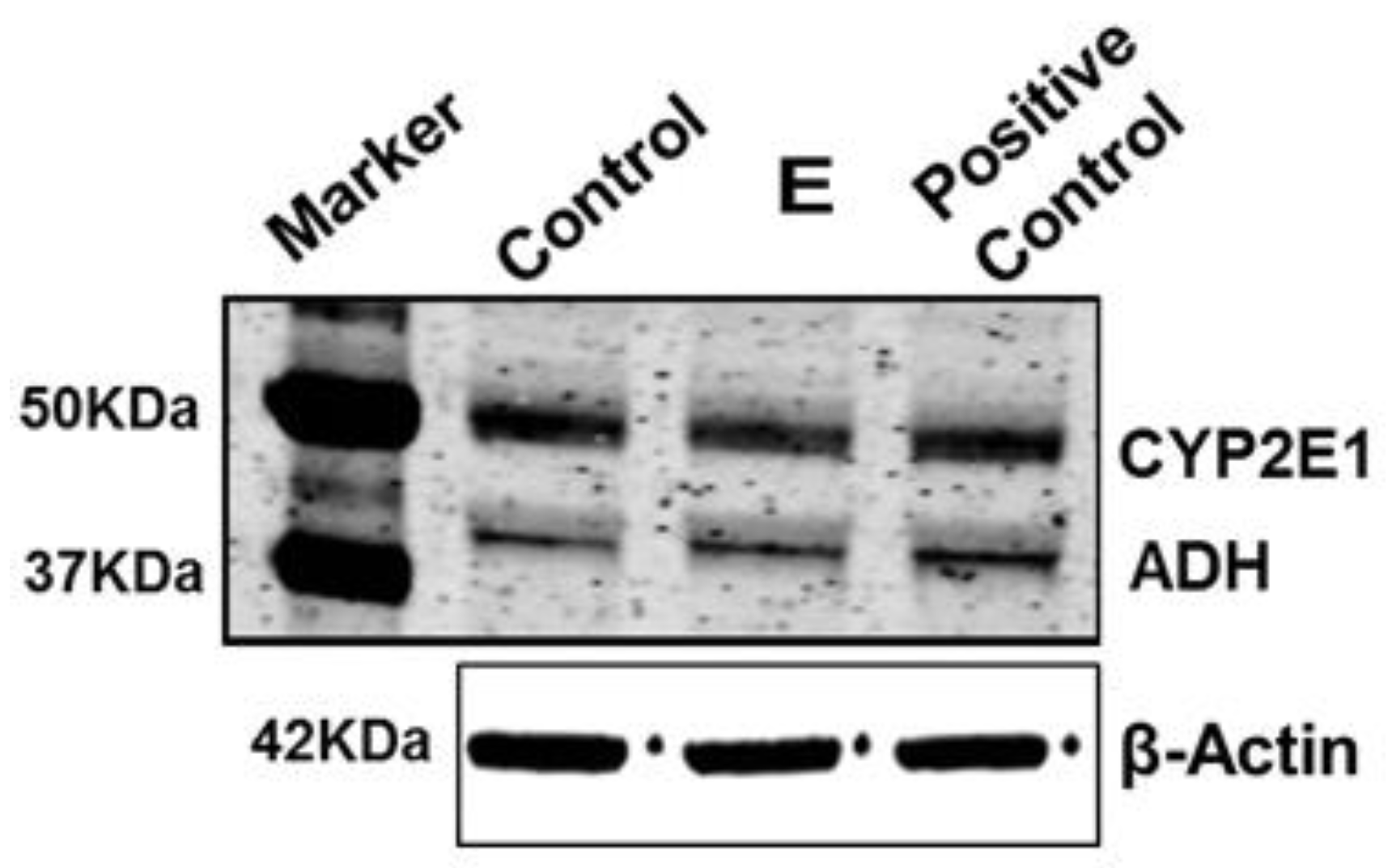
2.2. Huh7.5-CYP Cells and Treatments
2.3. Primary Human Macrophages
2.4. Hepatic Stellate Cells
2.5. Apoptotic Bodies Generation and Characterization of Apoptotic Cells
2.6. Macrophage and HSC Treatment with AB
2.7. RNA Isolation and Real-Time PCR
2.8. Immunoblotting (Western Blot)
2.9. Statistical Analyses
3. Results
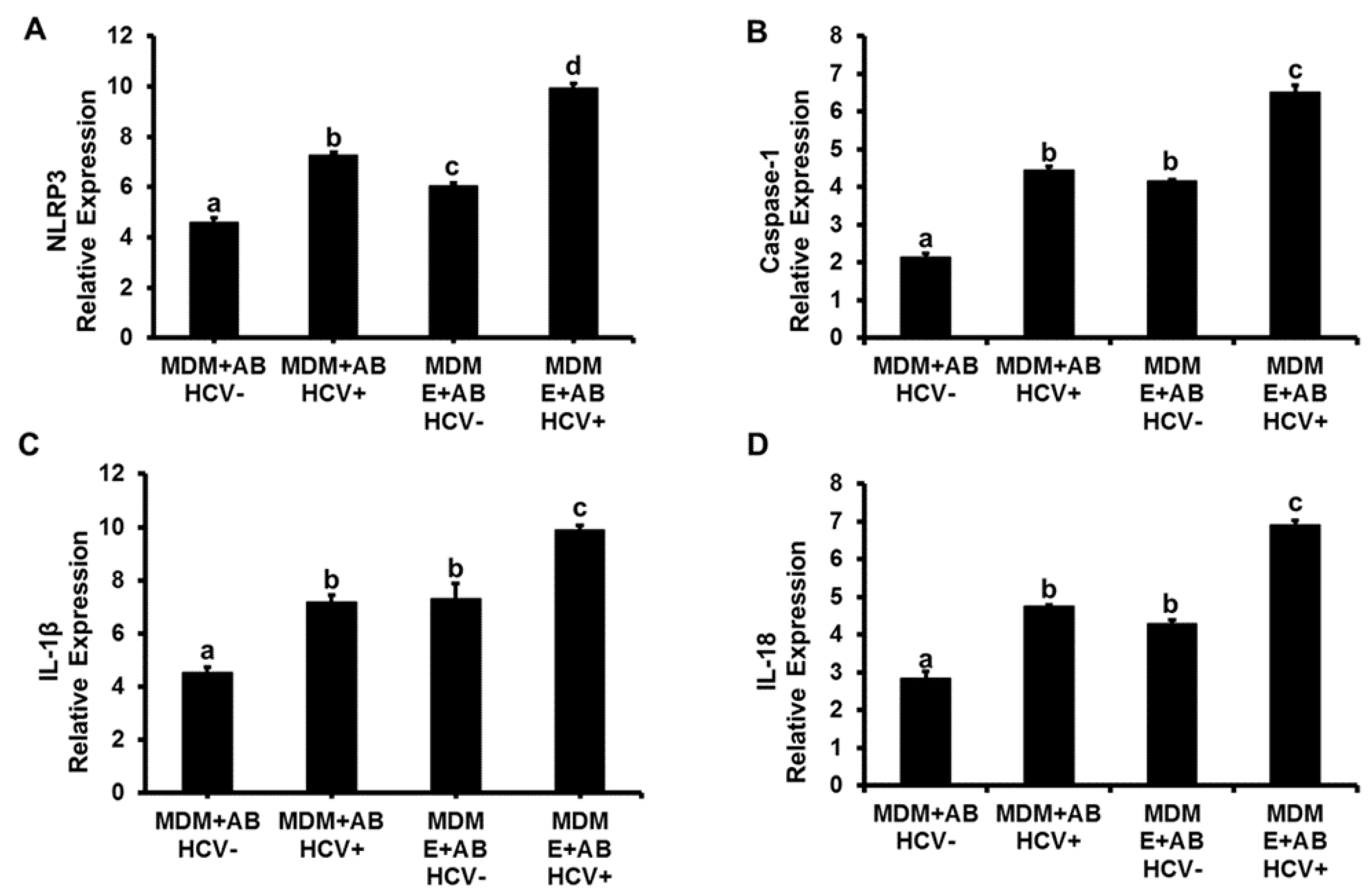
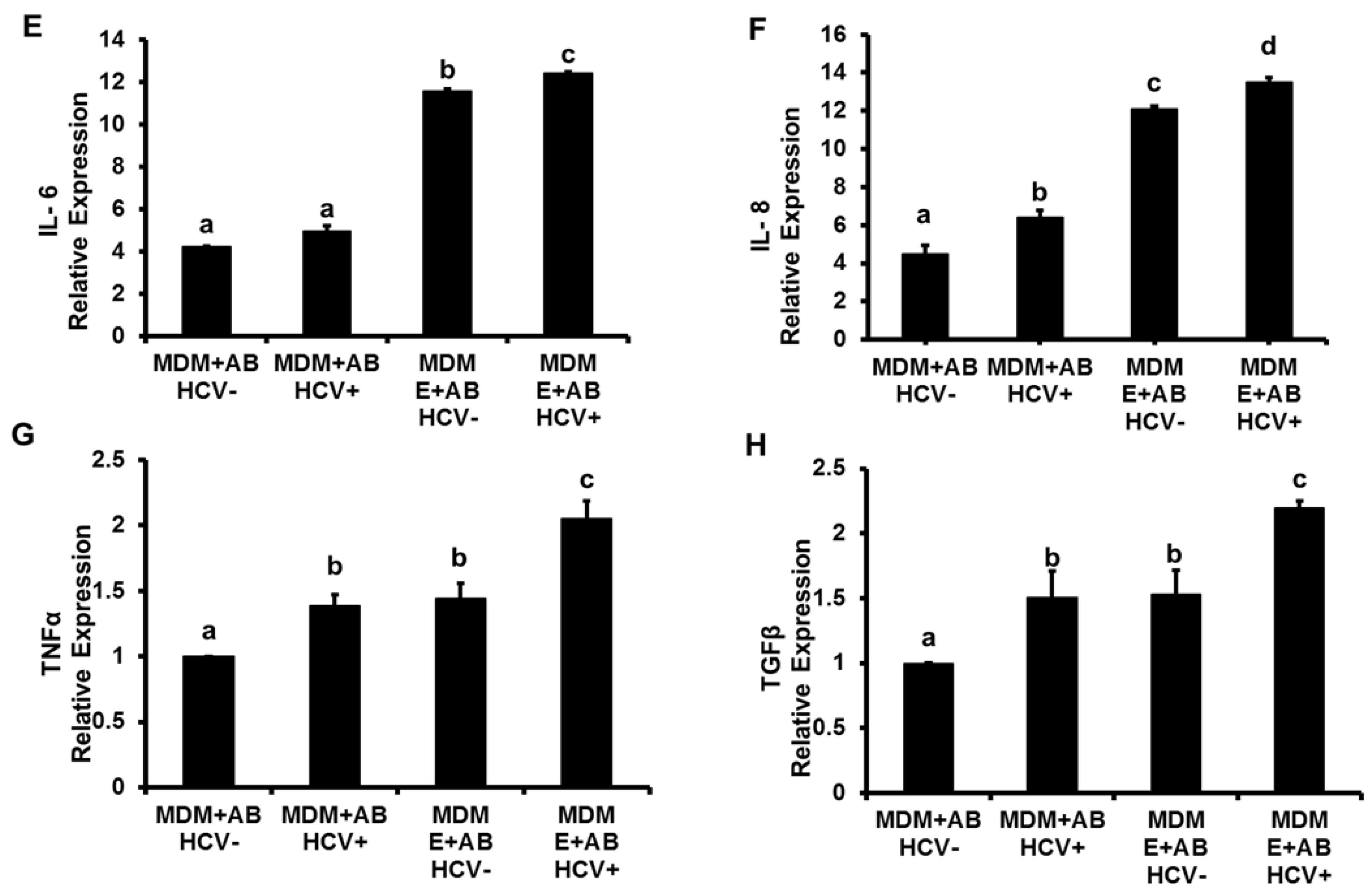
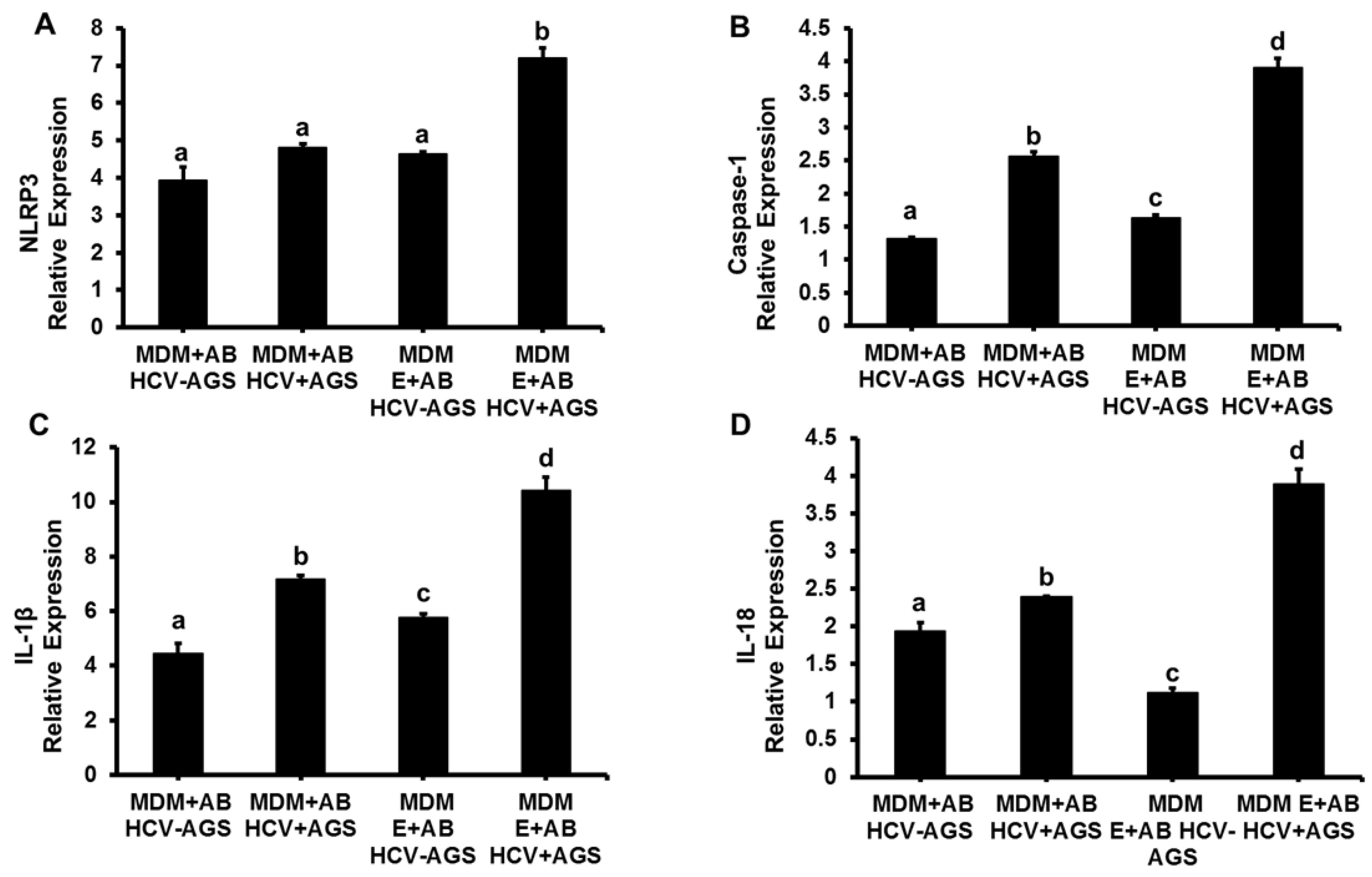
3.1. Induction of Inflammasome, Pro-Inflammatory Cytokines and TGFβ in MDMs Exposed to AB from HCV+ and HCV− RLW Cells
3.2. Effects of Ethanol Exposure to MDMs on AB-Induced Inflammasome Activation
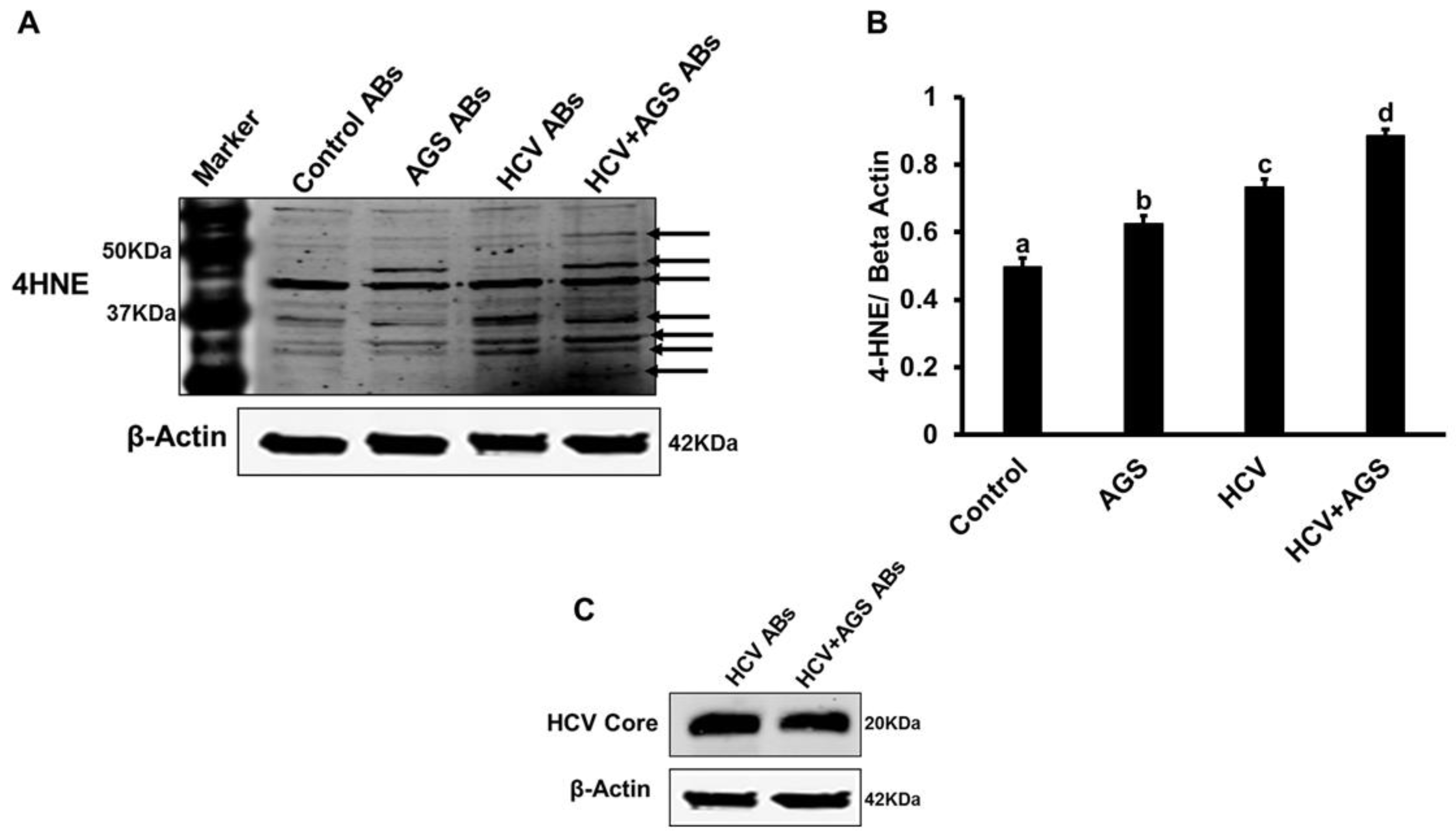
3.3. Effects of AGSExposure to RLW Cells on AB-Triggered Activation of Inflammasomes in MDMs
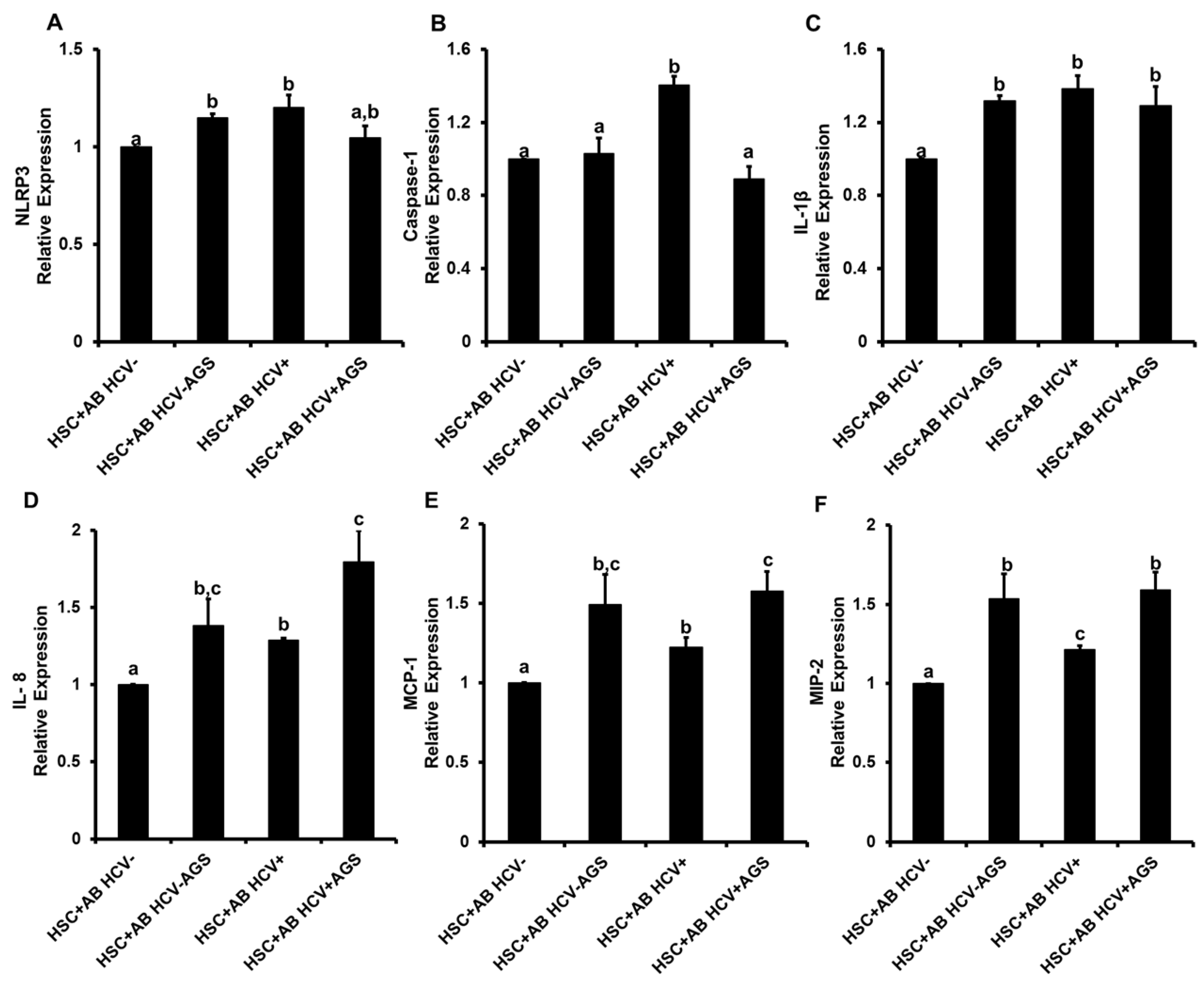
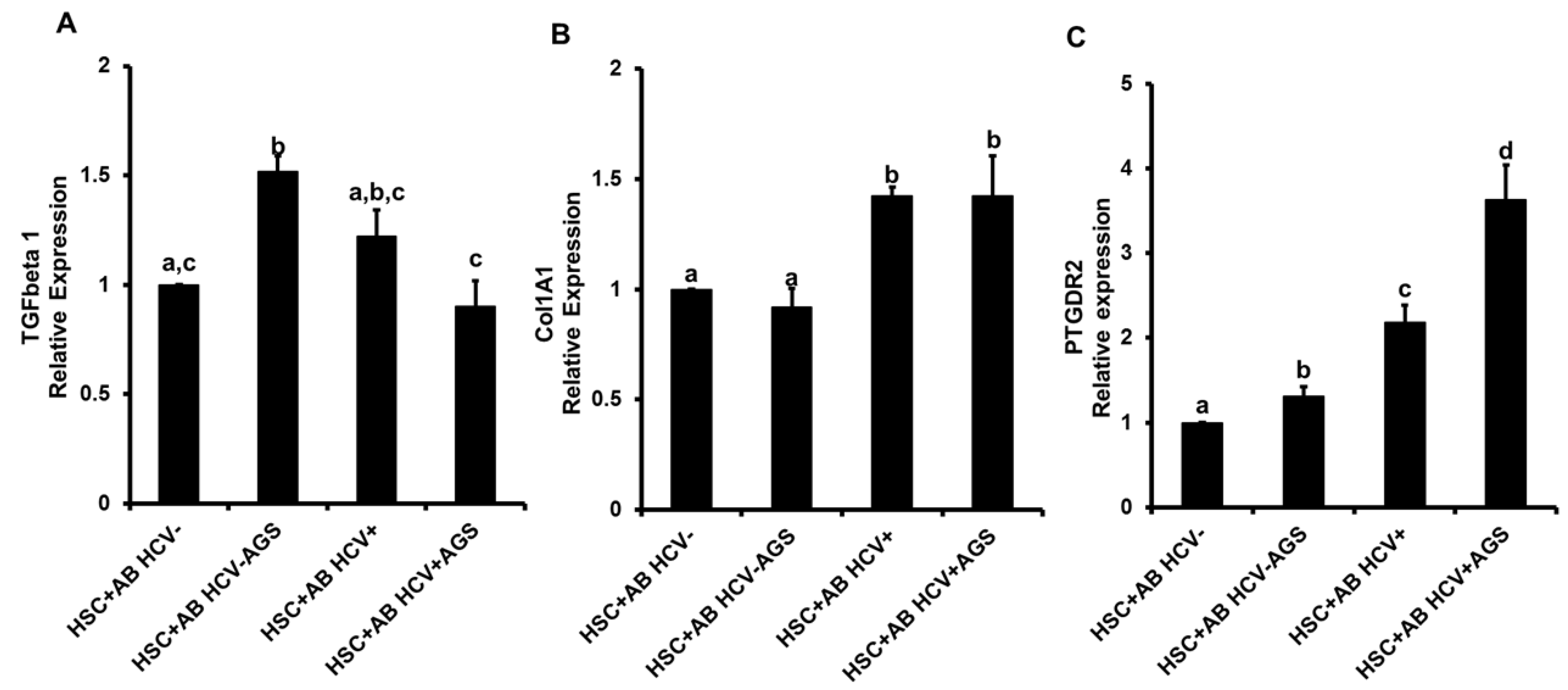
3.4. Induction of Inflammasome and Pro-Fibrotic Response in Hepatic Stellate Cells (HSC) Exposed to AB from HCV+ and HCV− RLW Cells
3.4.1. Pro-Inflammatory HSC Activation by AB
3.4.2. Pro-Fibrotic HSC Activation by AB
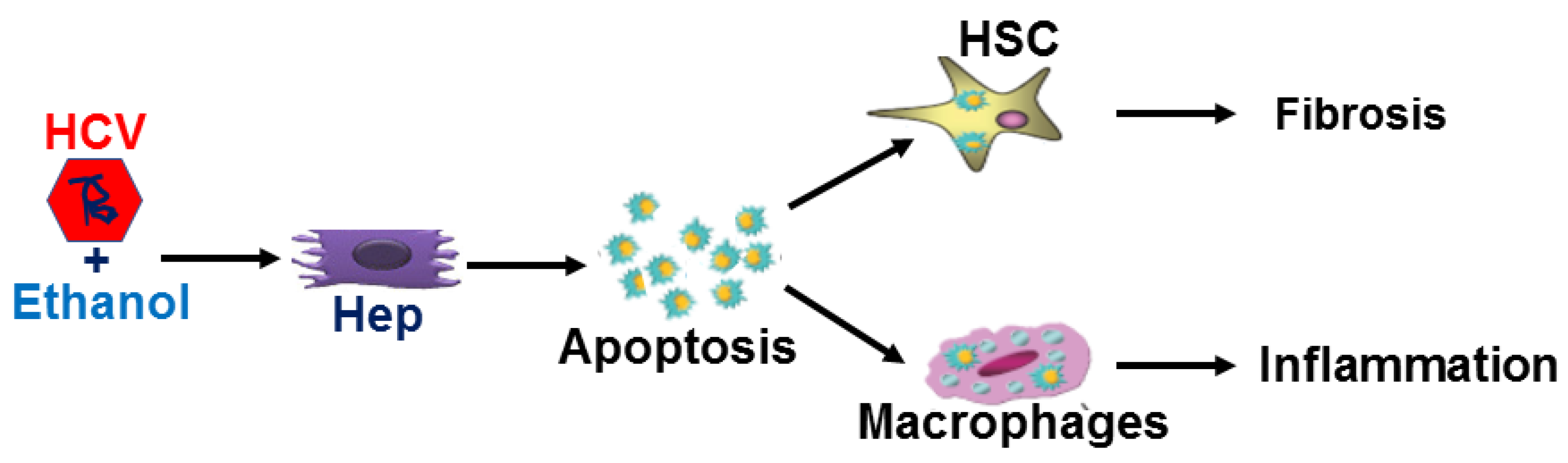
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of interest
References
