
Chromatin Remodeling and Transcriptional Control in Innate Immunity: Emergence of Akirin2 as a Novel Player

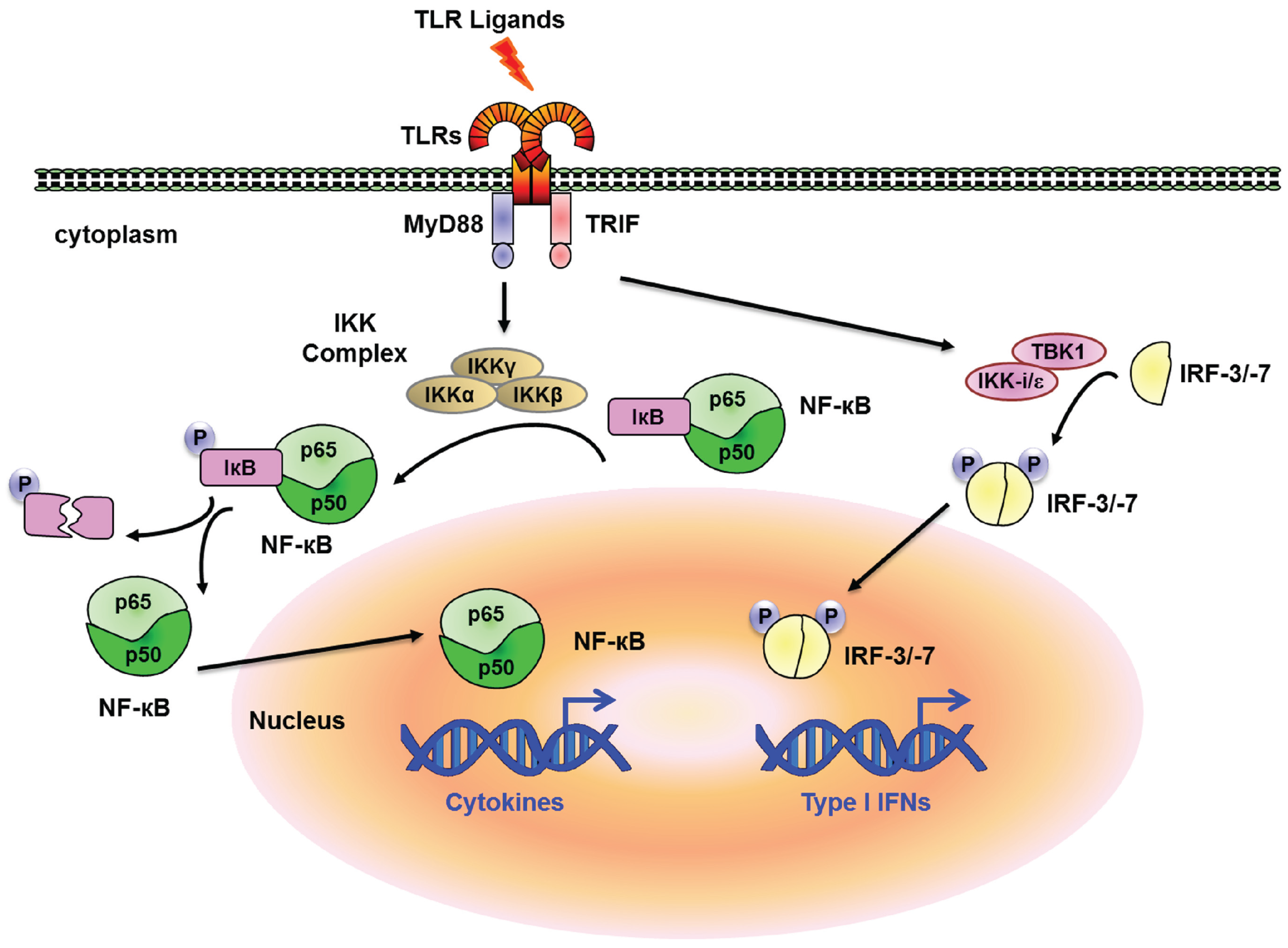
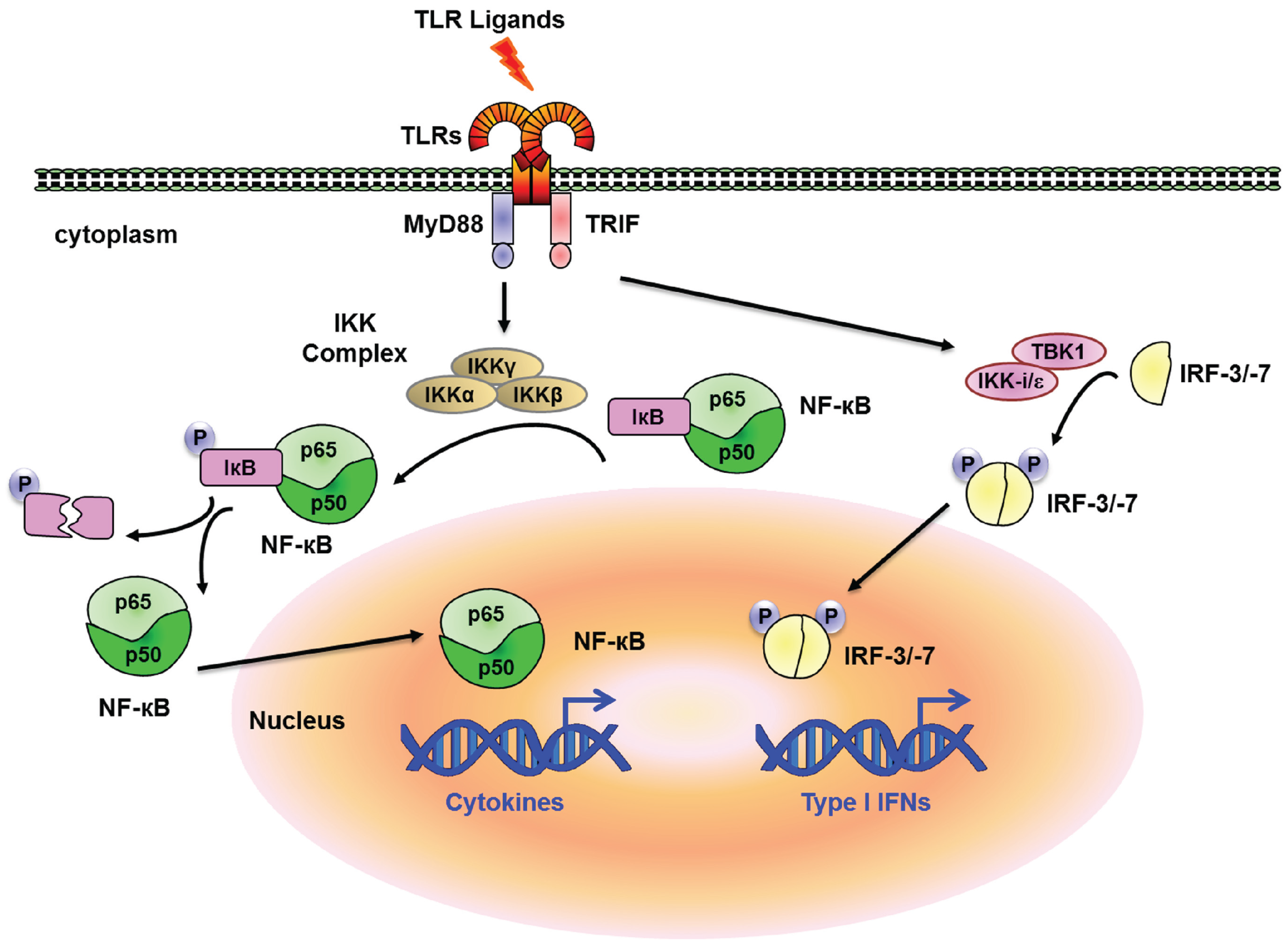{kind=link}
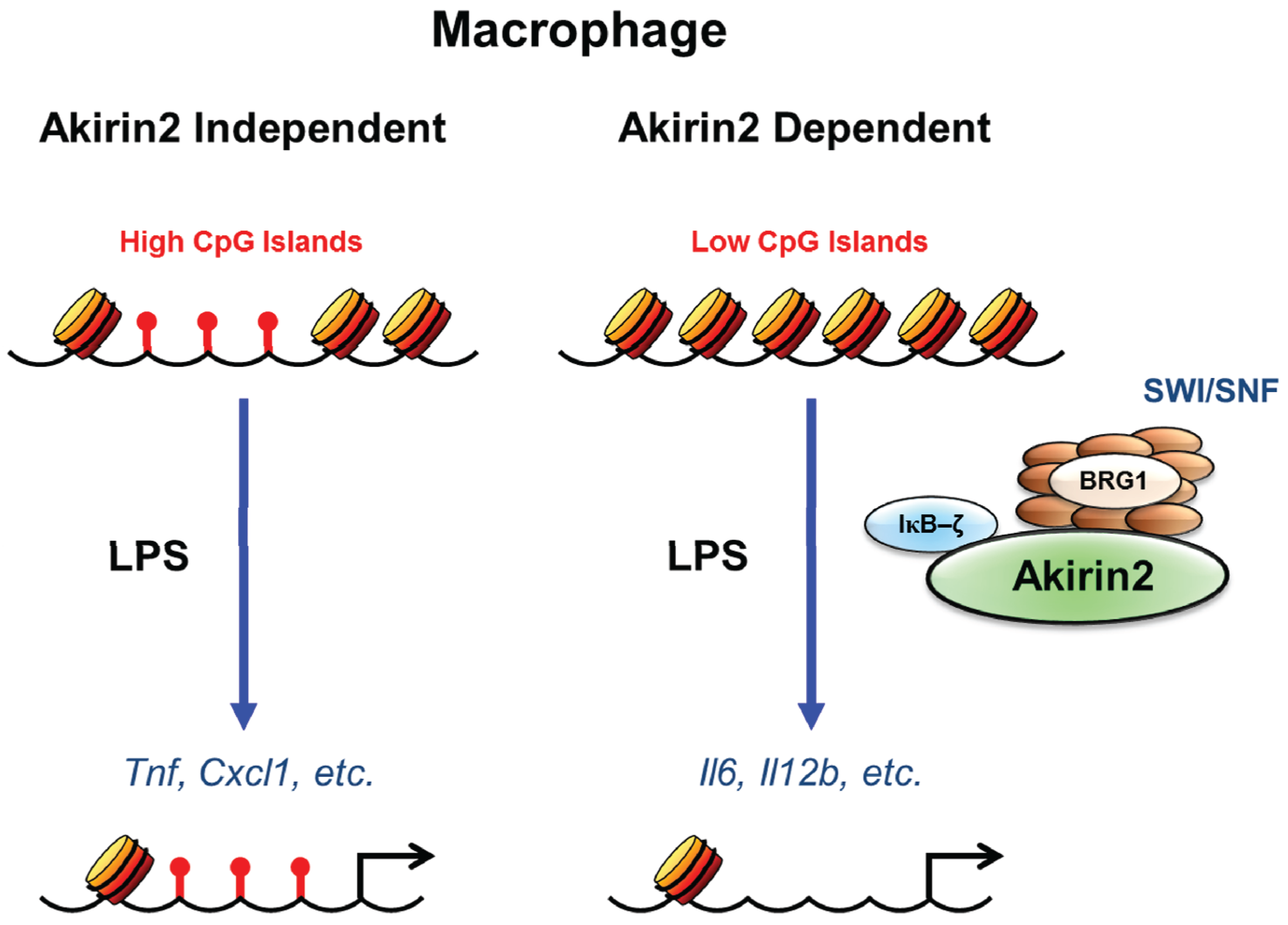{kind=link}
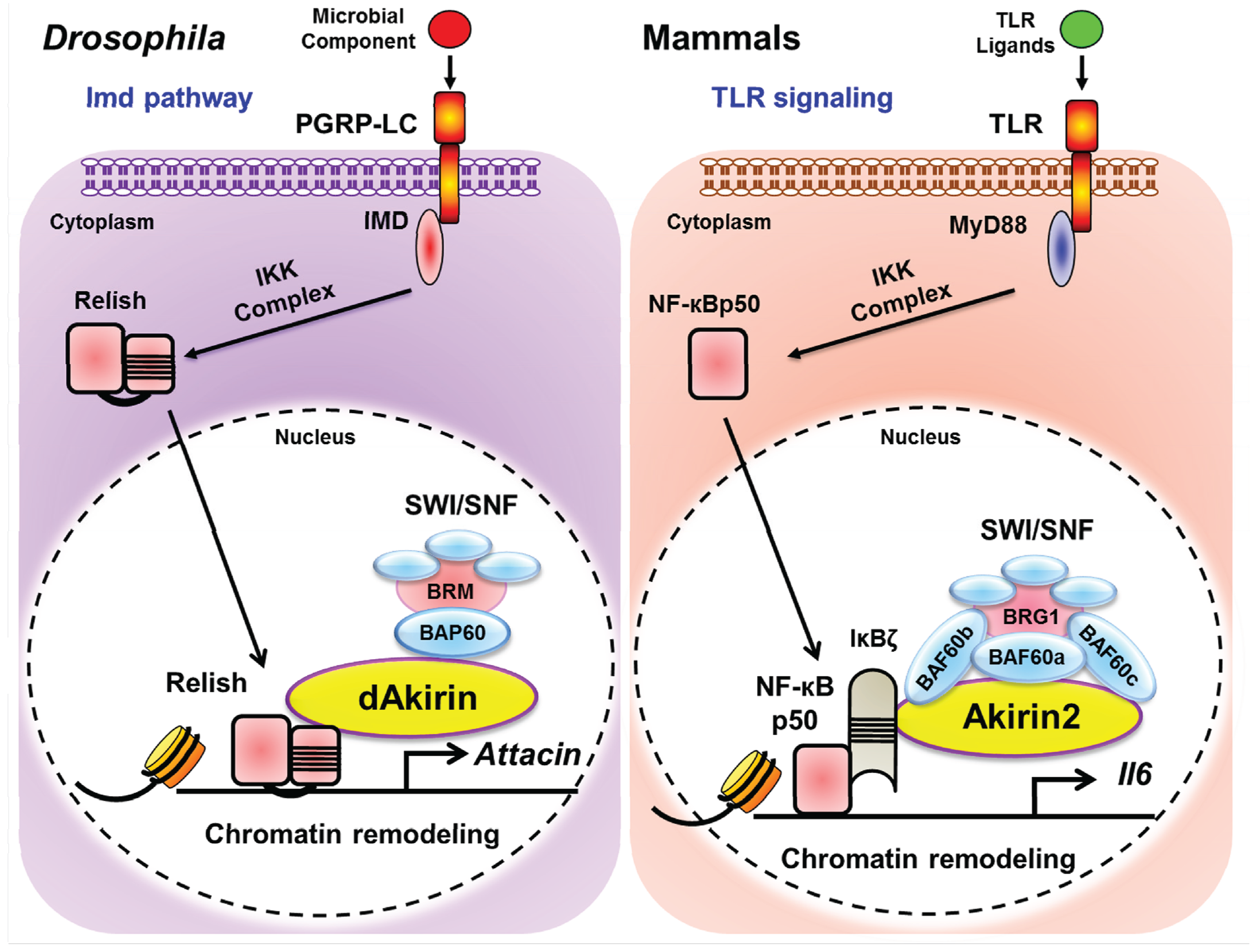{kind=link}
Abstract
:1. Introduction
2. Transcriptional Regulation and Chromatin Remodeling
2.1. Influences of Histone Modifications on Transcriptional Activation
2.2. Specificity of Chromatin Responses on Transcriptional Activation
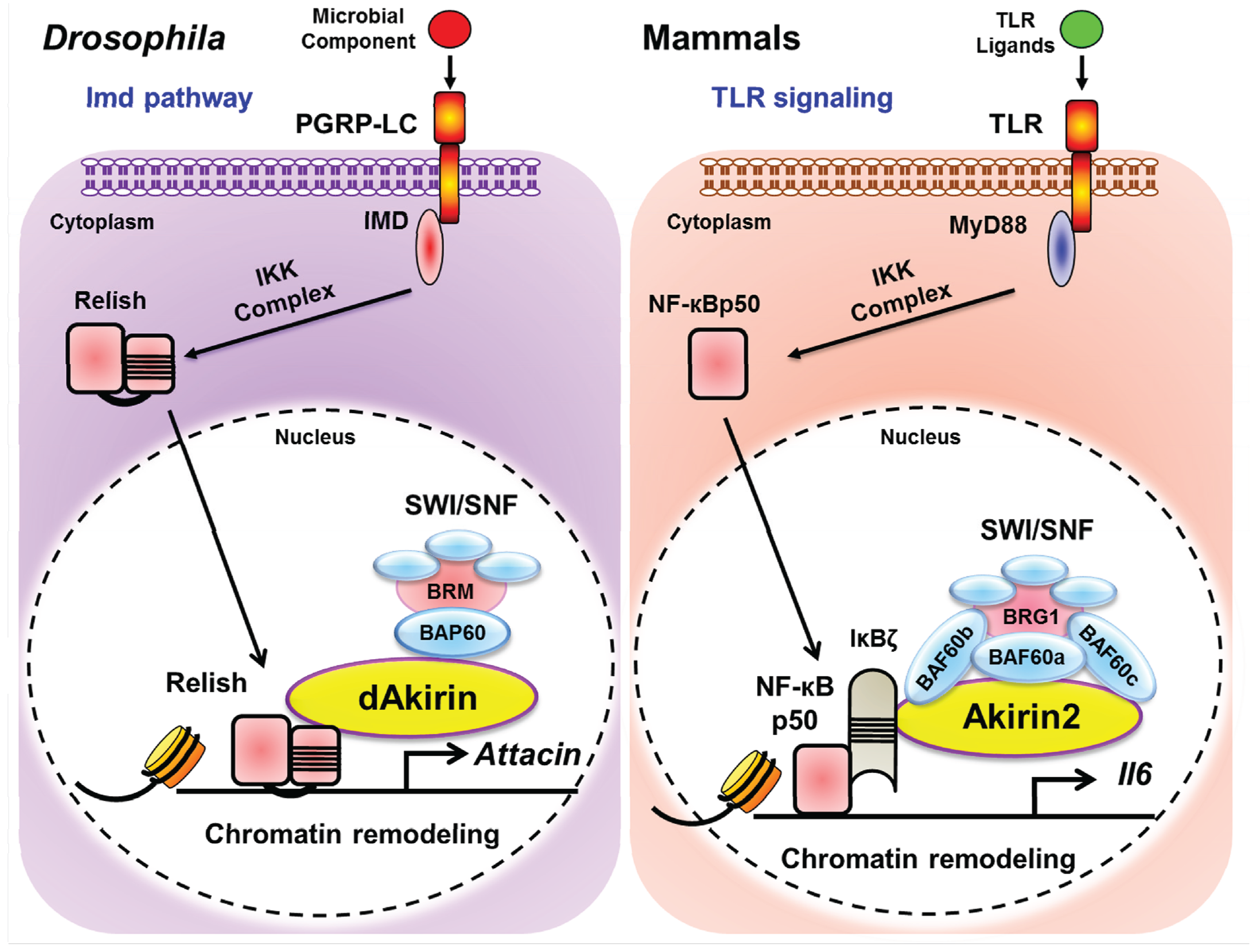
3. Akirin2 Mediated Transcriptional Control of NF-κB Dependent Genes
3.1. Identification of Akirin as a Protein Involved in NF-κB-Dependent Transcription
3.2. Identification of Akirin2 Binding Partners and their Role in Immune Regulation
3.3. SWI/SNF and CpG Island Dependency of Akirin Mediated Transcriptional Induction
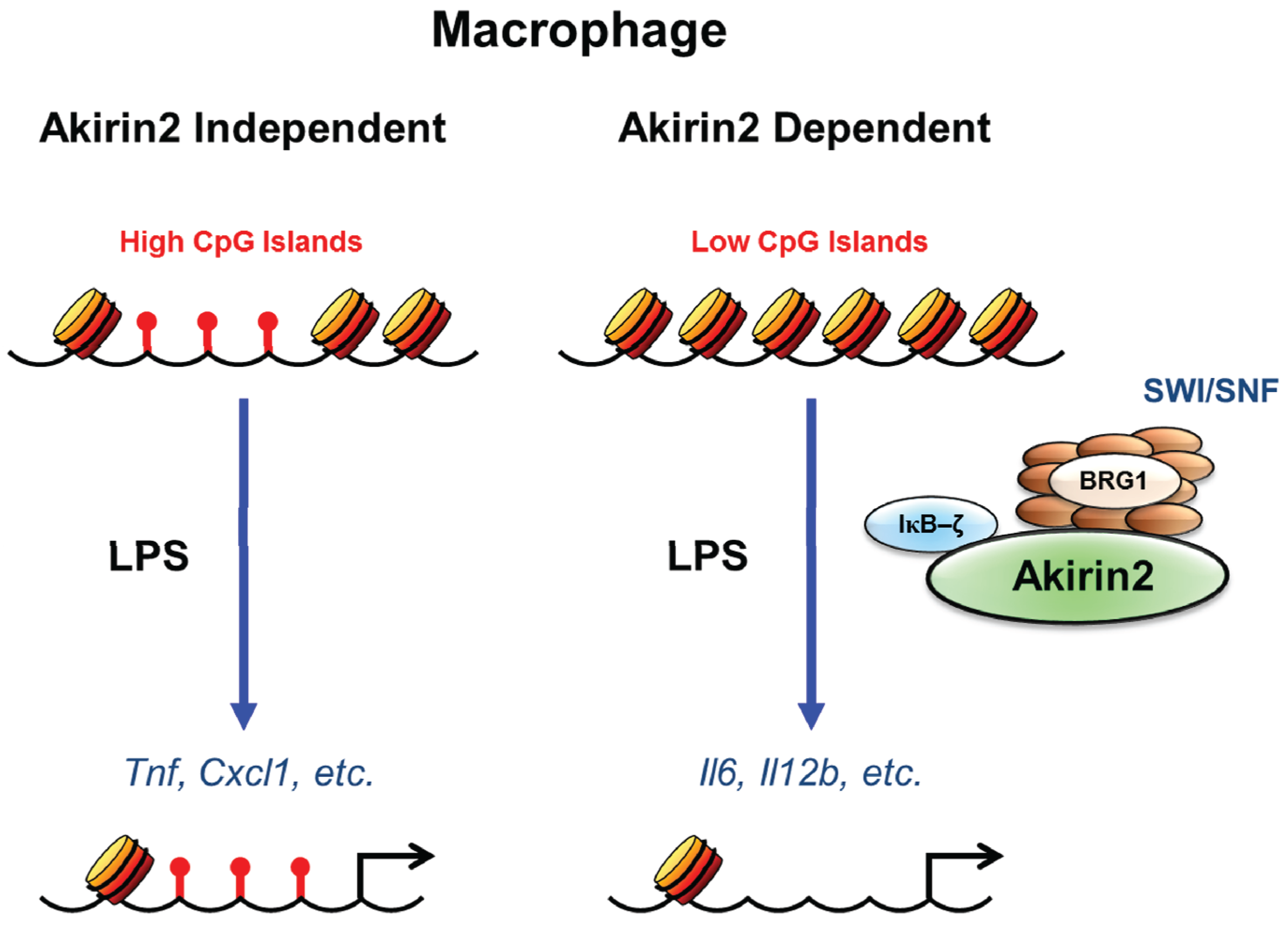
3.4. IFN-Inducible Gene Expression and Akirin2
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beutler, B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol. Rev. 2009, 227, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Ronald, P.C.; Beutler, B. Plant and animal sensors of conserved microbial signatures. Science 2010, 330, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Anderson, K.V. Drosophila: The genetics of innate immune recognition and response. Annu. Rev. Immunol. 2004, 22, 457–483. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, C.; Reichhart, J.M. Evolution and integration of innate immune systems from fruit flies to man: Lessons and questions. J. Endotoxin. Res. 2005, 11, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, K. Evolution of innate immunity: Clues from invertebrates via fish to mammals. