
Oxidative Stress and the Homeodynamics of Iron Metabolism



{kind=link}
{kind=link}
Abstract
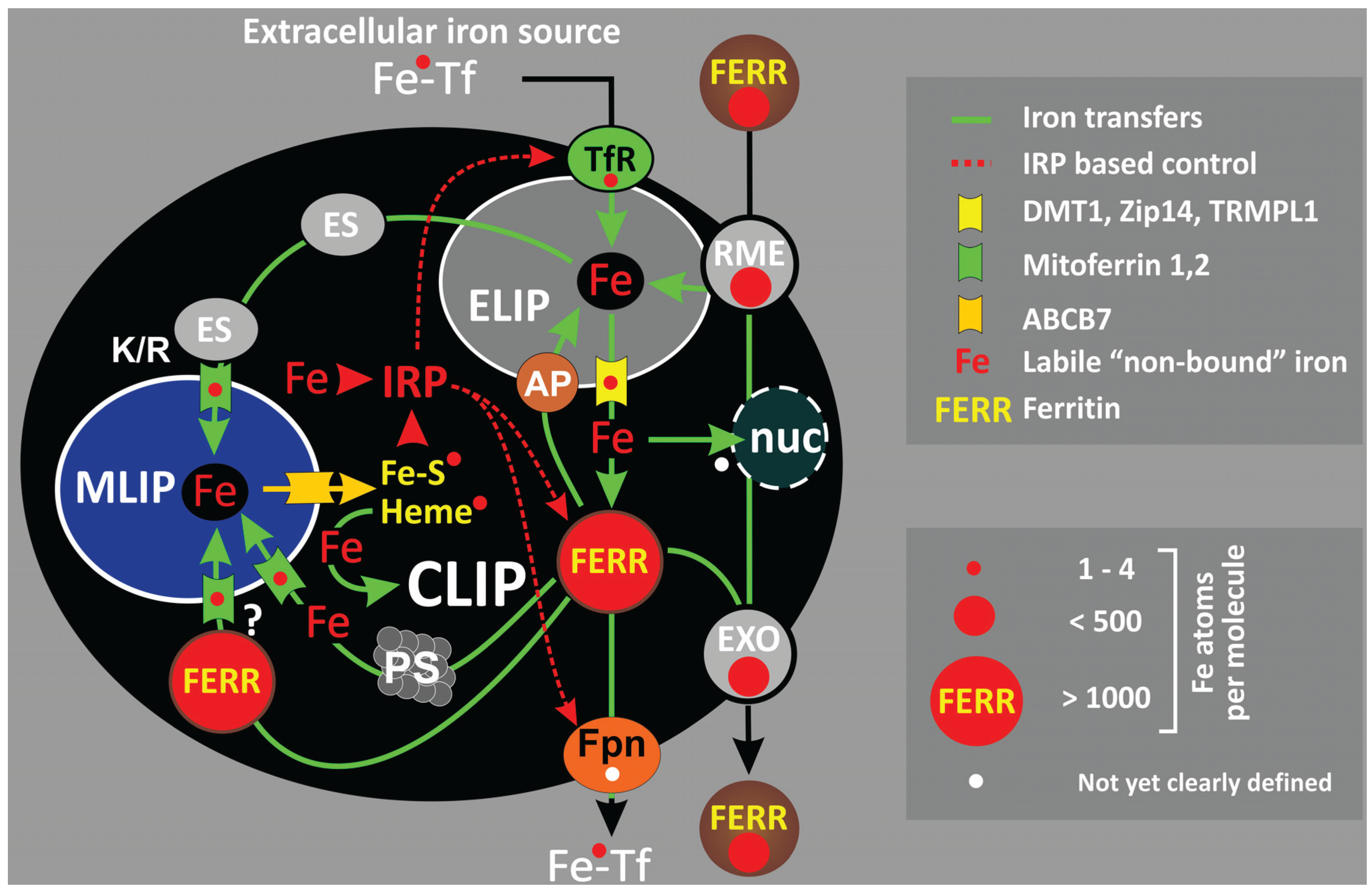
:1. Systemic and Cellular Iron Transfers
2. Cellular Iron Compartmentalization
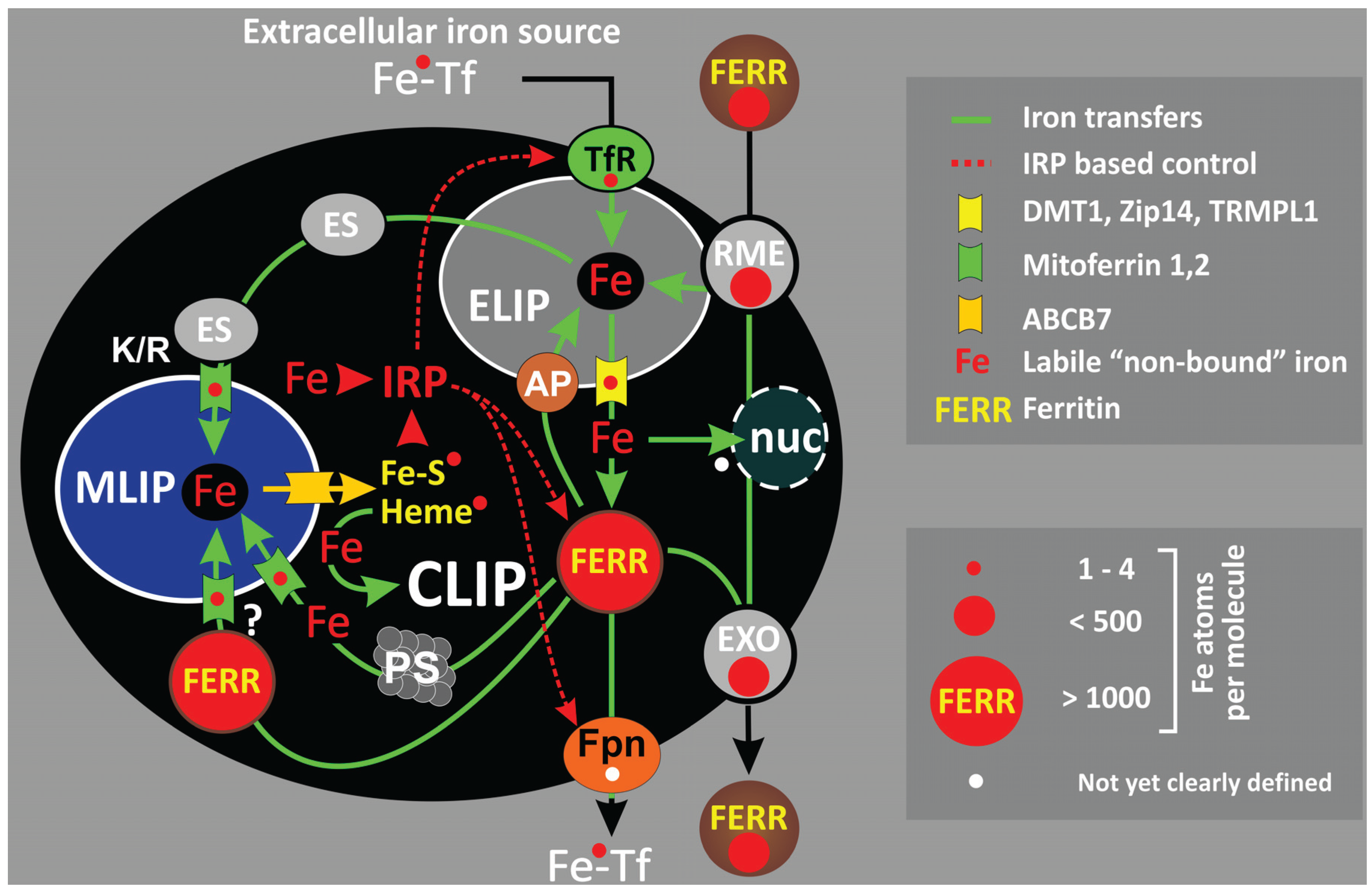
2.1. Endo-/Lysosomal Iron—ELIP
2.2. Ferritin—CLIP
2.3. The Mitochondrial Iron Pool—MLIP
2.4. Mitochondrial Iron Usage—Frataxin and Mitochondrial Ferritin
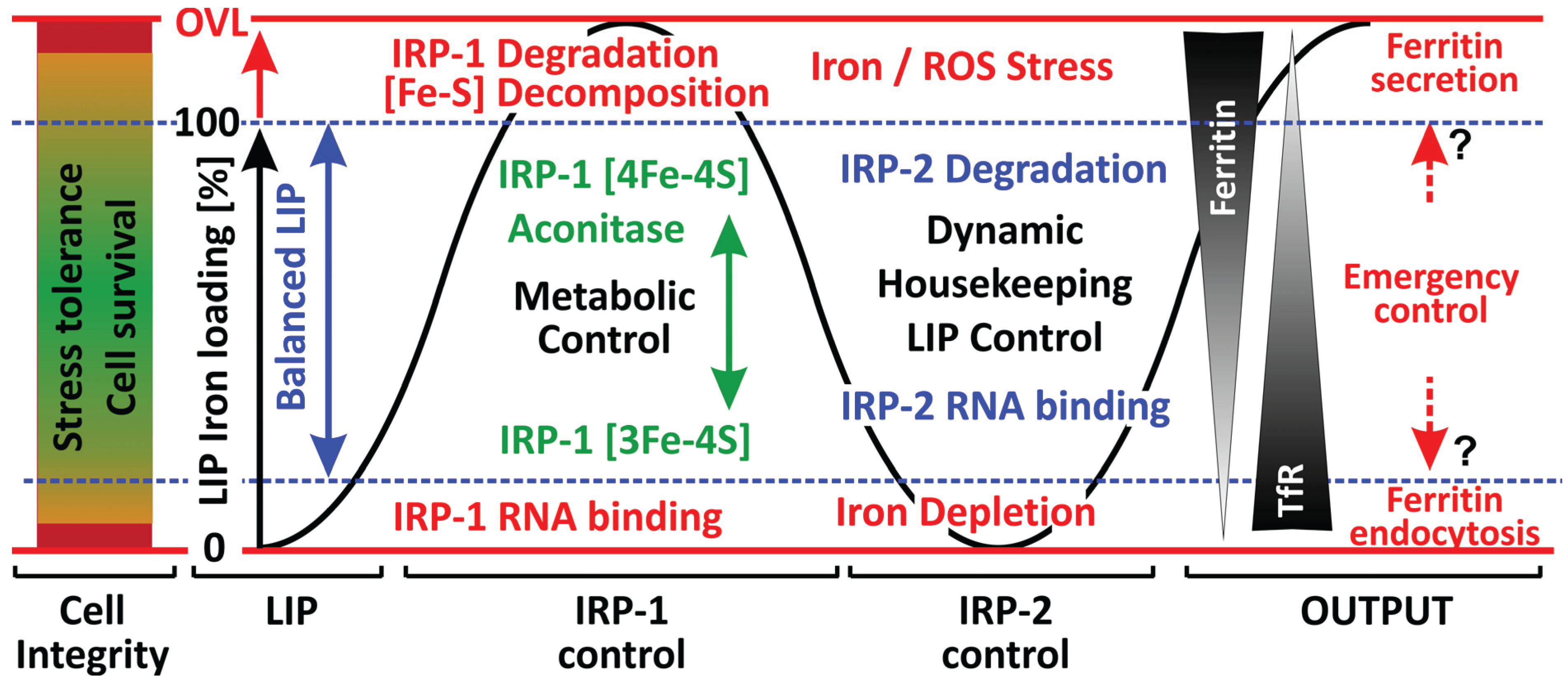
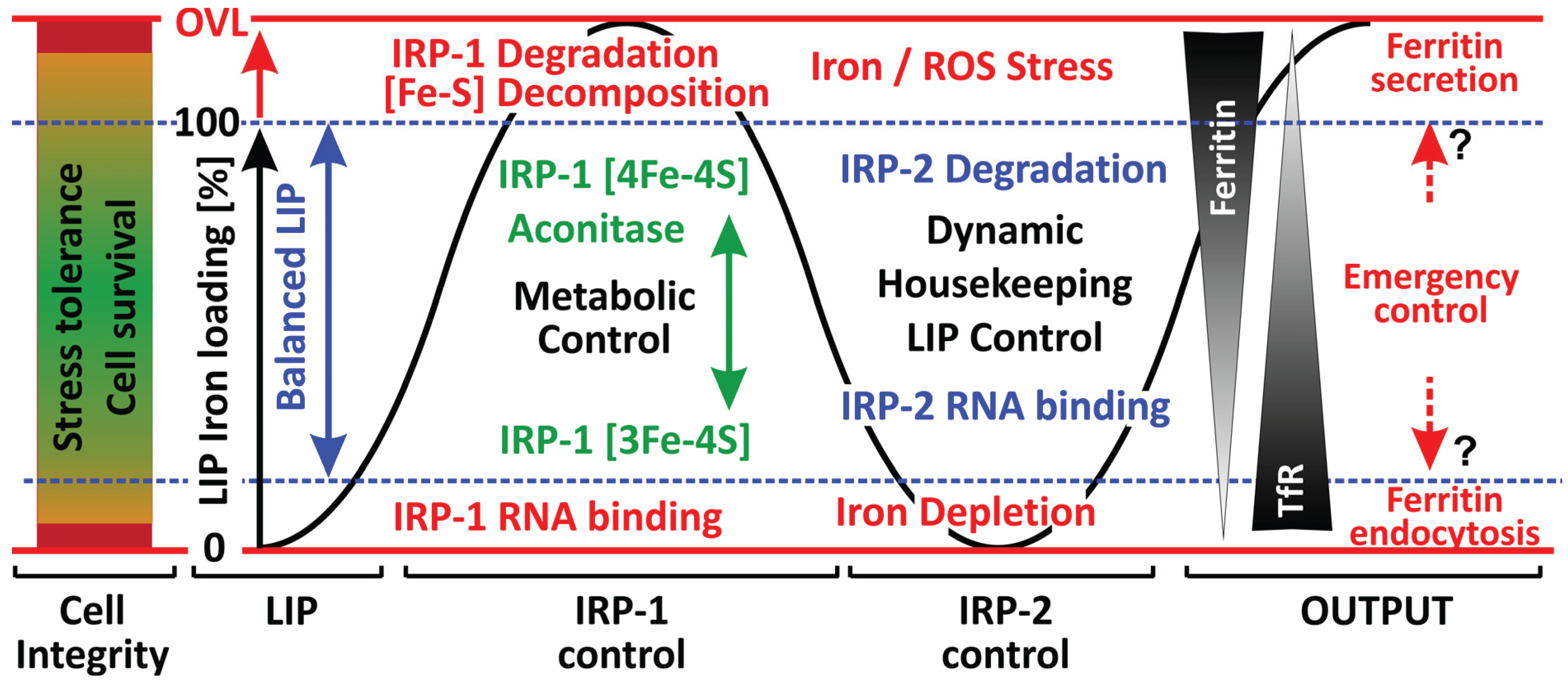
3. Iron Regulatory Proteins (IRP-1, IRP-2)
3.1. IRP-1: Redox-Based Control of Cellular Iron Homeodynamics
3.2. Fe-S Cluster Oxidation
3.3. IRP-2
3.4. NO Signaling and IRP Regulation
3.5. Additional Regulatory Roles of IRPs
4. Iron Homeodynamics under Stress Conditions—A Distinct Role for Ferritin?
5. Conclusions
Abbreviations
| CLIP | cytosolic labile iron pool |
| DMT1 | divalent metal transporter |
| ELC | endo-/lysosomal compartment |
| ELIP | endo-/lysosomal labile iron pool |
| Fpn | ferroportin |
| Ftx | frataxin |
| HNE | 4-hydroxy-nonenal |
| FRDA | Friedreich’s ataxia |
| IRE | iron regulatory elements |
| IRP | iron regulatory protein |
| ISCU | iron-sulfur cluster forming unit |
| LIP | labile iron pool |
| LMP | lysosomal membrane permeability |
| MLIP | mitochondrial labile iron pool |
| mtFER | mitochondrial ferritin |
| NTBI | non-transferrin bound iron |
| RME | receptor mediated endocytosis |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| TBI | transferrin-bound iron |
| TfR | transferrin receptor |
| TRPML1 | type IV mucolipidosis-associated protein |
Conflicts of Interest
References
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Garrick, M.D.; Dolan, K.G.; Horbinski, C.; Ghio, A.J.; Higgins, D.; Porubcin, M.; Moore, E.G.; Hainsworth, L.N.; Umbreit, J.N.; Conrad, M.E.; et al. DMT1: A mammalian transporter for multiple metals. Biometals 2003, 16, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. The iron transporter DMT1. Int. J. Biochem. Cell Biol. 1999, 31, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Garrick, M.D.; Garrick, L.M. Cellular iron transport. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 309–325. [Google Scholar] [CrossRef]
- Conrad, M.E.; Umbreit, J.N.; Moore, E.G.; Hainsworth, L.N.; Porubcin, M.; Simovich, M.J.; Nakada, M.T.; Dolan, K.; Garrick, M.D. Separate pathways for cellular uptake of ferric and ferrous iron. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G767–G774. [Google Scholar] [PubMed]
- Conrad, M.E.; Umbreit, J.N. Pathways of iron absorption. Blood Cells Mol. Dis. 2002, 29, 336–355. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal iron absorption. J. Trace Elem. Med. Biol. 2012, 26, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed]
- Pinilla-Tenas, J.J.; Sparkman, B.K.; Shawki, A.; Illing, A.C.; Mitchell, C.J.; Zhao, N.; Liuzzi, J.P.; Cousins, R.J.; Knutson, M.D.; Mackenzie, B. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell Physiol. 2011, 301, C862–C871. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Morgan, E.H.; Baker, E. Characterisation of citrate and iron citrate uptake by cultured rat hepatocytes. J. Hepatol. 1998, 29, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.; Baker, S.M.; Morgan, E.H. Characterisation of non-transferrin-bound iron (ferric citrate) uptake by rat hepatocytes in culture. Biochim. Biophys. Acta Gen. Subj. 1998, 1380, 21–30. [Google Scholar] [CrossRef]
- Graham, R.M.; Morgan, E.H.; Baker, E. Ferric citrate uptake by cultured rat hepatocytes is inhibited in the presence of transferrin. Eur. J. Biochem. 1998, 253, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Cighetti, G.; Duca, L.; Bortone, L.; Sala, S.; Nava, I.; Fiorelli, G.; Cappellini, M.D. Oxidative status and malondialdehyde in β-thalassaemia patients. Eur. J. Clin. Investig. 2002, 32, 55–60. [Google Scholar] [CrossRef]
- Porter, J.B.; Garbowski, M. The Pathophysiology of Transfusional Iron Overload. Hematol. Oncol. Clin. N. Am. 2014, 28, 683–701. [Google Scholar] [CrossRef]
- De Luca, C.; Filosa, A.; Grandinetti, M.; Maggio, F.; Lamba, M.; Passi, S. Blood antioxidant status and urinary levels of catecholamine metabolites in beta-thalassemia. Free Radic. Res. 1999, 30, 453–462. [Google Scholar]
- Esposito, B.P.; Breuer, W.; Sirankapracha, P.; Pootrakul, P.; Hershko, C.; Cabantchik, Z.I. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood 2003, 102, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Link, G.; Cabantchik, I. Pathophysiology of iron overload. Ann. NY Acad. Sci. 1998, 850, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.; Rowley, D.A.; Griffiths, E.; Halliwell, B. Low-molecular-weight iron complexes and oxygen radical reactions in idiopathic haemochromatosis. Clin. Sci. 1985, 68, 463–467. [Google Scholar] [PubMed]
- Sibille, J.C.; Kondo, H.; Aisen, P. Interactions between isolated hepatocytes and Kupffer cells in iron metabolism: A possible role for ferritin as an iron carrier protein. Hepatology 1988, 8, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Sibille, J.C.; Kondo, H.; Aisen, P. Uptake of ferritin and iron bound to ferritin by rat hepatocytes: Modulation by apotransferrin, iron chelators and chloroquine. Biochim. Biophys. Acta 1989, 1010, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Knovich, M.A.; Coffman, L.G.; Torti, F.M.; Torti, S.V. Serum ferritin: Past, present and future. Biochim. Biophys. Acta 2010, 1800, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Sibille, J.C.; Ciriolo, M.; Kondo, H.; Crichton, R.R.; Aisen, P. Subcellular localization of ferritin and iron taken up by rat hepatocytes. Biochem. J. 1989, 262, 685–688. [Google Scholar] [PubMed]
- Kalgaonkar, S.; Lonnerdal, B. Receptor-mediated uptake of ferritin-bound iron by human intestinal Caco-2 cells. J. Nutr. Biochem. 2009, 20, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Gelvan, D.; Fibach, E.; Meyron-Holtz, E.G.; Konijn, A.M. Ferritin uptake by human erythroid precursors is a regulated iron uptake pathway. Blood 1996, 88, 3200–3207. [Google Scholar] [PubMed]
- Meyron-Holtz, E.G.; Fibach, E.; Gelvan, D.; Konijn, A.M. Binding and uptake of exogenous isoferritins by cultured human erythroid precursor cells. Br. J. Haematol. 1994, 86, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Hulet, S.W.; Hess, E.J.; Debinski, W.; Arosio, P.; Bruce, K.; Powers, S.; Connor, J.R. Characterization and distribution of ferritin binding sites in the adult mouse brain. J. Neurochem. 1999, 72, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.K.; Kong, P.A.; Gao, J.; Li, F.Y.; Qian, Z.M. Expression of ferritin receptor in placental microvilli membrane in pregnant women with different iron status at mid-term gestation. Eur. J. Clin. Nutr. 2001, 55, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Mack, U.; Storey, E.L.; Powell, L.W.; Halliday, J.W. Characterization of the binding of ferritin to the rat liver ferritin receptor. Biochim. Biophys. Acta 1985, 843, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Mizumoto, K.; Kasuya, I.; Kino, K.; Sussman, H.H.; Tsunoo, H. Human placental ferritin receptor. Biochim. Biophys. Acta Gen. Subj. 1986, 884, 31–38. [Google Scholar] [CrossRef]
