The Functional Amyloid Curli Protects Escherichia coli against Complement-Mediated Bactericidal Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
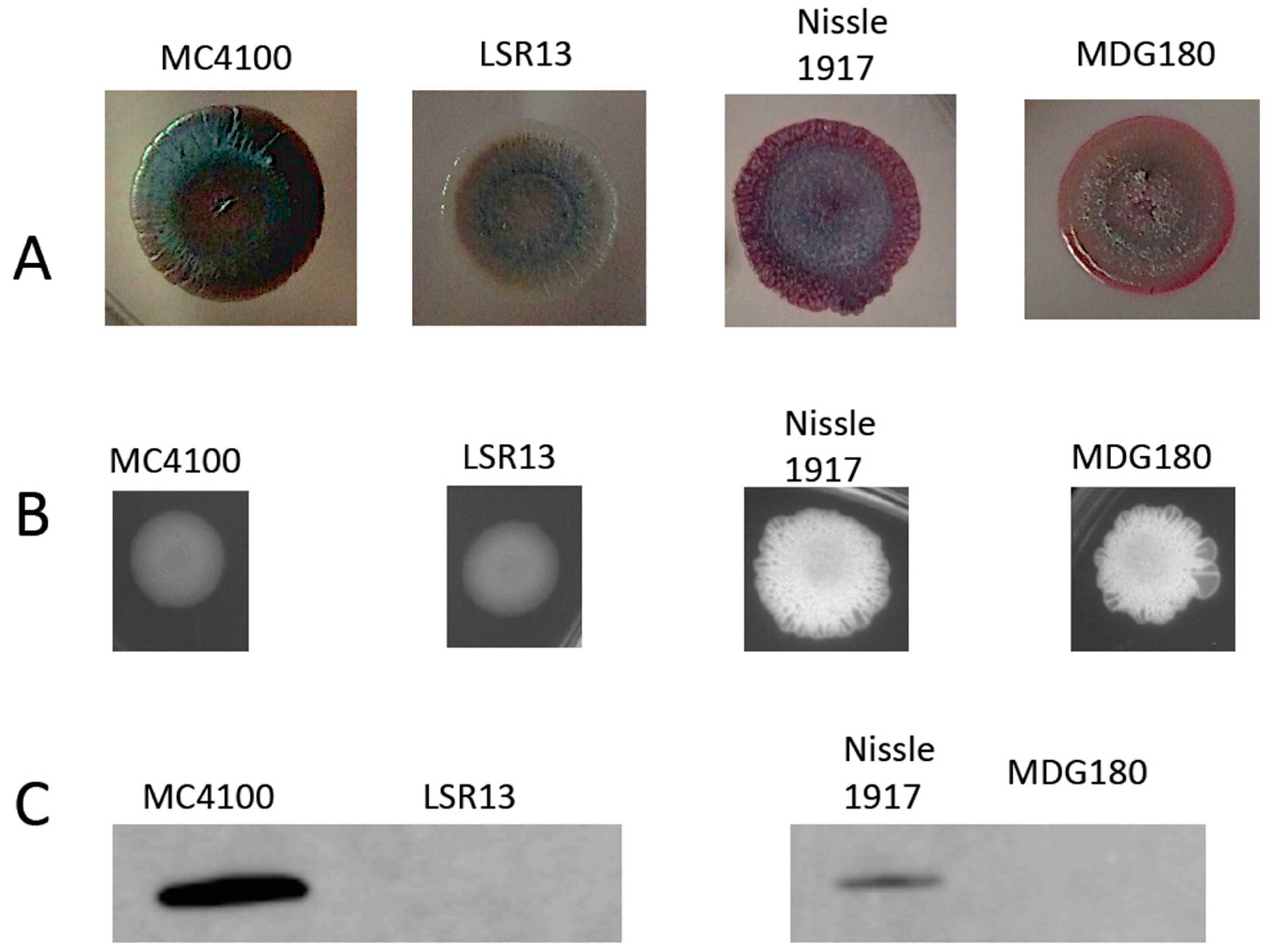
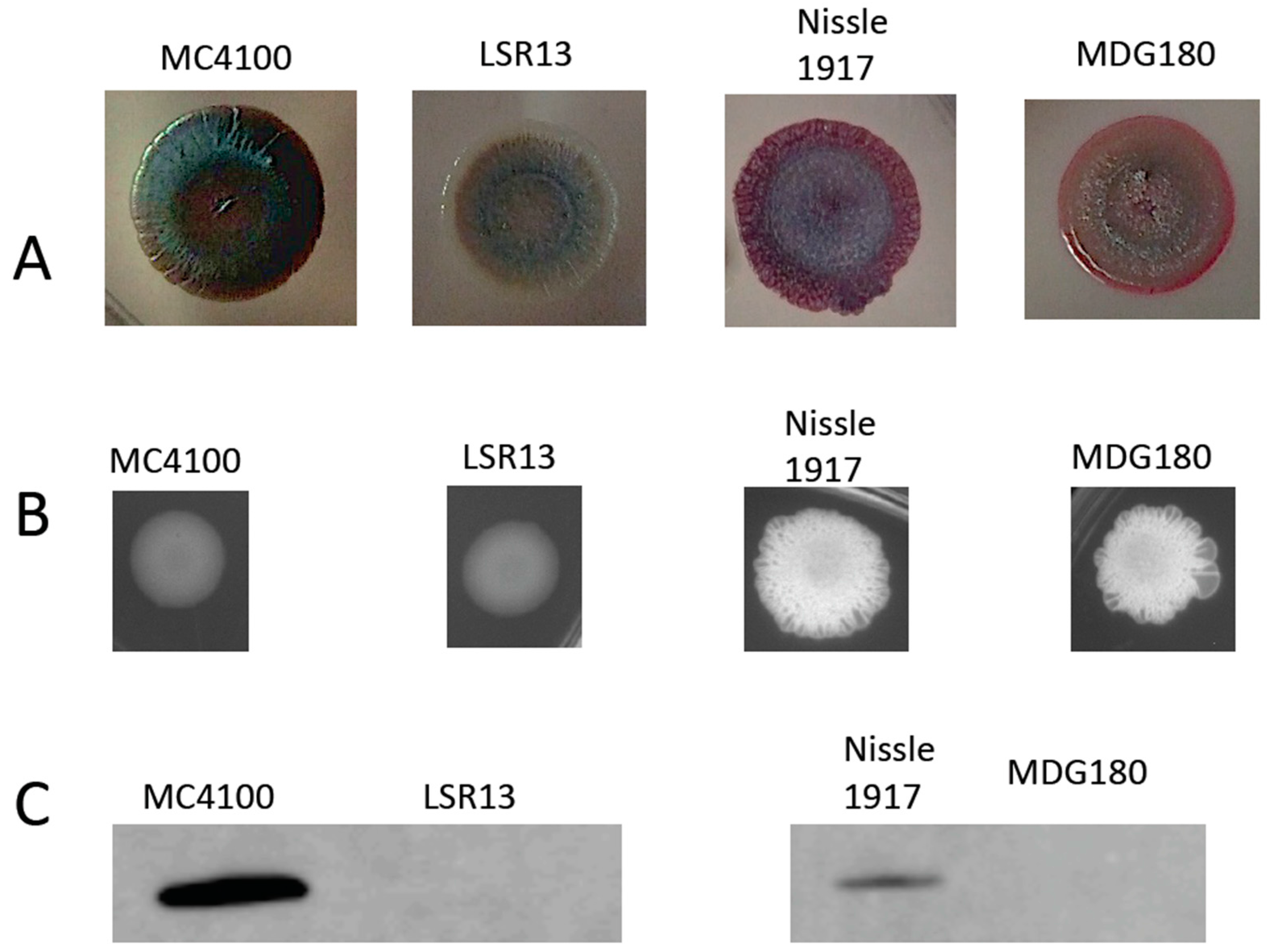
2.1. Characterization of Bacterial Strains
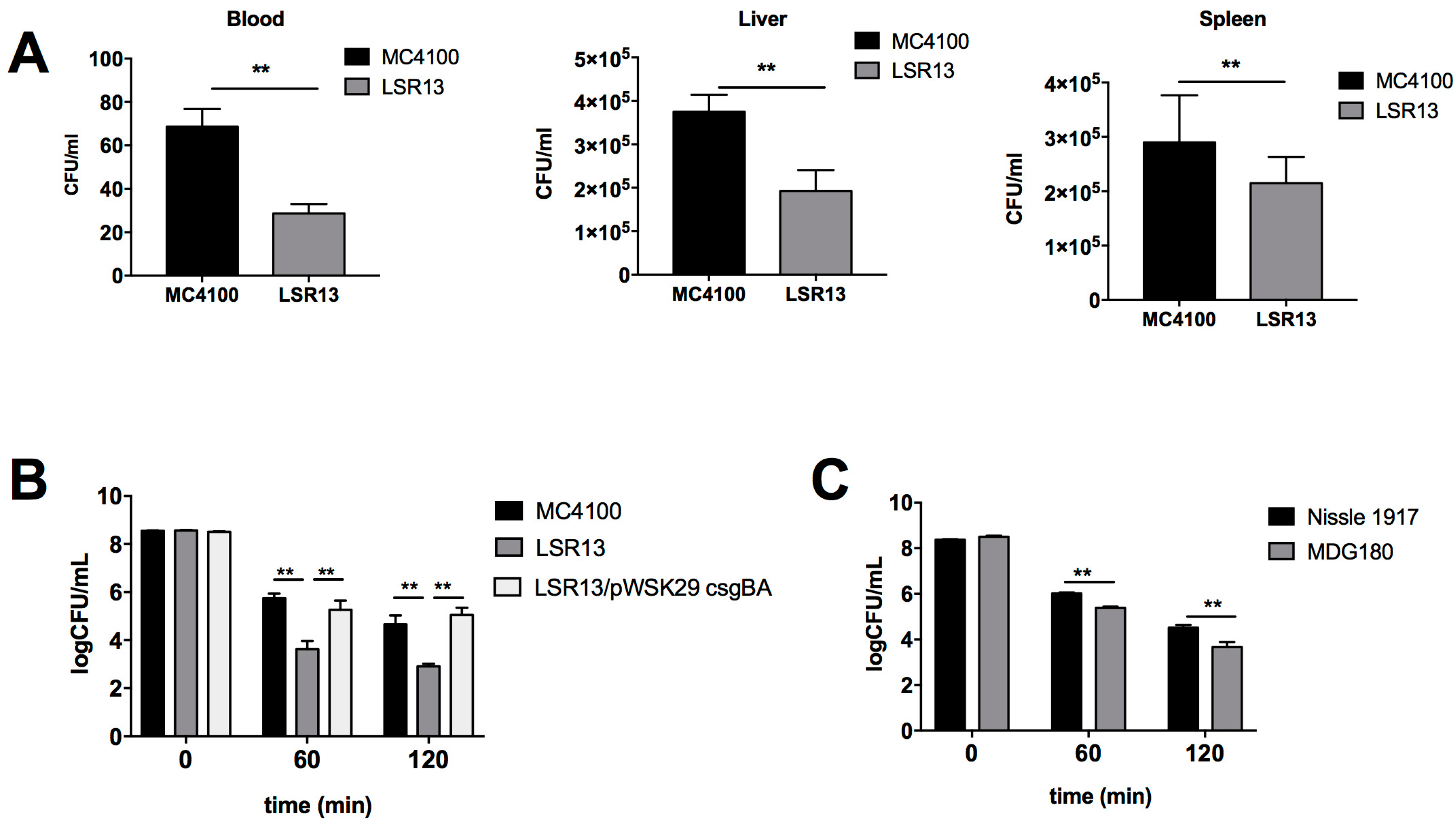
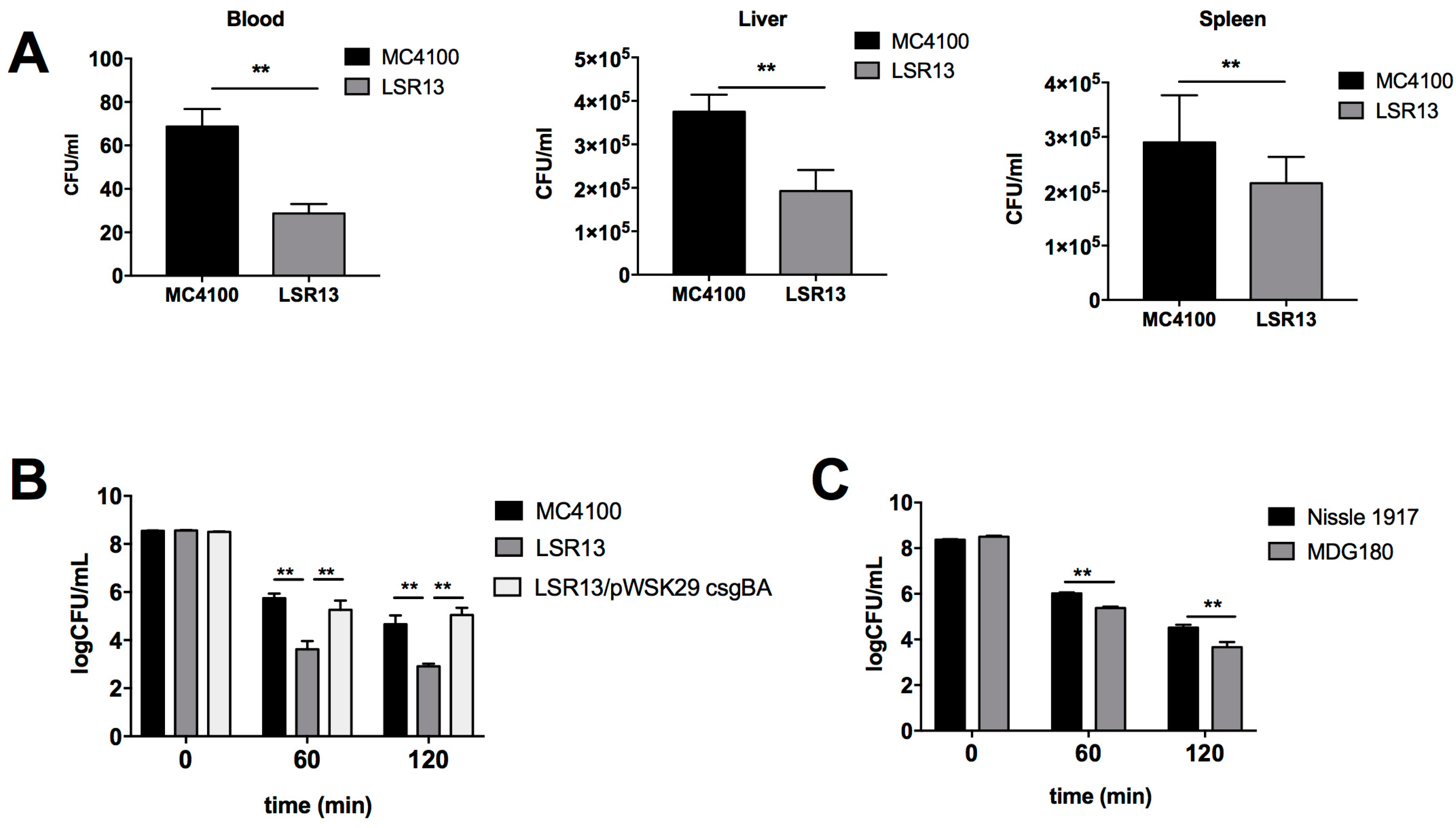
2.2. Curli Enhances Bacterial Cell Survival in an In Vivo Model
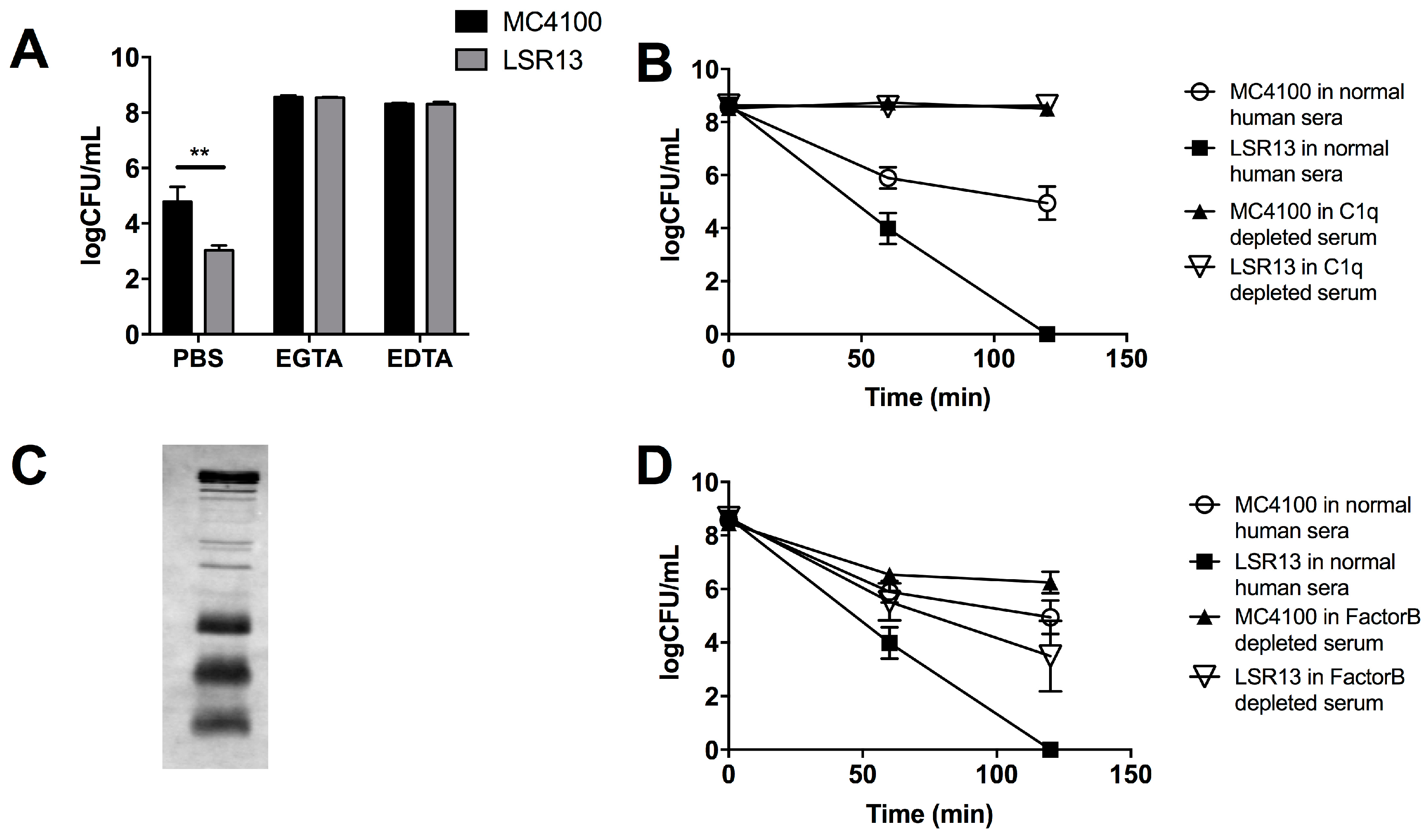
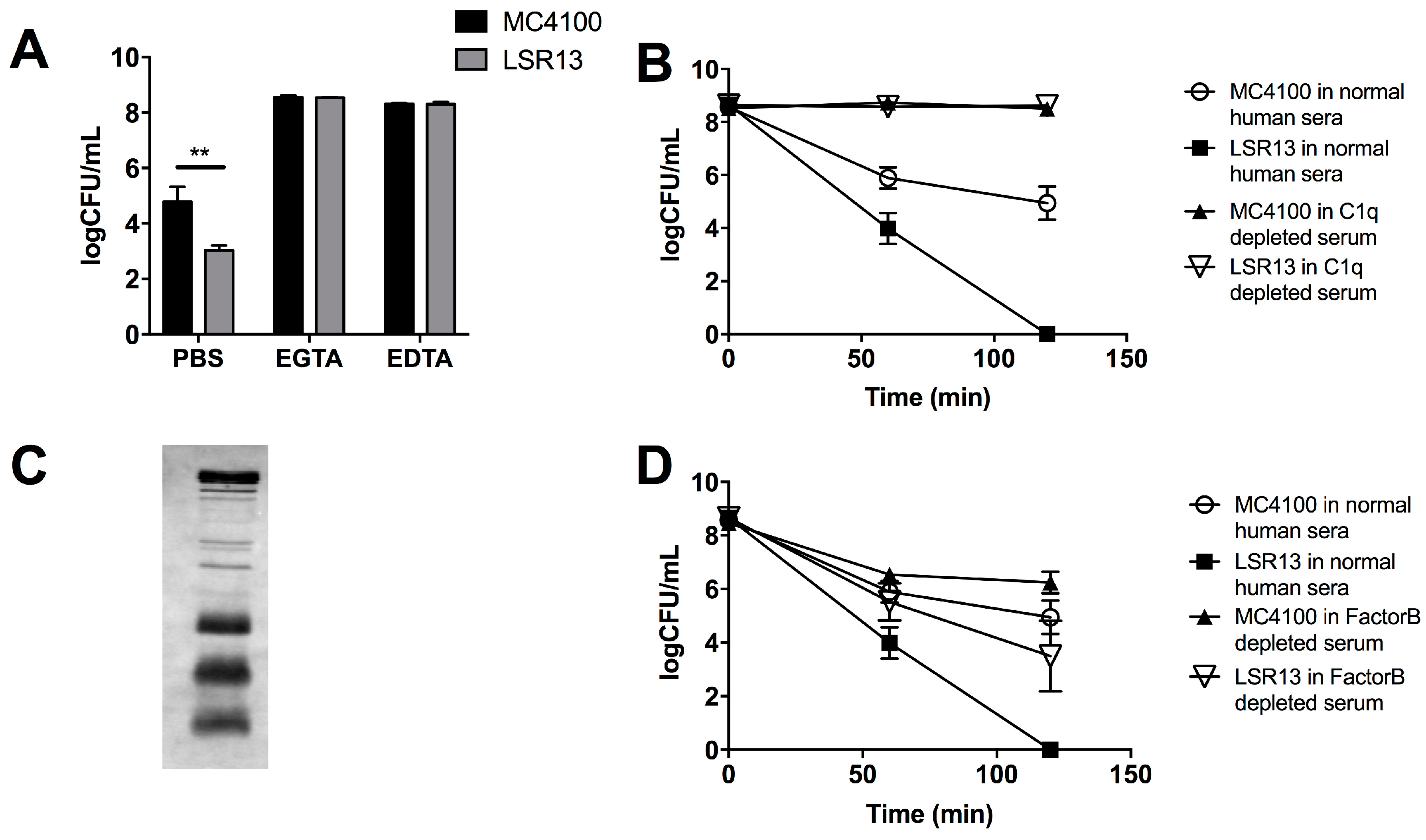
2.3. Curli Inhibits Complement-Mediated Killing of Bacterial Cells In Vitro
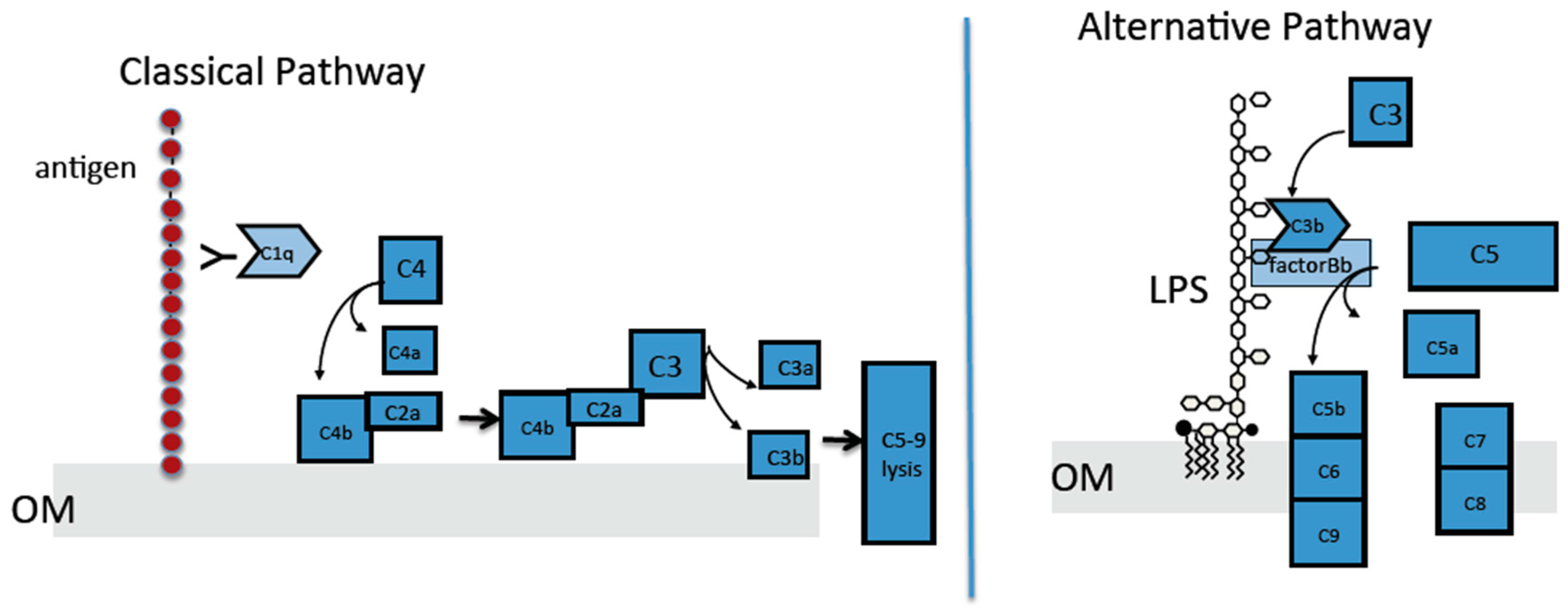
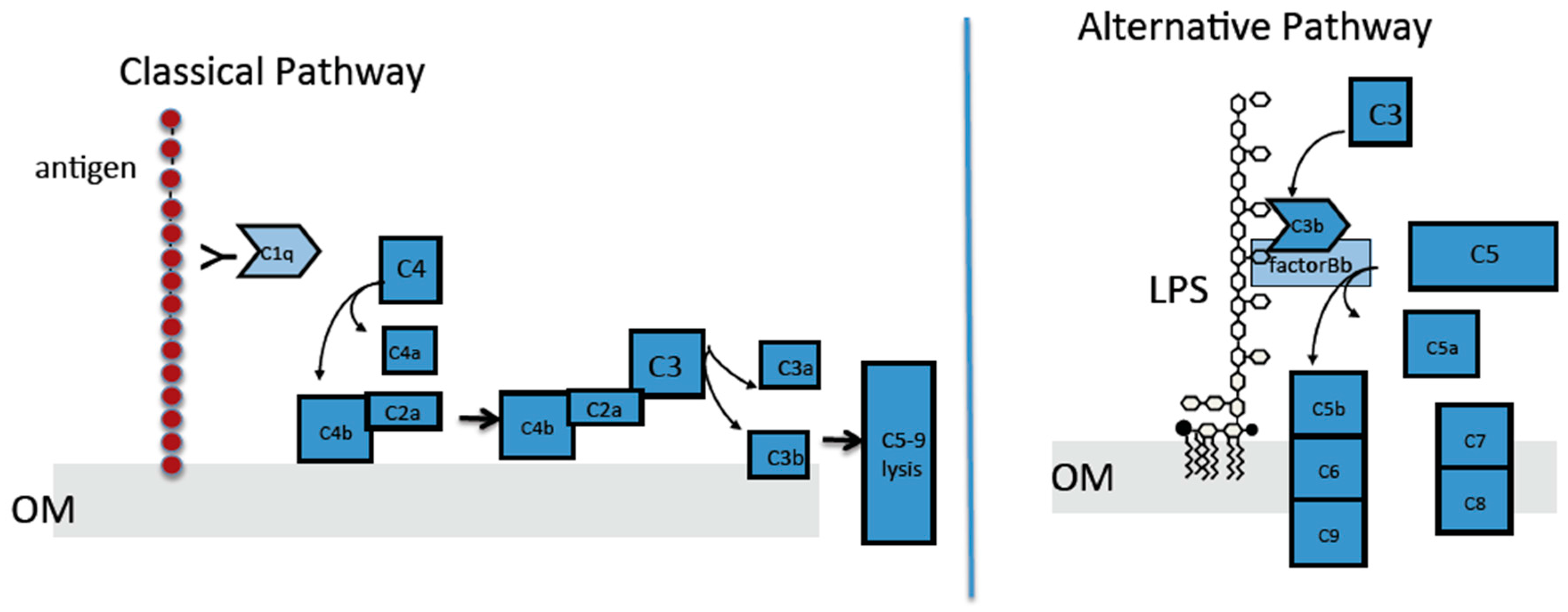
2.4. The Classical Complement Pathway Is Inhibited by Curli
2.5. The Alternative Complement Pathway Is Not Influenced by Curli
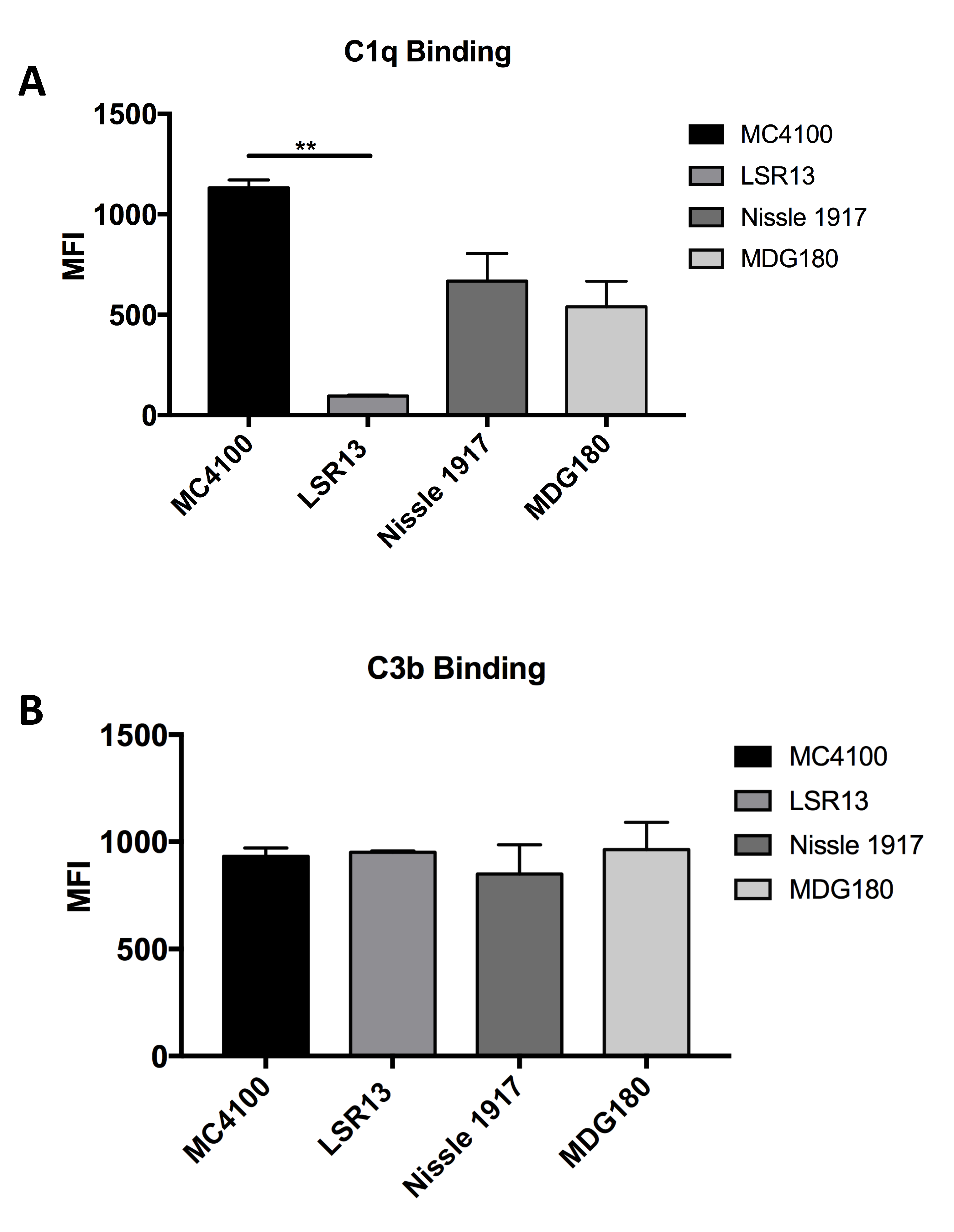
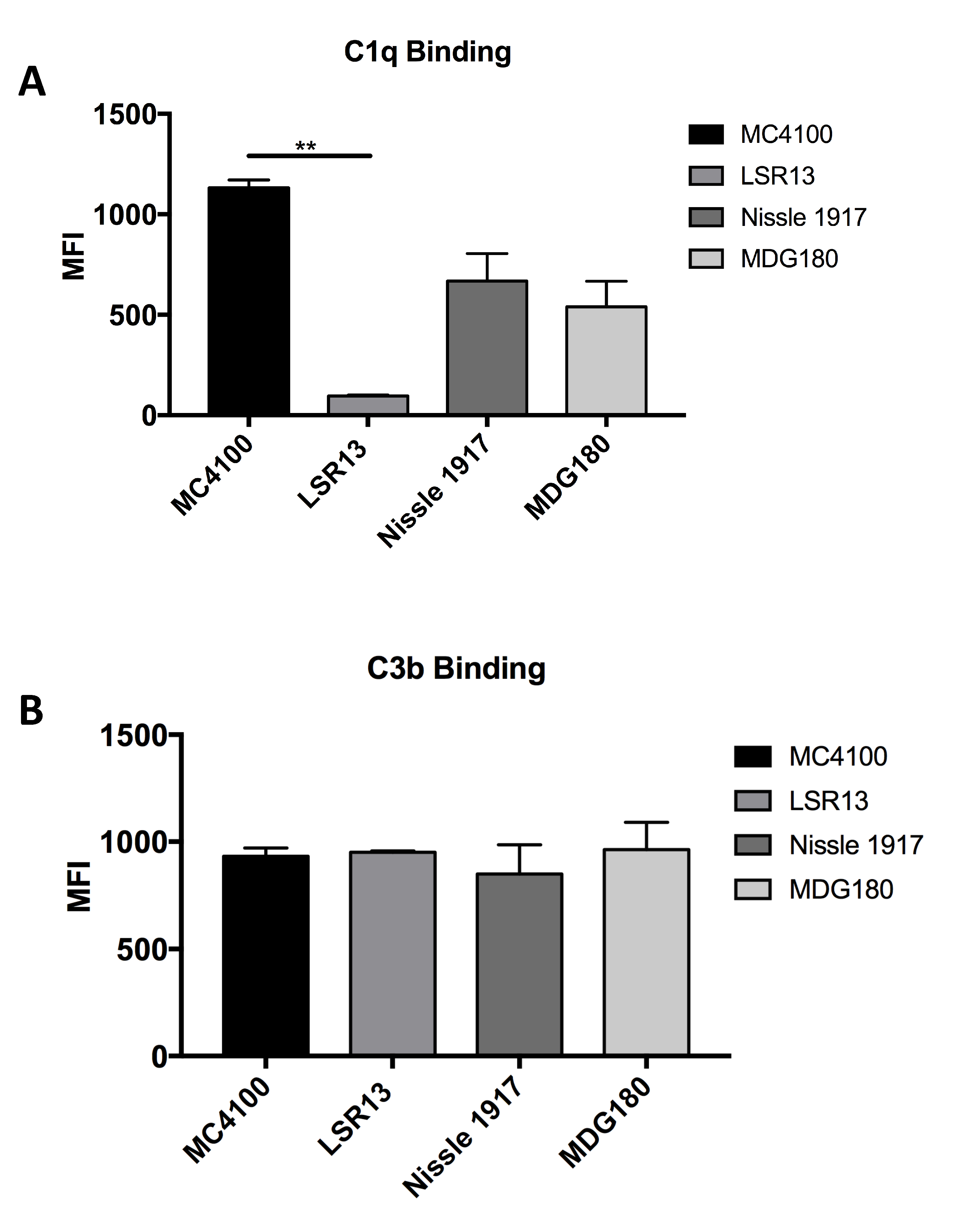
2.6. E. coli Cells Bind C1q but Not C3b
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Serum Sensitivity Assays
4.3. Inhibition of Complement Pathways
4.4. Western Blot
4.5. C1q and C3 Binding Assay
4.6. Animal Experiments
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bokranz, W.; Wang, X.; Tschäpe, H.; Römling, U. Expression of cellulose and curli fimbriae by Escherichia coli isolated from the gastrointestinal tract. J. Med. Microbiol. 