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M.; Medzhitov, R. Toll-like receptor signaling pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012, 13, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Vignali, M.; Hassan, A.H.; Neely, K.E.; Workman, J.L. ATP-dependent chromatin-remodeling complexes. Mol. Cell. Biol. 2000, 20, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Wade, P.A. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat. Rev. Cancer 2011, 11, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Conaway, R.C.; Conaway, J.W. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem. Sci. 2009, 34, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Vinayachandran, V.; Pugh, B.F. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell 2013, 154, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T.; Tarakhovsky, A.; Natoli, G. Chromatin contributions to the regulation of innate immunity. Annu. Rev. Immunol. 2014, 32, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T.; Natoli, G. Transcriptional control of inflammatory responses. Cold Spring Harb. Perspect. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Bonnay, F.; Nguyen, X.H.; Cohen-Berros, E.; Troxler, L.; Batsche, E.; Camonis, J.; Takeuchi, O.; Reichhart, J.M.; Matt, N. Akirin specifies NF-κB selectivity of drosophila innate immune response via chromatin remodeling. EMBO J. 2014, 33, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Tartey, S.; Matsushita, K.; Vandenbon, A.; Ori, D.; Imamura, T.; Mino, T.; Standley, D.M.; Hoffmann, J.A.; Reichhart, J.M.; Akira, S.; et al. Akirin2 is critical for inducing inflammatory genes by bridging IκB-ζ and the SWI/SNF complex. EMBO J. 2014, 33, 2332–2348. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.F.; Vakoc, C.R. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014, 30, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Roberts, C.W.M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Messner, S.; Hottiger, M.O. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011, 21, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Reinke, H.; Horz, W. Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol. Cell 2003, 11, 1599–1607. [Google Scholar] [CrossRef]
- Zhao, J.; Herrera-Diaz, J.; Gross, D.S. Domain-wide displacement of histones by activated heat shock factor occurs independently of SWI/SNF and is not correlated with RNA polymerase II density. Mol. Cell. Biol. 2005, 25, 8985–8999. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.; Neely, K.E.; Workman, J.L. Histone acetyltransferase complexes stabilize SWI/SNF binding to promoter nucleosomes. Cell 2001, 104, 817–827. [Google Scholar] [CrossRef]
- Ito, T.; Ikehara, T.; Nakagawa, T.; Kraus, W.L.; Muramatsu, M. P300-mediated acetylation facilitates the transfer of histone H2A-H2B dimers from nucleosomes to a histone chaperone. Genes Dev. 2000, 14, 1899–1907. [Google Scholar] [PubMed]
- Lundblad, J.R.; Kwok, R.P.; Laurance, M.E.; Harter, M.L.; Goodman, R.H. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature 1995, 374, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Sellers, W.R.; Livingston, D.M.; Eckner, R. E1A-associated p300 and creb-associated CBP belong to a conserved family of coactivators. Cell 1994, 77, 799–800. [Google Scholar] [CrossRef]
- Eckner, R.; Ewen, M.E.; Newsome, D.; Gerdes, M.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994, 8, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, R.J.; Schiltz, R.L.; Chakravarti, D.; Nash, A.; Nagy, L.; Privalsky, M.L.; Nakatani, Y.; Evans, R.M. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 1997, 90, 569–580. [Google Scholar] [CrossRef]