- Hulet, S.W.; Heyliger, S.O.; Powers, S.; Connor, J.R. Oligodendrocyte progenitor cells internalize ferritin via clathrin-dependent receptor mediated endocytosis. J. Neurosci. Res. 2000, 61, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Li, L.; Chung, D.-H.; Allen, C.D.C.; Torti, S.V.; Torti, F.M.; Cyster, J.G.; Chen, C.-Y.; Brodsky, F.M.; Niemi, E.C.; et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J. Exp. Med. 2005, 202, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Todorich, B.; Zhang, X.; Slagle-Webb, B.; Seaman, W.E.; Connor, J.R. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J. Neurochem. 2008, 107, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebron, J.A.; Björkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.H.; Etzerodt, A.; Svendsen, P.; Moestrup, S.K. The haptoglobin-CD163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef]
- Nairz, M.; Schroll, A.; Demetz, E.; Tancevski, I.; Theurl, I.; Weiss, G. “Ride on the ferrous wheel”—The cycle of iron in macrophages in health and disease. Immunobiology 2014, 220, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Abboud, S.; Haile, D.J. A Novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.; Barlow, D. The SLC40 basolateral iron transporter family (IREG1/ferroportin/MTP1). Pflüg. Arch. 2004, 447, 801–806. [Google Scholar] [CrossRef]
- Musci, G.; Polticelli, F.; Bonaccorsi di Patti, M.C. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J. Biol. Chem. 2014, 5, 204–215. [Google Scholar] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; de Lara, F.M.; Pendás, A.M.; Garabaya, C.; Rodríguez, F.; Astudillo, A.; Bernal, T.; Cabanillas, R.; López-Otín, C.; Velasco, G. Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008, 112, 2539–2545. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; She, E.; Gelbart, T.; Truksa, J.; Lee, P.; Xia, Y.; Khovananth, K.; Mudd, S.; Mann, N.; Moresco, E.M.Y.; et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science 2008, 320, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar] [CrossRef] [PubMed]
- Vulpe, C.D.; Kuo, Y.-M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Vashchenko, G.; MacGillivray, R. Multi-copper oxidases and human iron metabolism. Nutrients 2013, 5, 2289–2313. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; xEb; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Chaston, T.B.; Matak, P.; Pourvali, K.; Srai, S.K.; McKie, A.T.; Sharp, P.A. Hypoxia inhibits hepcidin expression in HuH7 hepatoma cells via decreased SMAD4 signaling. Am. J. Physiol. Cell Physiol. 2011, 300, C888–C895. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Invest. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Hintze, K.J.; McClung, J.P. Hepcidin: A critical regulator of iron metabolism during hypoxia. Adv. Hematol. 2011. [Google Scholar] [CrossRef]
- Song, N.; Wang, J.; Jiang, H.; Xie, J. Ferroportin1 and hephaestin overexpression attenuate iron-induced oxidative stress in MES23.5 dopaminergic cells. J. Cell. Biochem. 2010, 110, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. The ferroportin disease. Blood Cells Mol. Dis. 2004, 32, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hevi, S.; Chuck, S.L. Regulated secretion of glycosylated human ferritin from hepatocytes. Blood 2004, 103, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.N.; Eubanks, S.K.; Schaffer, K.J.; Zhou, C.Y.; Linder, M.C. Secretion of ferritin by rat hepatoma cells and its regulation by inflammatory cytokines and iron. Blood 1997, 90, 4979–4986. [Google Scholar] [PubMed]
- Cohen, L.A.; Gutierrez, L.; Weiss, A.; Leichtmann-Bardoogo, Y.; Zhang, D.-L.; Crooks, D.R.; Sougrat, R.; Morgenstern, A.; Galy, B.; Hentze, M.W.; et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010, 116, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Van Deurs, B.; Von Bulow, F.; Moller, M. Vesicular transport of cationized ferritin by the epithelium of the rat choroid plexus. J. Cell Biol. 1981, 89, 131–139. [Google Scholar]
- Fisher, J.; Devraj, K.; Ingram, J.; Slagle-Webb, B.; Madhankumar, A.B.; Liu, X.; Klinger, M.; Simpson, I.A.; Connor, J.R. Ferritin: A novel mechanism for delivery of iron to the brain and other organs. Am. J. Physiol. Cell Physiol. 2007, 293, C641–C649. [Google Scholar] [CrossRef] [PubMed]
- Meyron-Holtz, E.; Moshe-Belizowski, S.; Cohen, L. A possible role for secreted ferritin in tissue iron distribution. J. Neural Transm. 2011, 118, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Todorich, B.; Zhang, X.; Connor, J.R. H-ferritin is the major source of iron for oligodendrocytes. Glia 2011, 59, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Knovich, M.A.; Storey, J.A.; Coffman, L.G.; Torti, S.V.; Torti, F.M. Ferritin for the clinician. Blood Rev. 2009, 23, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Casgrain, A.; Collings, R.; Harvey, L.J.; Hooper, L.; Fairweather-Tait, S.J. Effect of iron intake on iron status: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2012, 96, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Yip, R. Changes in iron metabolism with age. In Iron Metabolism in Health and Disease; Brock, J.H., Halliday, J.W., Pippard, M.J., Powell, L.W.E., Eds.; WB Saunders: London, UK, 1994; pp. 428–448. [Google Scholar]
- Finch, C.A.; Bellotti, V.; Stray, S.; Lipschitz, D.A.; Cook, J.D.; Pippard, M.J.; Huebers, H.A. Plasma ferritin determination as a diagnostic tool. West. J. Med. 1986, 145, 657–663. [Google Scholar] [PubMed]
- Ferraro, S.; Mozzi, R.; Panteghini, M. Revaluating serum ferritin as a marker of body iron stores in the traceability era. Clin. Chem. Lab. Med. 2012, 50, 1911–1916. [Google Scholar] [PubMed]
- Karlsson, T.; Sjöö, F.; Kedinge-Cyrus, B.; Bäckström, B. Plasma soluble transferrin receptor assay when screening for iron-deficiency in an unselected population of elderly anaemic patients. J. Intern. Med. 2010, 267, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Alkhateeb, A.A.; Connor, J.R. The significance of ferritin in cancer: Anti-oxidation, inflammation and tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2013, 1836, 245–254. [Google Scholar] [CrossRef]
- Fan, K.; Gao, L.; Yan, X. Human ferritin for tumor detection and therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Fairweather-Tait, S.J.; Wawer, A.A.; Gillings, R.; Jennings, A.; Myint, P.K. Iron status in the elderly. Mech. Ageing Dev. 2014, 136–137, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Jenkitkasemwong, S.; Wang, C.-Y.; Mackenzie, B.; Knutson, M. Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals 2012, 25, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Gao, J.; Enns, C.A.; Knutson, M.D. ZRT/IRT-like protein 14 (ZIP14) promotes the cellular assimilation of iron from transferrin. J. Biol. Chem. 2010, 285, 32141–32150. [Google Scholar] [CrossRef] [PubMed]
- Zeevi, D.A.; Frumkin, A.; Bach, G. TRPML and lysosomal function. Biochim. Biophys. Acta 2007, 1772, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Berberat, P.O.; Katori, M.; Kaczmarek, E.; Anselmo, D.; Lassman, C.; Ke, B.; Shen, X.; Busuttil, R.W.; Yamashita, K.; Csizmadia, E.; et al. Heavy chain ferritin acts as an antiapoptotic gene that protects livers from ischemia reperfusion injury. FASEB J. 2003, 17, 1724–1726. [Google Scholar] [PubMed]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 589–599. [Google Scholar] [CrossRef]