2005, 54, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Garnett, J.; Martínez-Santos, V.; Saldaña, Z.; Pape, T.; Hawthorne, W.; Chan, J.; Simpson, P.; Cota, E.; Puente, J.; Girón, J.; et al. Structural insights into the biogenesis and biofilm formation by the Escherichia coli common pilus. Proc. Natl. Acad. Sci. USA 2012, 109, 3950–3955. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Hudault, S.; Guignot, J.; Servin, A. Escherichia coli strains colonising the gastrointestinal tract protect germfree mice against Salmonella typhimurium infection. Gut 2001, 49, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.-E.; Smalley, D.; Tucker, D.; Leatham, M.; Norris, W.; Stevenson, S.; Anderson, A.; Grissom, J.; Laux, D.; Cohen, P.; et al. Carbon nutrition of Escherichia coli in the mouse intestine. Proc. Natl. Acad. Sci. USA 2004, 101, 7427–7432. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gordon, J. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Osuchowski, M.F.; Valentine, C.; Kurosawa, S.; Remick, D.G. The pathogenesis of sepsis. Ann. Rev. Pathol. 2011, 6, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.; Slack, E.; Geuking, M.; McCoy, K. The mucosal firewalls against commensal intestinal microbes. Semin. Immunopathol. 2009, 31, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.; Nataro, J.; Mobley, H. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Croxen, M.; Finlay, B. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 2010, 8, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Russo, T.; Johnson, J. Medical and economic impact of extraintestinal infections due to Escherichia coli: Focus on an increasingly important endemic problem. Microbes Infect. 2003, 5, 449–456. [Google Scholar] [CrossRef]
- Morris, K.M.; Aden, D.P.; Knowles, B.B.; Colten, H.R. Complement biosynthesis by the human hepatoma-derived cell line HepG2. J. Clin. Investig. 1982, 70, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Eisen, D.; Minchinton, R. Impact of mannose-binding lectin on susceptibility to infectious diseases. Clin. Infect. Dis. 2003, 37, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Joiner, K.; Hammer, C.; Brown, E.; Frank, M. Studies on the mechanism of bacterial resistance to complement-mediated killing. II. C8 and C9 release C5b67 from the surface of Salmonella minnesota S218 because the terminal complex does not insert into the bacterial outer membrane. J. Exp. Med. 1982, 155, 809–828. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.; Young, L.; Adamu, S. Opsonization of various capsular (K) E. coli by the alternative complement pathway. Immunology 1983, 50, 497–502. [Google Scholar] [PubMed]