- Baumert, T.F.; Juhling, F.; Ono, A.; Hoshida, Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med. 2017, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Bruden, D.J.T.; McMahon, B.J.; Townshend-Bulson, L.; Gounder, P.; Gove, J.; Plotnik, J.; Homan, C.; Hewitt, A.; Barbour, Y.; Spradling, P.R.; et al. Risk of end-stage liver disease, hepatocellular carcinoma, and liver-related death by fibrosis stage in the hepatitis C Alaska cohort. Hepatology 2017, 66, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Vandenbulcke, H.; Moreno, C.; Colle, I.; Knebel, J.F.; Francque, S.; Serste, T.; George, C.; de Galocsy, C.; Laleman, W.; Delwaide, J.; et al. Alcohol intake increases the risk of HCC in hepatitis C virus-related compensated cirrhosis: A prospective study. J. Hepatol. 2016, 65, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Marcon, P.D.S.; Tovo, C.V.; Kliemann, D.A.; Fisch, P.; de Mattos, A.A. Incidence of hepatocellular carcinoma in patients with chronic liver disease due to hepatitis B or C and coinfected with the human immunodeficiency virus: A retrospective cohort study. World J. Gastroenterol. 2018, 24, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Sze, C.W.; Keng, C.T.; Al-Haddawi, M.; Liu, M.; Tan, S.Y.; Kwek, H.L.; Her, Z.; Chan, X.Y.; Barnwal, B.; et al. Hepatitis C virus mediated chronic inflammation and tumorigenesis in the humanised immune system and liver mouse model. PLoS ONE 2017, 12, e0184127. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, R.K.; Marquitan, G.; Schlattjan, M.; Sowa, J.P.; Bechmann, L.P.; Timm, J.; Roggendorf, M.; Gerken, G.; Friedman, S.L.; Canbay, A. Hepatocyte apoptotic bodies encasing nonstructural HCV proteins amplify hepatic stellate cell activation: Implications for chronic hepatitis c. J. Viral Hepat. 2011, 18, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Klevens, R.M.; Hu, D.J.; Jiles, R.; Holmberg, S.D. Evolving epidemiology of hepatitis C virus in the united states. Clin. Infect. Dis. 2012, 55 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef]
- Ganesan, M.; Natarajan, S.K.; Zhang, J.; Mott, J.L.; Poluektova, L.I.; McVicker, B.L.; Kharbanda, K.K.; Tuma, D.J.; Osna, N.A. Role of apoptotic hepatocytes in HCV dissemination: Regulation by acetaldehyde. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G930–G940. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Poluektova, L.Y.; Tuma, D.J.; Kharbanda, K.K.; Osna, N.A. Acetaldehyde disrupts interferon alpha signaling in hepatitis C virus-infected liver cells by up-regulating USP18. Alcohol. Clin. Exp. Res. 2016, 40, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Zhang, J.; Bronich, T.; Poluektova, L.I.; Donohue, T.M., Jr.; Tuma, D.J.; Kharbanda, K.K.; Osna, N.A. Acetaldehyde accelerates HCV-induced impairment of innate immunity by suppressing methylation reactions in liver cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G566–G577. [Google Scholar] [CrossRef] [PubMed]