- Breuer, W.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron. Int. J. Biochem. Cell Biol. 2008, 40, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 531, 81–92. [Google Scholar] [CrossRef]
- Petrat, F.; de Groot, H.; Sustmann, R.; Rauen, U. The chelatable iron pool in living cells: A methodically defined quantity. Biol. Chem. 2002, 383, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Rauen, U.; Springer, A.; Weisheit, D.; Petrat, F.; Korth, H.-G.; de Groot, H.; Sustmann, R. Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. ChemBioChem 2007, 8, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I.; Kakhlon, O.; Epsztejn, S.; Zanninelli, G.; Breuer, W. Intracellular and extracellular labile iron pools. Adv. Exp. Med. Biol. 2002, 509, 55–75. [Google Scholar] [PubMed]
- Petrat, F.; de Groot, H.; Rauen, U. Subcellular distribution of chelatable iron: A laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem. J. 2001, 356, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ulvik, R.J. Relevance of ferritin-binding sites on isolated mitochondria to the mobilization of iron from ferritin. Biochim. Biophys. Acta 1982, 715, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.S.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Redistribution of accumulated cell iron: A modality of chelation with therapeutic implications. Blood 2008, 111, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Lane, D.J.R.; Becker, E.M.; Huang, M.L.-H.; Whitnall, M.; Rahmanto, Y.S.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.; Ponka, P.; Vyoral, D. Distribution of iron in reticulocytes after inhibition of heme synthesis with succinylacetone: Examination of the intermediates involved in iron metabolism. Blood 1996, 87, 3477–3488. [Google Scholar] [PubMed]
- Ponka, P. Tissue-Specific regulation of iron metabolism and heme synthesis: Distinct control mechanisms in erythroid cells. Blood 1997, 89, 1–25. [Google Scholar] [PubMed]
- Shvartsman, M.; Kikkeri, R.; Shanzer, A.; Cabantchik, Z.I. Non-transferrin-bound iron reaches mitochondria by a chelator-inaccessible mechanism: Biological and clinical implications. Am. J. Physiol. Cell Physiol. 2007, 293, C1383–C1394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.-S.; Sheftel, A.D.; Ponka, P. Intracellular kinetics of iron in reticulocytes: Evidence for endosome involvement in iron targeting to mitochondria. Blood 2005, 105, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Gustafsson, B.; Brunk, U.T. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006, 273, 3106–3117. [Google Scholar] [CrossRef] [PubMed]
- Garner, B.; Roberg, K.; Brunk, U.T. Endogenous ferritin protects cells with iron-laden lysosomes against oxidative stress. Free Radic. Res. 1998, 29, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Gustafsson, B.; Brunk, U.T. Cell sensitivity to oxidative stress is influenced by ferritin autophagy. Free Radic. Biol. Med. 2011, 50, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Jaksch, H.; Lacher, H.; Ohlenschläger, I.; Uchida, K.; Eckl, P.M. Iron mediated oxidative stress plays an essential role in ferritin induced cell death. Free Radic. Biol. Med. 2010, 48, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Krenn, M.A.; Schürz, M.; Teufl, B.; Uchida, K.; Eckl, P.M.; Bresgen, N. Ferritin stimulated lipid peroxidation, lysosomal leak and macroautophagy promote lysosomal “metastability” in primary hepatocytes determining in vitro cell survival. Free Radic. Biol. Med. 2015, 80, 45–58. [Google Scholar] [CrossRef]
- Umbreit, J.N.; Conrad, M.E.; Moore, E.G.; Desai, M.P.; Turrens, J. Paraferritin: A protein complex with ferrireductase activity is associated with iron absorption in rats. Biochemistry 1996, 35, 6460–6469. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, J.N.; Conrad, M.E.; Hainsworth, L.N.; Simovich, M. The ferrireductase paraferritin contains divalent metal transporter as well as mobilferrin. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G534–G539. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Tao, Y.; Zhang, Z.; Guo, X.; An, P.; Shen, Y.; Wu, Q.; Yu, Y.; Wang, F. Metalloreductase Steap3 coordinates the regulation of iron homeostasis and inflammatory responses. Haematologica 2012, 97, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Jalanko, A.; Braulke, T. Neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 697–709. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Lipofuscin. Int. J. Biochem. Cell Biol. 2004, 36, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Bassi, M.T.; Manzoni, M.; Monti, E.; Pizzo, M.T.; Ballabio, A.; Borsani, G. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am. J. Hum. Genet. 2000, 67, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S., Jr.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Yamashita, Y.; Ohgawara, H.; Sekiguchi, M.; Satake, N.; Orino, K.; Yamamoto, S. Iron content of rat serum ferritin. J. Vet. Med. Sci. 2001, 63, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Worwood, M.; Dawkins, S.; Wagstaff, M.; Jacobs, A. The purification and properties of ferritin from human serum. Biochem. J. 1976, 157, 97–103. [Google Scholar] [PubMed]
- Adams, P.C.; Chau, L.A. Uptake of ferritin by isolated rat hepatocytes. Effect of metabolic inhibitors and iron. Clin. Investig. Med. 1993, 16, 15–21. [Google Scholar]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, J.; Kominami, E.; Ueno, T. Peroxisome degradation in mammals. IUBMB Life 2011, 63, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar]
- Tolkovsky, A.M. Mitophagy. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1508–1515. [Google Scholar] [CrossRef]
- Persson, H.L.; Kurz, T.; Eaton, J.W.; Brunk, U.T. Radiation-induced cell death: Importance of lysosomal destabilization. Biochem. J. 2005, 389, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, E. Autophagy: A cell repair mechanism that retards ageing and age-associated diseases and can be intensified pharmacologically. Mol. Asp. Med. 2006, 27, 403–410. [Google Scholar] [CrossRef]
- Zhang, Y.; Mikhael, M.; Xu, D.; Li, Y.; Soe-Lin, S.; Ning, B.; Li, W.; Nie, G.; Zhao, Y.; Ponka, P. Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit. Antioxid. Redox Signal. 2010, 13, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Brunk, U.T. Autophagy of HSP70 and chelation of lysosomal iron in a non-redox-active form. Autophagy 2009, 5, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Persson, H.L.; Eaton, J.W.; Brunk, U.T. Intralysosomal iron: A major determinant of oxidant-induced cell death. Free Radic. Biol. Med. 2003, 34, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.L.; Nilsson, K.J.; Brunk, U.T. Novel cellular defenses against iron and oxidation: Ferritin and autophagocytosis preserve lysosomal stability in airway epithelium. Redox Rep. 2001, 6, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Cadenas, E. Cellular titration of apoptosis with steady state concentrations of H2O2: Submicromolar levels of H2O2 induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radic. Biol. Med. 2001, 30, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Cadenas, E.; Brunk, U.T. Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem. J. 2001, 356, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.; Ghassemifar, R.; Brunk, U.T. Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem. J. 1997, 29, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Zdolsek, J.M.; Svensson, I. Effect of reactive oxygen species on lysosomal membrane integrity. A study on a lysosomal fraction. Virchows Arch. B 1993, 64, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.L.; Yu, Z.; Tirosh, O.; Eaton, J.W.; Brunk, U.T. Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radic. Biol. Med. 2003, 34, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Eaton, J.W.; Persson, H.L. The radioprotective agent, amifostine, suppresses the reactivity of intralysosomal iron. Redox Rep. 2003, 8, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ollinger, K.; Brunk, U.T. Cellular injury induced by oxidative stress is mediated through lysosomal damage. Free Radic. Biol. Med. 1995, 19, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Neuzil, J.; Eaton, J.W. Lysosomal involvement in apoptosis. Redox Rep. 2001, 6, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Baird, S.K.; Kurz, T.; Brunk, U.T. Metallothionein protects against oxidative stress-induced lysosomal destabilization. Biochem. J. 2006, 394, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Carlsson, F.; Laskar, A.; Yuan, X.M.; Li, W. Lysosomal membrane permeabilization causes oxidative stress and ferritin induction in macrophages. FEBS Lett. 2011, 585, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Yokota, M.; Drysdale, J.W. Characterization of serum ferritin in iron overload: Possible identity to natural apoferritin. Br. J. Haematol. 1977, 36, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Adelman, T.G.; Drysdale, J.W. On ferritin heterogeneity. Further evidence for heteropolymers. J. Biol. Chem. 1978, 253, 4451–4458. [Google Scholar] [PubMed]
- Bomford, A.; Berger, M.; Lis, Y.; Williams, R. The iron content of human liver and spleen isoferritins correlates with their isoelectric point and subunit composition. Biochem. Biophys. Res. Commun. 1978, 83, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Arosio, P.; Chasteen, N.D. Iron(II) and hydrogen peroxide detoxification by human H-chain ferritin. An EPR spin-trapping study. Biochemistry 2006, 45, 3429–3436. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Yewdall, S.J.; Harrison, P.M.; Santambrogio, P.; Cozzi, A.; Rovida, E.; Albertini, A.; Arosio, P. Evidence of H- and L-chains have co-operative roles in the iron-uptake mechanism of human ferritin. Biochem. J. 1992, 288, 591–596. [Google Scholar] [PubMed]
- Papaefthymiou, G.C. The Mossbauer and magnetic properties of ferritin cores. Biochim. Biophys. Acta 2010, 1800, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Linder, M. Mobilization of stored iron in mammals: A Review. Nutrients 2013, 5, 4022–4050. [Google Scholar] [CrossRef] [PubMed]
- Watt, R.K.; Hilton, R.J.; Graff, D.M. Oxido-reduction is not the only mechanism allowing ions to traverse the ferritin protein shell. Biochim. Biophys. Acta 2010, 1800, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Cassanelli, S.; Moulis, J. Sulfide is an efficient iron releasing agent for mammalian ferritins. Biochim. Biophys. Acta 2001, 1547, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Maywald, M.; Schmittel, M.; Riederer, P.; Gerlach, M. In vitro studies of ferritin iron release and neurotoxicity. J. Neurochem. 1998, 70, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.C.; Rawal, M.; Wagner, B.A.; Du, J.; Cullen, J.J.; Buettner, G.R. Pharmacological ascorbate and ionizing radiation (IR) increase labile iron in pancreatic cancer. Redox Biol. 2014, 2, 22–27. [Google Scholar] [CrossRef]
- Kidane, T.Z.; Sauble, E.; Linder, M.C. Release of iron from ferritin requires lysosomal activity. Am. J. Physiol. Cell Physiol. 2006, 291, C445–C455. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Vaughn, M.B.; Li, L.; Bagley, D.; Musci, G.; Ward, D.M.; Kaplan, J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 2006, 25, 5396–5404. [Google Scholar] [CrossRef] [PubMed]
- Konijn, A.M.; Glickstein, H.; Vaisman, B.; Meyron-Holtz, E.G.; Slotki, I.N.; Cabantchik, Z.I. The cellular labile iron pool and intracellular ferritin in K562 cells. Blood 1999, 94, 2128–2134. [Google Scholar] [PubMed]
- Truty, J.; Malpe, R.; Linder, M.C. Iron prevents ferritin turnover in hepatic cells. J. Biol. Chem. 2001, 276, 48775–48780. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Specific iron chelators determine the route of ferritin degradation. Blood 2009, 114, 4546–4551. [Google Scholar] [CrossRef] [PubMed]
- Mehlhase, J.; Sandig, G.; Pantopoulos, K.; Grune, T. Oxidation-induced ferritin turnover in microglial cells: Role of proteasome. Free Radic. Biol. Med. 2005, 38, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, A.; Santambrogio, P.; Levi, S.; Arosio, P. Iron detoxifying activity of ferritin. Effects of H and L human apoferritins on lipid peroxidation in vitro. FEBS Lett. 1990, 277, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Santambrogio, P.; Cozzi, A.; Rovida, E.; Corsi, B.; Tamborini, E.; Spada, S.; Albertini, A.; Arosio, P. The role of the L-chain in ferritin iron incorporation. Studies of homo and heteropolymers. J. Mol. Biol. 1994, 238, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Dewaele, S.; Braeckman, B.; Desmyter, L.; Verstraelen, J.; Borgonie, G.; Vanfleteren, J.; Contreras, R. A high-throughput screening system for genes extending life-span. Exp. Gerontol. 2003, 38, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y.; Akebi, N.; Lam, T.; Nakabeppu, Y.; Torti, S.; Torti, F. FER-1, an enhancer of the ferritin H gene and a target of E1A-mediated transcriptional repression. Mol. Cell. Biol. 1995, 15, 5152–5164. [Google Scholar] [PubMed]
- Kwak, E.L.; Larochelle, D.A.; Beaumont, C.; Torti, S.V.; Torti, F.M. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J. Biol. Chem. 1995, 270, 15285–15293. [Google Scholar] [CrossRef] [PubMed]
- Hintze, K.J.; Theil, E.C. DNA and mRNA elements with complementary responses to hemin, antioxidant inducers, and iron control ferritin-L expression. Proc. Natl. Acad. Sci. USA 2005, 102, 15048–15052. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y. JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene 2005, 24, 7567–7578. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Bridges, K.R.; Durmowicz, G.P.; Glass, J.; Auron, P.E.; Munro, H.N. Translational control during the acute phase response. Ferritin synthesis in response to interleukin-1. J. Biol. Chem. 1990, 265, 14572–14578. [Google Scholar] [PubMed]
- Ikegami, Y.; Inukai, K.; Imai, K.; Sakamoto, Y.; Katagiri, H.; Kurihara, S.; Awata, T.; Katayama, S. Adiponectin upregulates ferritin heavy chain in skeletal muscle cells. Diabetes 2009, 58, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Papa, S.; Bubici, C.; Zazzeroni, F.; Franzoso, G. In the crosshairs: NF-κB targets the JNK signaling cascade. Curr. Med. Chem. Anti Inflamm. Anti Allergy Agents 2005, 4, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y.; Miller, L.L.; Miller, S.C.; Torti, S.V.; Torti, F.M. Tumor necrosis factor-alpha and interleukin 1-alpha regulate transferrin receptor in human diploid fibroblasts. Relationship to the induction of ferritin heavy chain. J. Biol. Chem. 1991, 266, 7257–7261. [Google Scholar] [PubMed]