- Horwitz, M.; Silverstein, S. Influence of the Escherichia coli capsule on complement fixation and on phagocytosis and killing by human phagocytes. J. Clin. Investig. 1980, 65, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Rautemaa, R.; Jarvis, G.; Marnila, P.; Meri, S. Acquired resistance of Escherichia coli to complement lysis by binding of glycophosphoinositol-anchored protectin (CD59). Infect. Immun. 1998, 66, 1928–1933. [Google Scholar] [PubMed]
- Prasadarao, N.; Blom, A.; Villoutreix, B.; Linsangan, L. A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J. Immunol. 2002, 169, 6352–6360. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.; Manning, P.; Achtman, M. Promoter-distal region of the tra operon of F-like sex factor R100 in Escherichia coli K-12. J. Bacteriol. 1982, 150, 89–99. [Google Scholar] [PubMed]
- Pramoonjago, P.; Kaneko, M.; Kinoshita, T.; Ohtsubo, E.; Taketa, J.; Hong, K.; Inagi, R.; Inoue, K. Role of TraT protein, an anticomplementary protein produced in Escherichia coli by R100 factor, in serum resistance. J. Immunol. 1992, 148, 827–836. [Google Scholar] [PubMed]
- Ogata, R.; Winters, C.; Levine, R. Nucleotide sequence analysis of the complement resistance gene from plasmid R100. J. Bacteriol. 1982, 151, 819–827. [Google Scholar] [PubMed]