- Spencer, T.E.; Jenster, G.; Burcin, M.M.; Allis, C.D.; Zhou, J.; Mizzen, C.A.; McKenna, N.J.; Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 1997, 389, 194–198. [Google Scholar] [PubMed]
- Torchia, J.; Glass, C.; Rosenfeld, M.G. Co-activators and co-repressors in the integration of transcriptional responses. Curr. Opin. Cell Biol. 1998, 10, 373–383. [Google Scholar] [CrossRef]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Selective transcription in response to an inflammatory stimulus. Cell 2010, 140, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G. Maintaining cell identity through global control of genomic organization. Immunity 2010, 33, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Transcriptional regulation in the innate immune system. Curr. Opin. Immunol. 2012, 24, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G. Control of NF-kappaB-dependent transcriptional responses by chromatin organization. Cold Spring Harb. Perspect. Biol. 2009. [Google Scholar] [CrossRef]
- Smale, S.T. Hierarchies of NF-kappaB target-gene regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Lomvardas, S.; Parekh, B.; Yie, J.; Maniatis, T.; Thanos, D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell 2000, 103, 667–678. [Google Scholar] [CrossRef]
- Saccani, S.; Pantano, S.; Natoli, G. Two waves of nuclear factor kappab recruitment to target promoters. J. Exp. Med. 2001, 193, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Fowler, T.; Sen, R.; Roy, A.L. Regulation of primary response genes. Mol. Cell 2011, 44, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Herschman, H.R. Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 1991, 60, 281–319. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Matsushita, K.; Gesellchen, V.; El Chamy, L.; Kuttenkeuler, D.; Takeuchi, O.; Hoffmann, J.A.; Akira, S.; Boutros, M.; Reichhart, J.M. Akirins are highly conserved nuclear proteins required for NF-kappaB-dependent gene expression in drosophila and mice. Nat. Immunol. 2008, 9, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Macqueen, D.J.; Johnston, I.A. Evolution of the multifaceted eukaryotic akirin gene family. BMC Evol. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Nowak, S.J.; Aihara, H.; Gonzalez, K.; Nibu, Y.; Baylies, M.K. Akirin links twist-regulated transcription with the Brahma chromatin remodeling complex during embryogenesis. PLoS Genet. 2012, 8, e1002547. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.; Salerno, M.S.; Thomas, M.; Davies, T.; Berry, C.; Dyer, K.; Bracegirdle, J.; Watson, T.; Dziadek, M.; Kambadur, R.; et al. Mighty is a novel promyogenic factor in skeletal myogenesis. Exp. Cell Res. 2008, 314, 1013–1029. [Google Scholar] [CrossRef] [PubMed]
- Mohrmann, L.; Langenberg, K.; Krijgsveld, J.; Kal, A.J.; Heck, A.J.; Verrijzer, C.P. Differential targeting of two distinct SWI/SNF-related drosophila chromatin-remodeling complexes. Mol. Cell. Biol. 2004, 24, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Moshkin, Y.M.; Mohrmann, L.; van Ijcken, W.F.; Verrijzer, C.P. Functional differentiation of SWI/SNF remodelers in transcription and cell cycle control. Mol. Cell. Biol. 2007, 27, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.L.; Mercola, M. BAF60 A, B, and Cs of muscle determination and renewal. Genes Dev. 2012, 26, 2673–2683. [Google Scholar] [CrossRef] [PubMed]