- Miller, L.L.; Miller, S.C.; Torti, S.V.; Tsuji, Y.; Torti, F.M. Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1991, 88, 4946–4950. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, M.; Young, S.P. Modulation of iron metabolism in monocyte cell line U937 by inflammatory cytokines: Changes in transferrin uptake, iron handling and ferritin mRNA. Biochem. J. 1993, 296, 175–181. [Google Scholar] [PubMed]
- Hirayama, M.; Kohgo, Y.; Kondo, H.; Shintani, N.; Fujikawa, K.; Sasaki, K.; Kato, J.; Niitsu, Y. Regulation of iron metabolism in HepG2 cells: A possible role for cytokines in the hepatic deposition of iron. Hepatology 1993, 18, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Yamaguchi, M.; Bannai, S. Regulation of ferritin synthesis in macrophages by oxygen and a sulfhydryl-reactive agent. Biochem. Biophys. Res. Commun. 1994, 201, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Dawson, G. Hypoxia specifically and reversibly induces the synthesis of ferritin in oligodendrocytes and human oligodendrogliomas. J. Neurochem. 1994, 63, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef] [PubMed]
- Faniello, M.C.; Di Sanzo, M.; Quaresima, B.; Baudi, F.; di Caro, V.; Cuda, G.; Morrone, G.; del Sal, G.; Spinelli, G.; Venuta, S.; et al. p53-Mediated downregulation of H ferritin promoter transcriptional efficiency via NF-Y. Int. J. Biochem. Cell Biol. 2008, 40, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, W.; Tsuji, Y.; Torti, S.V.; Torti, F.M. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008, 283, 33911–33918. [Google Scholar] [CrossRef] [PubMed]
- Sammarco, M.C.; Ditch, S.; Banerjee, A.; Grabczyk, E. Ferritin L and H subunits are differentially regulated on a post-transcriptional level. J. Biol. Chem. 2008, 283, 4578–4587. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y.; Ayaki, H.; Whitman, S.P.; Morrow, C.S.; Torti, S.V.; Torti, F.M. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol. Cell. Biol. 2000, 20, 5818–5827. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Tacchini, L.; Pogliaghi, G.; Anzon, E.; Tomasi, A.; Bernelli-Zazzera, A. Induction of ferritin synthesis by oxidative stress. J. Biol. Chem. 1995, 270, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Tacchini, L.; Recalcati, S.; Azzimonti, B.; Minotti, G.; Bernelli-Zazzera, A. Effect of reactive oxygen species on iron regulatory protein activity. Ann. NY Acad. Sci. 1998, 851, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.; Richter, A.; Gunkel, N.; Riedel, D.; Polycarpou-Schwarz, M.; Hentze, S.; Falkenhahn, M.; Stremmel, W.; Ansorge, W.; Hentze, M.W. Relationships and distinctions in iron-regulatory networks responding to interrelated signals. Blood 2003, 101, 3690–3698. [Google Scholar] [CrossRef] [PubMed]
- Koorts, A.M.; Viljoen, M. Ferritin and ferritin isoforms II: Protection against uncontrolled cellular proliferation, oxidative damage and inflammatory processes. Arch. Physiol. Biochem. 2007, 113, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Koc, M.; Taysi, S.; Sezen, O.; Bakan, N. Levels of some acute-phase proteins in the serum of patients with cancer during radiotherapy. Biol. Pharm. Bull. 2003, 26, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Maria, T.G.; Vasileios, K.E.; Panagiotis, P.S.; Kostas, S.N. Changes of acute-phase protein levels in the serum of lung cancer patients following radiotherapy. Int. J. Clin. Exp. Med. 2013, 6, 50–56. [Google Scholar] [PubMed]
- Koorts, A.M.; Viljoen, M. Acute Phase Proteins: Ferritin and Ferritin Isoforms, Acute Phase Proteins—Regulation and Functions of Acute Phase Proteins. In Acute Phase Proteins—Regulation and Functions of Acute Phase Proteins; Francesco, V., Ed.; InTech: Rijeka, Croatia, 2011; pp. 153–184. [Google Scholar]
- Breda, L.; Nozzi, M.; De Sanctis, S.; Chiarelli, F. Laboratory tests in the diagnosis and follow-up of pediatric rheumatic diseases: An update. Semin. Arthritis Rheum. 2010, 40, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Lacroix, L.; Durmowitz, G.; Kasschau, K.; Andriotakis, J.; Bridges, K. The Role of Cytokines in the Regulation of Ferritin Expression. In Progress in Iron Research; Hershko, C., Konijn, A., Aisen, P., Eds.; Springer: Berlin/Heidelberg, Germany, 1994; Volume 356, pp. 127–132. [Google Scholar]
- Ajioka, R.S.; Phillips, J.D.; Kushner, J.P. Biosynthesis of heme in mammals. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 723–736. [Google Scholar] [CrossRef]
- Wegiel, B.; Nemeth, Z.; Correa-Costa, M.; Bulmer, A.C.; Otterbein, L.E. Heme oxygenase-1: A metabolic nike. Antioxid. Redox Signal. 2013, 20, 1709–1722. [Google Scholar] [CrossRef]
- Soe-Lin, S.; Apte, S.S.; Andriopoulos, B.; Andrews, M.C.; Schranzhofer, M.; Kahawita, T.; Garcia-Santos, D.; Ponka, P. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5960–5965. [Google Scholar] [CrossRef] [PubMed]
- Soe-Lin, S.; Apte, S.S.; Mikhael, M.R.; Kayembe, L.K.; Nie, G.; Ponka, P. Both Nramp1 and DMT1 are necessary for efficient macrophage iron recycling. Exp. Hematol. 2010, 38, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Seixas, E.; Gozzelino, R.; Chora, Â.; Ferreira, A.; Silva, G.; Larsen, R.; Rebelo, S.; Penido, C.; Smith, N.R.; Coutinho, A.; et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. USA 2009, 106, 15837–15842. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassu, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Soares, M.P. Coupling heme and iron metabolism via ferritin H chain. Antioxid. Redox Signal. 2013, 20, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Vile, G.F.; Basu-Modak, S.; Waltner, C.; Tyrrell, R.M. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc. Natl. Acad. Sci. USA 1994, 91, 2607–2610. [Google Scholar] [CrossRef] [PubMed]
- Vile, G.F.; Tyrrell, R.M. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J. Biol. Chem. 1993, 268, 14678–14681. [Google Scholar] [PubMed]
- Gozzelino, R.; Andrade, B.B.; Larsen, R.; Luz, N.F.; Vanoaica, L.; Seixas, E.; Coutinho, A.; Cardoso, S.; Rebelo, S.; Poli, M.; et al. Metabolic adaptation to tissue iron overload confers tolerance to malaria. Cell Host Microbe 2012, 12, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Hamza, I.; Dailey, H.A. One ring to rule them all: Trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1617–1632. [Google Scholar] [CrossRef]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Mühlenhoff, U. The role of mitochondria in cellular iron–sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1491–1508. [Google Scholar] [CrossRef]
- Lill, R.; Mühlenhoff, U. Maturation of iron-sulfur proteins in eukaryotes: Mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 2008, 77, 669–700. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Mühlenhoff, U. Iron–sulfur-protein biogenesis in eukaryotes. Trends Biochem. Sci. 2005, 30, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Ulvik, R.J. Ferritin iron as substrate for synthesis of protoheme in intact rat liver mitochondria. FEBS Lett. 1981, 132, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef] [PubMed]