- Ogata, R.T.; Levine, R.P. Characterization of complement resistance in Escherichia coli conferred by the antibiotic resistance plasmid R100. J. Immunol. 1980, 125, 1494–1498. [Google Scholar] [PubMed]
- Liu, Y.-F.; Yan, J.-J.; Lei, H.-Y.; Teng, C.-H.; Wang, M.-C.; Tseng, C.-C.; Wu, J.-J. Loss of outer membrane protein C in Escherichia coli contributes to both antibiotic resistance and escaping antibody-dependent bactericidal activity. Infect. Immun. 2012, 80, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Roantree, R.; Rantz, L. A study of the relationship of the normal bactericidal activity of human serum to bacterial infection. J. Clin. Investig. 1960, 39, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.; Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 230, 13–18. [Google Scholar] [CrossRef]
- Anderson, G.; O’Toole, G. Innate and induced resistance mechanisms of bacterial biofilms. Curr. Top. Microbiol. Immunol. 2008, 322, 85–105. [Google Scholar] [PubMed]
- Barnhart, M.; Chapman, M. Curli biogenesis and function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Zhou, Y.; Salgado, P.; Patwardhan, A.; McGuffie, M.; Pape, T.; Grabe, G.; Ashman, E.; Constable, S.; Simpson, P.; et al. Atomic resolution insights