- Aruin, A.S.; Kanekar, N.; Lee, Y.J.; Ganesan, M. Enhancement of anticipatory postural adjustments in older adults as a result of a single session of ball throwing exercise. Exp. Brain Res. 2015, 233, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Cederbaum, A.I. NRF2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology 2006, 43, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.; Clemens, D.; Donohue, T. Ethanol reciprocally regulates CYP2E1 and proteasome activities in double-recombinant HepG2 cells treated with interferon gamma. Alcohol Clin. ExpRes. 2002, 26, 60A (abstact). [Google Scholar]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: A clear and present danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef] [PubMed]
- Schiff, E.R. Hepatitis C and alcohol. Hepatology 1997, 26, 39S–42S. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Hepatitis C and alcohol. J. Clin. Gastroenterol. 2003, 36, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; El Khobar, K.; Chin, R.; Earnest-Silveira, L.; Angus, P.W.; Bock, C.T.; Nachbur, U.; Silke, J.; Torresi, J. Hepatitis C virus-induced hepatocyte cell death and protection by inhibition of apoptosis. J. Gen. Virol. 2014, 95, 2204–2215. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Tikhanovich, I.; Vangimalla, S.S.; Dagur, R.S.; Wang, W.; Poluektova, L.I.; Sun, Y.; Mercer, D.F.; Tuma, D.; Weinman, S.A.; et al. DemethylaseJMJD6 as a new regulator of interferon signaling: Effects of HCV and ethanol metabolism. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Norkina, O.; Kodys, K.; Catalano, D.; Bakis, G.; Marshall, C.; Mandrekar, P.; Szabo, G. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology 2007, 133, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Chattergoon, M.A.; Latanich, R.; Quinn, J.; Winter, M.E.; Buckheit, R.W., 3rd; Blankson, J.N.; Pardoll, D.; Cox, A.L. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal toll-like receptors without induction of type 1 interferon. PLoS Pathog 2014, 10, e1004082. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, Y.; Ye, J. Inhibition of hepatitis C virus replication by peroxidation of arachidonate and restoration by vitamin E. Proc. Natl. Acad. Sci. USA 2007, 104, 18666–18670. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; McGivern, D.R.; Wauthier, E.; Yi, M.; Madden, V.J.; Welsch, C.; Antes, I.; Wen, Y.; Chugh, P.E.; McGee, C.E.; et al. Regulation of the hepatitis C virus RNA replicase by endogenous lipid peroxidation. Nat. Med. 2014, 20, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.N. Ethanol metabolism by macrophages: Possible role in organ damage. Addict. Biol. 1998, 3, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Hutson, J.L.; Wickramasinghe, S.N. Expression of CYP2E1 by human monocyte-derived macrophages. J. Pathol. 1999, 188, 197–200. [Google Scholar] [CrossRef]
- Kessova, I.G.; Cederbaum, A.I. The effect of CYP2E1-dependent oxidant stress on activity of proteasomes in HEPG2 cells. J. Pharmacol. Exp. Ther. 2005, 315, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guan, S.; Acharya, P.; Koop, D.R.; Liu, Y.; Liao, M.; Burlingame, A.L.; Correia, M.A. Ubiquitin-dependent proteasomal degradation of human liver cytochrome p450 2e1: Identification of sites targeted for phosphorylation and ubiquitination. J. Biol. Chem. 2011, 286, 9443–9456. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr.; McVicker, D.L.; Kharbanda, K.K.; Chaisson, M.L.; Zetterman, R.K. Ethanol administration alters the proteolytic activity of hepatic lysosomes. Alcohol. Clin. Exp. Res. 1994, 18, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M., Jr. Cyp2e1-catalyzed alcohol metabolism: Role of oxidant generation in interferon signaling, antigen presentation and autophagy. Sub-Cell. Biochem. 2013, 67, 177–197. [Google Scholar]