- Amigo, J.D.; Yu, M.; Troadec, M.-B.; Gwynn, B.; Cooney, J.D.; Lambert, A.J.; Chi, N.C.; Weiss, M.J.; Peters, L.L.; Kaplan, J.; et al. Identification of distal cis-regulatory elements at mouse mitoferrin loci using zebrafish transgenesis. Mol. Cell. Biol. 2011, 31, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Invernizzi, R.; Bergamaschi, G.; Levi, S.; Corsi, B.; Travaglino, E.; Rolandi, V.; Biasiotto, G.; Drysdale, J.; Arosio, P. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood 2003, 101, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Gonzalez, M.J.; Trapero-Marugan, M.; Jones, E.A.; Moreno-Otero, R. Liver disease and erythropoietic protoporphyria: A concise review. World J. Gastroenterol. 2010, 16, 4526–4531. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. ExpNeurol. 2013, 72, 78–90. [Google Scholar] [CrossRef]
- Liochev, S.I.; Fridovich, I. Superoxide and iron: Partners in crime. IUBMB Life 1999, 48, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Schenker, S.; Frosto, T.A.; Henderson, G.I. Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE): Role of HNE adduct formation with the enzyme subunits. Biochim. Biophys. Acta Gen. Subj. 1998, 1380, 336–344. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Biologically relevant metal ion-dependent hydroxyl radical generation An update. FEBS Lett. 1992, 307, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Gustafsson, B.; Brunk, U.T. Mitochondrial damage and intralysosomal degradation in cellular aging. Mol. Aspects Med. 2006, 27, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Gustafsson, B.; Brunk, U.T. The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem. Biol. Interact. 2006, 163, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kispal, G.; Csere, P.; Prohl, C.; Lill, R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999, 18, 3981–3989. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, N.; Miki, T.; Senbongi, H.; Yokoi, N.; Yano, H.; Miyazaki, M.; Nakajima, N.; Iwanaga, T.; Yokoyama, Y.; Shibata, T.; et al. MTABC3, a novel mitochondrial ATP-binding cassette protein involved in iron homeostasis. J. Biol. Chem. 2000, 275, 17536–17540. [Google Scholar] [CrossRef] [PubMed]
- Bekri, S.; Kispal, G.; Lange, H.; Fitzsimons, E.; Tolmie, J.; Lill, R.; Bishop, D.F. Human ABC7 transporter: Gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 2000, 96, 3256–3264. [Google Scholar] [PubMed]
- Csere, P.; Lill, R.; Kispal, G. Identification of a human mitochondrial ABC transporter, the functional orthologue of yeast Atm1p. FEBS Lett. 1998, 441, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Kispal, G.; Csere, P.; Guiard, B.; Lill, R. The ABC transporter Atm1p is required for mitochondrial iron homeostasis. FEBS Lett. 1997, 418, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Koc, S.; Harris, J.W. Sideroblastic anemias: Variations on imprecision in diagnostic criteria, proposal for an extended classification of sideroblastic anemias. Am. J. Hematol. 1998, 57, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Taketani, S.; Adachi, Y.; Kohno, H.; Ikehara, S.; Tokunaga, R.; Ishii, T. Molecular characterization of a newly identified heme-binding protein induced during differentiation of urine erythroleukemia cells. J. Biol. Chem. 1998, 273, 31388–31394. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Napierala, M.; Puccio, H. Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis. Model. Mech. 2012, 5, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Puccio, H. Frataxin: A protein in search for a function. J. Neurochem. 2013, 126, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Adamec, J.; Rusnak, F.; Owen, W.G.; Naylor, S.; Benson, L.M.; Gacy, A.M.; Isaya, G. Iron-dependent self-assembly of recombinant yeast frataxin: implications for Friedreich ataxia. Am. J. Hum. Genet. 2000, 67, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Layer, G.; Ollagnier-de Choudens, S.; Sanakis, Y.; Fontecave, M. Iron-Sulfur cluster biosynthesis: characterization of Escherichia coli CYaY as an iron donor for the assembly of [2Fe-2S] clusters in the scaffold IscU. J. Biol. Chem. 2006, 281, 16256–16263. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Koutnikova, H.; Campuzano, V.; Foury, F.; Dolle, P.; Cazzalini, O.; Koenig, M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 1997, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Cossee, M.; Campuzano, V.; Koutnikova, H.; Fischbeck, K.; Mandel, J.-L.; Koenig, M.; Bidichandani, S.I.; Patel, P.I.; Molte, M.D.; Canizares, J.; et al. Frataxin fracas. Nat. Genet. 1997, 15, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, J.B.; Cote, M.; Lemieux, B. The cardiomyopathy of Friedreich’s ataxia morphological observations in 3 cases. Can. J. Neurol. Sci. 1980, 7, 389–396. [Google Scholar] [PubMed]
- Rotig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhofer-Donzé, M.; Reutenauer, L.; Puccio, H. Mammalian Frataxin: An essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLOS ONE 2011, 6, e16199. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Iannuzzi, C.; Prischi, F.; Pastore, C.; Iametti, S.; Martin, S.R.; Bonomi, F.; Pastore, A. Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009, 16, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-L.; Barondeau, D.P. Human frataxin Is an Allosteric Switch That Activates the Fe-S Cluster Biosynthetic Complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Frataxin-mediated Iron Delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [PubMed]
- Sellers, V.M.; Wu, C.-K.; Dailey, T.A.; Dailey, H.A. Human ferrochelatase: Characterization of Substrate-iron binding and proton-abstracting residues. Biochemistry 2001, 40, 9821–9827. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-K.; Dailey, H.A.; Rose, J.P.; Burden, A.; Sellers, V.M.; Wang, B.-C. The 2.0 Å structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Mol. Biol. 2001, 8, 156–160. [Google Scholar] [CrossRef]
- Busi, M.V.; Gomez-Casati, D.F. Exploring frataxin function. IUBMB Life 2012, 64, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Seznec, H.; Gansmuller, A.; Carelle, N.; Weber, P.; Metzger, D.; Rustin, P.; Koenig, M.; Puccio, H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 2004, 24, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Terman, A.; Brunk, U.T. Autophagy, ageing and apoptosis: The role of oxidative stress and lysosomal iron. Arch. Biochem. Biophys. 2007, 462, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.L.; Homayoun, S.; Hart, P.E.; Schapira, A.H.V.; Cooper, J.M. Role of oxidative damage in Friedreich’s ataxia. Neurochem. Res. 2004, 29, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Dehmer, T.; Schöls, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, S.; Sliwa, D.; Rustin, P.; Camadro, J.-M.; Santos, R. Oxidative stress induces mitochondrial fragmentation in frataxin-deficient cells. Biochem. Biophys. Res. Commun. 2012, 418, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Colman, M.J.R.; Gómez-Casati, D.F.; Lamattina, L.; Zabaleta, E.J. Nitric oxide accumulation is required to protect against iron-mediated oxidative stress in frataxin-deficient Arabidopsis plants. FEBS Lett. 2009, 583, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Calmels, N.; Schmucker, S.; Wattenhofer-Donzé, M.; Martelli, A.; Vaucamps, N.; Reutenauer, L.; Messaddeq, N.; Bouton, C.; Koenig, M.; Puccio, H. The First cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia. PLOS ONE 2009, 4, e6379. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Puccio, H. Dysregulation of cellular iron metabolism in Friedreich ataxia: From primary iron-sulfur cluster deficit to mitochondrial iron accumulation. Front. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Afford, S.C.; Randhawa, S.; Eliopoulos, A.G.; Hubscher, S.G.; Young, L.S.; Adams, D.H. CD40 activation induces apoptosis in cultured human hepatocytes via induction of cell surface fas ligand expression and amplifies Fas-mediated hepatocyte death during allograft rejection. J. Exp. Med. 1999, 189, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Seguin, A.; Sutak, R.; Bulteau, A.-L.; Garcia-Serres, R.; Oddou, J.-L.; Lefevre, S.; Santos, R.; Dancis, A.; Camadro, J.-M.; Latour, J.-M.; et al. Evidence that yeast frataxin is not an iron storage protein in vivo. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 531–538. [Google Scholar] [CrossRef]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Chang, Y.-Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Levi, S.; Arosio, P. Mitochondrial ferritin. Int. J. Biochem. Cell Biol. 2004, 36, 1887–1889. [Google Scholar] [CrossRef] [PubMed]