into curli fiber biogenesis. Structure 2011, 19, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Gerstel, U.; Römling, U. Oxygen tension and nutrient starvation are major signals that regulate agfD promoter activity and expression of the multicellular morphotype in Salmonella typhimurium. Environ. Microbiol. 2001, 3, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Kai-Larsen, Y.; Lüthje, P.; Chromek, M.; Peters, V.; Wang, X.; Holm, A.; Kádas, L.; Hedlund, K.-O.; Johansson, J.; Chapman, M.; et al. Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL-37. PLoS Pathog. 2010, 6, e1001010. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.; Thate, T.; Craig, N. Definition of the Escherichia coli MC4100 genome by use of a DNA array. J. Bacteriol. 2003, 185, 2017–2021. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Puchtler, H.; Sweat, F.; Levine, M. On the binding of Congo red by amyloid. J. Histochem. Cytochem. 1962, 10, 355–364. [Google Scholar] [CrossRef]
- Zogaj, X.; Bokranz, W.; Nimtz, M.; Romling, U. Production of cellulose and curli fimbriae by members of the family Enterobacteriaceae isolated from the human gastrointestinal tract. Infect. Immun. 2003, 71, 4151–4158. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Brauner, A.; Li, Y.; Normark, S. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. J. Infect. Dis. 2000, 181, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Yan, Z.; Hansson, G.; Thorén, P.; Normark, S. Activation of inducible nitric oxide synthase/nitric oxide by curli fibers leads to a fall in blood pressure during systemic Escherichia coli infection in mice. J. Infect. Dis. 2001, 183, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Tükel, C.; Raffatellu, M.; Humphries, A.; Wilson, R.; Andrews-Polymenis, H.; Gull, T.; Figueiredo, J.; Wong, M.; Michelsen, K.; Akçelik, M.; et al. CsgA is a pathogen-associated molecular pattern of Salmonella enterica serotype Typhimurium that is recognized by Toll-like receptor 2. Mol. Microbiol. 2005, 58, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Tükel, C.; Wilson, R.; Nishimori, J.; Pezeshki, M.; Chromy, B.; Bäumler, A. Responses to amyloids of microbial and host origin are mediated through toll-like receptor 2. Cell Host Microbe 2009, 6, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Tükel, C.; Nishimori, J.; Wilson, R.; Winter, M.; Keestra, A.; van Putten, J.; Bäumler, A. Toll-like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms. Cell. Microbiol. 2010, 12, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, S.G. Temple University: Philadelphia PA. unpublished work.