- Forcales, S.V.; Albini, S.; Giordani, L.; Malecova, B.; Cignolo, L.; Chernov, A.; Coutinho, P.; Saccone, V.; Consalvi, S.; Williams, R.; et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 2012, 31, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, J.K.; Bruneau, B.G. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 2009, 459, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wong, R.H.; Tang, T.; Hudak, C.S.; Yang, D.; Duncan, R.E.; Sul, H.S. Phosphorylation and recruitment of BAF60c in chromatin remodeling for lipogenesis in response to insulin. Mol. Cell 2013, 49, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Lores, P.; Visvikis, O.; Luna, R.; Lemichez, E.; Gacon, G. The SWI/SNF protein BAF60b is ubiquitinated through a signalling process involving rac GTPase and the ring finger protein UnKempt. FEBS J. 2010, 277, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Muta, T.; Takeshige, K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J. Biol. Chem. 2001, 276, 27657–27662. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Yamazaki, S.; Uematsu, S.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Kuwata, H.; Takeuchi, O.; Takeshige, K.; et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature 2004, 430, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Kayama, H.; Ramirez-Carrozzi, V.R.; Yamamoto, M.; Mizutani, T.; Kuwata, H.; Iba, H.; Matsumoto, M.; Honda, K.; Smale, S.T.; Takeda, K. Class-specific regulation of pro-inflammatory genes by MyD88 pathways and IkappaBzeta. J. Biol. Chem. 2008, 283, 12468–12477. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Satoh, T.; Kato, H.; Matsushita, K.; Kumagai, Y.; Vandenbon, A.; Tani, T.; Muta, T.; Akira, S.; Takeuchi, O. IkappaBzeta is essential for natural killer cell activation in response to IL-12 and IL-18. Proc. Natl. Acad. Sci. USA 2010, 107, 17680–17685. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Iwai, Y.; Oh-Hora, M.; Yamamoto, M.; Morio, T.; Aoki, K.; Ohya, K.; Jetten, A.M.; Akira, S.; Muta, T.; et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 2010, 464, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Okuma, A.; Hoshino, K.; Ohba, T.; Fukushi, S.; Aiba, S.; Akira, S.; Ono, M.; Kaisho, T.; Muta, T. Enhanced apoptosis by disruption of the STAT3-IkappaB-zeta signaling pathway in epithelial cells induces Sjogren’s syndrome-like autoimmune disease. Immunity 2013, 38, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, M.; Yamazaki, S.; Eto-Kimura, A.; Takeshige, K.; Muta, T. Positive and negative regulation of nuclear factor-kappaB-mediated transcription by IkappaB-zeta, an inducible nuclear protein. J. Biol. Chem. 2005, 280, 7444–7451. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Chi, T. A BAF-centred view of the immune system. Nat. Rev. Immunol. 2004, 4, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Graumann, J.; Wu, G.; Arauzo-Bravo, M.J.; Han, D.W.; Greber, B.; Gentile, L.; Mann, M.; Scholer, H.R. Chromatin-remodeling components of the BAF complex facilitate reprogramming. Cell 2010, 141, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Carrozzi, V.R.; Braas, D.; Bhatt, D.M.; Cheng, C.S.; Hong, C.; Doty, K.R.; Black, J.C.; Hoffmann, A.; Carey, M.; Smale, S.T. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 2009, 138, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Carrozzi, V.R.; Nazarian, A.A.; Li, C.C.; Gore, S.L.; Sridharan, R.; Imbalzano, A.N.; Smale, S.T. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006, 20, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Nanty, L.; Carbajosa, G.; Heap, G.A.; Ratnieks, F.; van Heel, D.A.; Down, T.A.; Rakyan, V.K. Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 2011, 21, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Tartey, S.; Matsushita, K.; Imamura, T.; Wakabayashi, A.; Ori, D.; Mino, T.; Takeuchi, O. Essential function for the nuclear protein Akirin2 in B cell activation and humoral immune responses. J. Immunol. 2015, 195, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Cui, K.; Murray, D.M.; Ling, C.; Xue, Y.; Gerstein, A.; Parsons, R.; Zhao, K.; Wang, W. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 2005, 19, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Qian, F.; Hu, Y.; Ang, C.; Li, Z.; Wen, Z. Chromatin-remodelling factor brg1 selectively activates a subset of interferon-alpha-inducible genes. Nat. Cell Biol. 2002, 4, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kang, H.; Liu, R.; Chen, X.; Zhao, K. Maximal induction of a subset of interferon target genes requires the chromatin-remodeling activity of the BAF complex. Mol. Cell. Biol. 2002, 22, 6471–6479. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tartey, S.; Takeuchi, O. Chromatin Remodeling and Transcriptional Control in Innate Immunity: Emergence of Akirin2 as a Novel Player. Biomolecules 2015, 5, 1618-1633. https://doi.org/10.3390/biom5031618
Tartey S, Takeuchi O. Chromatin Remodeling and Transcriptional Control in Innate Immunity: Emergence of Akirin2 as a Novel Player. Biomolecules. 2015; 5(3):1618-1633. https://doi.org/10.3390/biom5031618
Chicago/Turabian StyleTartey, Sarang, and Osamu Takeuchi. 2015. "Chromatin Remodeling and Transcriptional Control in Innate Immunity: Emergence of Akirin2 as a Novel Player" Biomolecules 5, no. 3: 1618-1633. https://doi.org/10.3390/biom5031618
APA StyleTartey, S., & Takeuchi, O. (2015). Chromatin Remodeling and Transcriptional Control in Innate Immunity: Emergence of Akirin2 as a Novel Player. Biomolecules, 5(3), 1618-1633. https://doi.org/10.3390/biom5031618