- Langlois d'Estaintot, B.; Santambrogio, P.; Granier, T.; Gallois, B.; Chevalier, J.M.; Precigoux, G.; Levi, S.; Arosio, P. Crystal structure and biochemical properties of the human mitochondrial ferritin and its mutant Ser144Ala. J. Mol. Biol. 2004, 340, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Santambrogio, P.; Levi, S.; Arosio, P.; Chasteen, N.D. Unique iron binding and oxidation properties of human mitochondrial ferritin: A comparative analysis with human H-chain ferritin. J. Mol. Biol. 2005, 347, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial ferritin: A new player in iron metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Corsi, B.; Cozzi, A.; Arosio, P.; Drysdale, J.; Santambrogio, P.; Campanella, A.; Biasiotto, G.; Albertini, A.; Levi, S. Human mitochondrial ferritin expressed in HeLa cells incorporates iron and affects cellular iron metabolism. J. Biol. Chem. 2002, 277, 22430–22437. [Google Scholar] [CrossRef] [PubMed]
- Nie, G.; Sheftel, A.D.; Kim, S.F.; Ponka, P. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood 2005, 105, 2161–2167. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Nie, G.; Li, Y.; Soe-Lin, S.; Tao, Y.; Cao, Y.; Zhang, Z.; Liu, N.; Ponka, P.; Zhao, B. Overexpression of mitochondrial ferritin sensitizes cells to oxidative stress via an iron-mediated mechanism. Antioxid. Redox Signal. 2009, 11, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Erba, B.G.; Campanella, A.; Cozzi, A.; Causarano, V.; Cremonesi, L.; Galli, A.; Della Porta, M.G.; Invernizzi, R.; Levi, S. Overexpression of mitochondrial ferritin affects the JAK2/STAT5 pathway in K562 cells and causes mitochondria iron accumulation. Haematologica 2011, 96, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Michael, S.; Petrocine, S.; Qian, J.; Lamarche, J.; Knutson, M.; Garrick, M.; Koeppen, A. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum 2006, 5, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Rovelli, E.; Santambrogio, P.; Cozzi, A.; Taroni, F.; Levi, S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 2009, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zanella, I.; Derosas, M.; Corrado, M.; Cocco, E.; Cavadini, P.; Biasiotto, G.; Poli, M.; Verardi, R.; Arosio, P. The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 90–98. [Google Scholar] [CrossRef]
- Campanella, A.; Isaya, G.; O'Neill, H.A.; Santambrogio, P.; Cozzi, A.; Arosio, P.; Levi, S. The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Hum. Mol. Genet. 2004, 13, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.; Stehling, O.; Lill, R. Iron–sulfur proteins in health and disease. Trends Endocrinol. Metab. 2010, 21, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Mettert, E.L.; Kiley, P.J. Fe-S proteins that regulate gene expression. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1284–1293. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.-J.; Lee, S.-M.; Son, B.-G.; Lee, S.-H.; Ryoo, Z.Y.; Chang, K.-T.; Park, J.-W.; Park, D.-C.; Song, B.J.; Veech, R.L.; et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J. Biol. Chem. 2004, 279, 39968–39974. [Google Scholar] [CrossRef] [PubMed]
- Wallander, M.L.; Leibold, E.A.; Eisenstein, R.S. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 668–689. [Google Scholar] [CrossRef]
- Walden, W.E.; Selezneva, A.I.; Dupuy, J.; Volbeda, A.; Fontecilla-Camps, J.C.; Theil, E.C.; Volz, K. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science 2006, 314, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, R.S.; Ross, K.L. Novel roles for iron regulatory proteins in the adaptive response to iron deficiency. J. Nutr. 2003, 133, 1510S–1516S. [Google Scholar] [PubMed]
- Tong, W.-H.; Rouault, T.A. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006, 3, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Hentze, M.W. Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J. 1995, 14, 2917–2924. [Google Scholar] [PubMed]
- Caltagirone, A.; Weiss, G.; Pantopoulos, K. Modulation of cellular iron metabolism by hydrogen peroxide: Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J. Biol. Chem. 2001, 276, 19738–19745. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R.; Raineri, I.; Epstein, L.B.; White, C.W. Superoxide radical and iron modulate aconitase activity in mammalian cells. J. Biol. Chem. 1995, 270, 13399–13405. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Klausner, R.D. Iron-sulfur clusters as biosensors of oxidants and iron. Trends Biochem. Sci. 1996, 21, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S.; Pietrangelo, A.; Minotti, G. The iron regulatory proteins: Targets and modulators of free radical reactions and oxidative damage. Free Radic. Biol. Med. 2002, 32, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Day, B.J.; Crapo, J.D.; Fridovich, I.; McNamara, J.O. Requirement for superoxide in excitotoxic cell death. Neuron 1996, 16, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Imlay, J.A. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J. Biol. Chem. 2007, 282, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Flint, D.H.; Tuminello, J.F.; Emptage, M.H. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar] [PubMed]
- Kennedy, M.C.; Emptage, M.H.; Dreyer, J.L.; Beinert, H. The role of iron in the activation-inactivation of aconitase. J. Biol. Chem. 1983, 258, 11098–11105. [Google Scholar] [PubMed]
- Singh, S.P.; Niemczyk, M.; Saini, D.; Awasthi, Y.C.; Zimniak, L.; Zimniak, P. Role of the electrophilic lipid peroxidation product 4-hydroxynonenal in the development and maintenance of obesity in mice. Biochemistry 2008, 47, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Yarian, C.S.; Rebrin, I.; Sohal, R.S. Aconitase and ATP synthase are targets of malondialdehyde modification and undergo an age-related decrease in activity in mouse heart mitochondria. Biochem. Biophys. Res. Commun. 2005, 330, 151–156. [Google Scholar] [CrossRef] [PubMed]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. alpha-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Yang, E.S.; Park, J.W. Inactivation of NADP+-dependent isocitrate dehydrogenase by lipid peroxidation products. Free Radic. Res. 2004, 38, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Lashin, O.M.; Szweda, P.A.; Szweda, L.I.; Romani, A.M.P. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic. Biol. Med. 2006, 40, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Mansouri, A.; Haouzi, D.; Fromenty, B. Hepatotoxicity due to mitochondrial dysfunction. Cell Biol. Toxicol. 1999, 15, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Simpson, D.C.; Gronert, S. Carbonylation of mitochondrial aconitase with 4-hydroxy-2-(E)-nonenal: Localization and relative reactivity of addition sites. Biochim. Biophys. Acta Proteins Proteomics 2013, 1834, 1144–1154. [Google Scholar] [CrossRef]