- Osna, N.A.; White, R.L.; Todero, S.; McVicker, B.L.; Thiele, G.M.; Clemens, D.L.; Tuma, D.J.; Donohue, T.M., Jr. Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology 2007, 45, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; White, R.L.; Krutik, V.M.; Wang, T.; Weinman, S.A.; Donohue, T.M., Jr. Proteasome activation by hepatitis c core protein is reversed by ethanol-induced oxidative stress. Gastroenterology 2008, 134, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Lefkowitz, D.L.; Lefkowitz, S.S. The effects of alcohol on murine macrophage function. Life Sci 1993, 52, 1585–1593. [Google Scholar] [CrossRef]
- Karavitis, J.; Kovacs, E.J. Macrophage phagocytosis: Effects of environmental pollutants, alcohol, cigarette smoke, and other external factors. J. Leukoc. Biol. 2011, 90, 1065–1078. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Mikami, K.; Shah, V.H.; Torok, N.J. Leptin induces phagocytosis of apoptotic bodies by hepatic stellate cells via a rho guanosine triphosphatase-dependent mechanism. Hepatology 2008, 48, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Mikami, K.; Venugopal, S.; Li, Y.; Torok, N.J. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the jak/stat and akt/nf-kappab-dependent pathways. J. Hepatol. 2009, 51, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Invest. 2003, 83, 655–663. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganesan, M.; Poluektova, L.Y.; Enweluzo, C.; Kharbanda, K.K.; Osna, N.A. Hepatitis C Virus-Infected Apoptotic Hepatocytes Program Macrophages and Hepatic Stellate Cells for Liver Inflammation and Fibrosis Development: Role of Ethanol as a Second Hit. Biomolecules 2018, 8, 113. https://doi.org/10.3390/biom8040113
Ganesan M, Poluektova LY, Enweluzo C, Kharbanda KK, Osna NA. Hepatitis C Virus-Infected Apoptotic Hepatocytes Program Macrophages and Hepatic Stellate Cells for Liver Inflammation and Fibrosis Development: Role of Ethanol as a Second Hit. Biomolecules. 2018; 8(4):113. https://doi.org/10.3390/biom8040113
Chicago/Turabian StyleGanesan, Murali, Larisa Y. Poluektova, Chijioke Enweluzo, Kusum K. Kharbanda, and Natalia A. Osna. 2018. "Hepatitis C Virus-Infected Apoptotic Hepatocytes Program Macrophages and Hepatic Stellate Cells for Liver Inflammation and Fibrosis Development: Role of Ethanol as a Second Hit" Biomolecules 8, no. 4: 113. https://doi.org/10.3390/biom8040113
APA StyleGanesan, M., Poluektova, L. Y., Enweluzo, C., Kharbanda, K. K., & Osna, N. A. (2018). Hepatitis C Virus-Infected Apoptotic Hepatocytes Program Macrophages and Hepatic Stellate Cells for Liver Inflammation and Fibrosis Development: Role of Ethanol as a Second Hit. Biomolecules, 8(4), 113. https://doi.org/10.3390/biom8040113