- Wilson, R.P.; Winter, S.E.; Spees, A.M.; Winter, M.G.; Nishimori, J.H.; Sanchez, J.F.; Nuccio, S.P.; Crawford, R.W.; Tukel, C.; Baumler, A.J. The Vi capsular polysaccharide prevents complement receptor 3-mediated clearance of Salmonella enterica serotype Typhi. Infect. Immun. 2011, 79, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Sim, R.; Kishore, U.; Villiers, C.; Marche, P.; Mitchell, D. C1q binding and complement activation by prions and amyloids. Immunobiology 2007, 212, 355–417. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Pop, M.; Deboy, R.; Eckburg, P.; Turnbaugh, P.; Samuel, B.; Gordon, J.; Relman, D.; Fraser-Liggett, C.; Nelson, K. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.; Ley, R.; Hamady, M.; Fraser-Liggett, C.; Knight, R.; Gordon, J. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D. Regulation by membrane sialic acid of beta1H-dependent decay-dissociation of amplification C3 convertase of the alternative complement pathway. Proc. Natl. Acad. Sci. USA 1978, 75, 1971–1975. [Google Scholar] [CrossRef] [PubMed]
- Pluschke, G.; Mayden, J.; Achtman, M.; Levine, R. Role of the capsule and the O antigen in resistance of O18:K1 Escherichia coli to complement-mediated killing. Infect. Immun. 1983, 42, 907–913. [Google Scholar] [PubMed]
- Costerton, J.; Stewart, P.; Greenberg, E. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Kristian, S.; Birkenstock, T.; Sauder, U.; Mack, D.; Götz, F.; Landmann, R. Biofilm formation induces C3a release and protects Staphylococcus epidermidis from IgG and complement deposition and from neutrophil-dependent killing. J. Infect. Dis. 2008, 197, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-C.; Tseng, C.-C.; Chen, C.-Y.; Wu, J.-J.; Huang, J.-J. The role of bacterial virulence and host factors in patients with Escherichia coli bacteremia who have acute cholangitis or upper urinary tract infection. Clin. Infect. Dis. 2002, 35, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Zanardo, L.; Galvão, N.; Carvalho, I.; Nero, L.; Moreira, M. Escherichia coli from clinical mastitis: Serotypes and virulence factors. J. Vet. Diagn. Investig. 2011, 23, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Devine, D.; Roberts, A. K1, K5 and O antigens of Escherichia coli in relation to serum killing via the classical and alternative complement pathways. J. Med. Microbiol. 1994, 41, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Tacnet-Delorme, P.; Chevallier, S.; Arlaud, G. Beta-amyloid fibrils activate the C1 complex of complement under physiological conditions: Evidence for a binding site for A beta on the C1q globular regions. J. Immunol. 2001, 167, 6374–6381. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Cooper, N.R.; Webster, S.; Schultz, J.; McGeer, P.L.; Styren, S.D.; Civin, W.H.; Brachova, L.; Bradt, B.; Ward, P.; et al. Complment activation by beta-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10016–10020. [Google Scholar] [CrossRef] [PubMed]
- Trouw, L.; Nielsen, H.; Minthon, L.; Londos, E.; Landberg, G.; Veerhuis, R.; Janciauskiene, S.; Blom, A. C4b-binding protein in Alzheimer’s disease: Binding to Abeta1-42 and to dead cells. Mol. Immunol. 2008, 45, 3649–3660. [Google Scholar] [CrossRef] [PubMed]
- De Beer, F.; Baltz, M.; Holford, S.; Feinstein, A.; Pepys, M. Fibronectin and C4-binding protein are selectively bound by aggregated amyloid P component. J. Exp. Med. 1981, 154, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.; Jonsson, A.; Normark, S. Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli. Nature 1989, 338, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Kibbey, M.; Jucker, M.; Weeks, B.; Neve, R.; van Nostrand, W.; Kleinman, H. Beta-amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc. Natl. Acad. Sci. USA 1993, 90, 10150–10153. [Google Scholar] [CrossRef] [PubMed]
- Sjöbring, U.; Pohl, G.; Olsén, A. Plasminogen, absorbed by Escherichia coli expressing curli or by Salmonella enteritidis expressing thin aggregative fimbriae, can be activated by simultaneously captured tissue-type plasminogen activator (t-PA). Mol. Microbiol. 1994, 14, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Tucker, H.; Kihiko, M.; Caldwell, J.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.; Rydel, R.; et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [PubMed]
- Maas, C.; Schiks, B.; Strangi, R.; Hackeng, T.; Bouma, B.; Gebbink, M.; Bouma, B. Identification of fibronectin type I domains as amyloid-binding modules on tissue-type plasminogen activator and three homologs. Amyloid 2008, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Collinson, S.; Emödy, L.; Müller, K.; Trust, T.; Kay, W. Purification and characterization of thin, aggregative fimbriae from Salmonella enteritidis. J. Bacteriol. 1991, 173, 4773–4781. [Google Scholar] [CrossRef] [PubMed]
- Humphries, A.; Raffatellu, M.; Winter, S.; Weening, E.; Kingsley, R.; Droleskey, R.; Zhang, S.; Figueiredo, J.; Khare, S.; Nunes, J.; et al. The use of flow cytometry to detect expression of subunits encoded by 11 Salmonella enterica serotype Typhimurium fimbrial operons. Mol. Microbiol. 2003, 48, 1357–1376. [Google Scholar] [CrossRef] [PubMed]
- Collinson, S.; Doig, P.; Doran, J.; Clouthier, S.; Trust, T.; Kay, W. Thin, aggregative fimbriae mediate binding of Salmonella enteritidis to fibronectin. J. Bacteriol. 1993, 175, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Römling, U.; Bokranz, W.; Rabsch, W.; Zogaj, X.; Nimtz, M.; Tschäpe, H. Occurrence and regulation of the multicellular morphotype in Salmonella serovars important in human disease. Int. J. Med. Microbiol. 2003, 293, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.P. Comparison of ethyleneglycoltetraacetic acid and its magnesium salt as reagent for studying alternative complement pathway function. Infect. Immun. 1977, 16, 124–128. [Google Scholar] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biesecker, S.G.; Nicastro, L.K.; Wilson, R.P.; Tükel, Ç. The Functional Amyloid Curli Protects Escherichia coli against Complement-Mediated Bactericidal Activity. Biomolecules 2018, 8, 5. https://doi.org/10.3390/biom8010005
Biesecker SG, Nicastro LK, Wilson RP, Tükel Ç. The Functional Amyloid Curli Protects Escherichia coli against Complement-Mediated Bactericidal Activity. Biomolecules. 2018; 8(1):5. https://doi.org/10.3390/biom8010005
Chicago/Turabian StyleBiesecker, Steven G., Lauren K. Nicastro, R. Paul Wilson, and Çağla Tükel. 2018. "The Functional Amyloid Curli Protects Escherichia coli against Complement-Mediated Bactericidal Activity" Biomolecules 8, no. 1: 5. https://doi.org/10.3390/biom8010005
APA StyleBiesecker, S. G., Nicastro, L. K., Wilson, R. P., & Tükel, Ç. (2018). The Functional Amyloid Curli Protects Escherichia coli against Complement-Mediated Bactericidal Activity. Biomolecules, 8(1), 5. https://doi.org/10.3390/biom8010005