- Brazzolotto, X.; Gaillard, J.; Pantopoulos, K.; Hentze, M.W.; Moulis, J.-M. Human cytoplasmic aconitase (iron regulatory protein 1) is converted into its [3Fe-4S] form by hydrogen peroxide in vitro but is not activated for iron-responsive element binding. J. Biol. Chem. 1999, 274, 21625–21630. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Hentze, M.W. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proc. Natl. Acad. Sci. USA 1998, 95, 10559–10563. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Mueller, S.; Atzberger, A.; Ansorge, W.; Stremmel, W.; Hentze, M.W. Differences in the regulation of iron regulatory protein-1 (IRP-1) by extra- and intracellular oxidative stress. J. Biol. Chem. 1997, 272, 9802–9808. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.M.; Anderson, S.A.; Steffen, D.W.; Carpenter, T.B.; Kennedy, M.C.; Walden, W.E.; Eisenstein, R.S. Novel role of phosphorylation in Fe-S cluster stability revealed by phosphomimetic mutations at Ser-138 of iron regulatory protein 1. Proc. Natl. Acad. Sci. USA 1998, 95, 15235–15240. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, R.S.; Tuazon, P.T.; Schalinske, K.L.; Anderson, S.A.; Traugh, J.A. Iron-responsive element-binding protein. Phosphorylation by protein kinase C. J. Biol. Chem. 1993, 268, 27363–27370. [Google Scholar] [PubMed]
- Clarke, S.L.; Vasanthakumar, A.; Anderson, S.A.; Pondarre, C.; Koh, C.M.; Deck, K.M.; Pitula, J.S.; Epstein, C.J.; Fleming, M.D.; Eisenstein, R.S. Iron-responsive degradation of iron-regulatory protein 1 does not require the Fe-S cluster. EMBO J. 2006, 25, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Neonaki, M.; Graham, D.C.; White, K.N.; Bomford, A. Down-regulation of liver iron-regulatory protein 1 in haemochromatosis. Biochem. Soc. Trans. 2002, 30, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Goessling, L.S.; Daniels-McQueen, S.; Bhattacharyya-Pakrasi, M.; Lin, J.J.; Thach, R.E. Enhanced degradation of the ferritin repressor protein during induction of ferritin messenger RNA translation. Science 1992, 256, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Ronchi, R.; Salvatorelli, E.; Menna, P.; Cairo, G. Doxorubicin irreversibly inactivates iron regulatory proteins 1 and 2 in cardiomyocytes. Cancer Res. 2001, 61, 8422–8428. [Google Scholar] [PubMed]
- Cairo, G.; Recalcati, S. Iron-regulatory proteins: Molecular biology and pathophysiological implications. Expert Rev. Mol. Med. 2007, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Starzyński, R.R.; Lipiński, P.; Drapier, J.-C.; Diet, A.; Smuda, E.; Bartłomiejczyk, T.; Gralak, M.A.; Kruszewski, M. Down-regulation of iron regulatory protein 1 activities and expression in superoxide dismutase 1 knock-out mice is not associated with alterations in iron metabolism. J. Biol. Chem. 2005, 280, 4207–4212. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T. Lysosomal iron, iron chelation, and cell death. Antioxid. Redox Signal. 2013, 18, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. Lysosomal labilization. IUBMB Life 2006, 58, 531–539. [Google Scholar] [CrossRef] [PubMed]
- McGahan, M.C.; Harned, J.; Mukunnemkeril, M.; Goralska, M.; Fleisher, L.; Ferrell, J.B. Iron alters glutamate secretion by regulating cytosolic aconitase activity. Am. J. Physiol. Cell Physiol. 2005, 288, C1117–C1124. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jo, S.H.; Lee, S.M.; Koh, H.J.; Song, H.; Park, J.W.; Lee, W.H.; Huh, T.L. Role of NADP+-dependent isocitrate dehydrogenase (NADP+-ICDH) on cellular defence against oxidative injury by γ-rays. Int. J. Radiat. Biol. 2004, 80, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system Xc: Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Lall, M.M.; Ferrell, J.; Nagar, S.; Fleisher, L.N.; McGahan, M.C. Iron regulates L-cystine uptake and glutathione levels in lens epithelial and retinal pigment epithelial cells by its effect on cytosolic aconitase. Investig. Ophthalmol. Vis. Sci. 2008, 49, 310–319. [Google Scholar] [CrossRef]
- Guo, B.; Yu, Y.; Leibold, E.A. Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J. Biol. Chem. 1994, 269, 24252–24260. [Google Scholar] [PubMed]
- Samaniego, F.; Chin, J.; Iwai, K.; Rouault, T.A.; Klausner, R.D. Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. Structure, function, and post-translational regulation. J. Biol. Chem. 1994, 269, 30904–30910. [Google Scholar] [PubMed]
- Hanson, E.S.; Foot, L.M.; Leibold, E.A. Hypoxia post-translationally activates iron-regulatory protein 2. J. Biol. Chem. 1999, 274, 5047–5052. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Phillips, J.D.; Yu, Y.; Leibold, E.A. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J. Biol. Chem. 1995, 270, 21645–21651. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Drake, S.K.; Wehr, N.B.; Weissman, A.M.; LaVaute, T.; Minato, N.; Klausner, R.D.; Levine, R.L.; Rouault, T.A. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: Implications for degradation of oxidized proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 4924–4928. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Sierzputowska-Gracz, H.; Gdaniec, Z.; Theil, E.C. Internal loop/bulge and hairpin loop of the iron-responsive element of ferritin mRNA contribute to maximal iron regulatory protein 2 binding and translational regulation in the iso-iron-responsive element/iso-iron regulatory protein family. Biochemistry 2000, 39, 6235–6242. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ponka, P. Nitrogen monoxide-mediated control of ferritin synthesis: Implications for macrophage iron homeostasis. Proc. Natl. Acad. Sci. USA 2002, 99, 12214–12219. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wu, J.; Leibold, E.A.; Walden, W.E.; Theil, E.C. Loops and bulge/loops in iron-responsive element isoforms influence iron regulatory protein binding. J. Biol. Chem. 1998, 273, 23637–23640. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.S.; Rawlins, M.L.; Leibold, E.A. Oxygen and iron regulation of iron regulatory protein 2. J. Biol. Chem. 2003, 278, 40337–40342. [Google Scholar] [CrossRef] [PubMed]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Rouault, T.A. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science 2004, 306, 2087–2090. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.S.; Schalinske, K.L.; Eisenstein, R.S. Dietary iron intake modulates the activity of iron regulatory proteins and the abundance of ferritin and mitochondrial aconitase in rat liver. J. Nutr. 1997, 127, 238–248. [Google Scholar] [PubMed]
- Schalinske, K.L.; Eisenstein, R.S. Phosphorylation and activation of both iron regulatory proteins 1 and 2 in HL-60 cells. J. Biol. Chem. 1996, 271, 7168–7176. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, M.A.; Grebien, F.; Gehart, H.; Schifrer, M.; Artaker, M.; Kovacic, B.; Beug, H.; Moriggl, R.; Müllner, E.W. Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1. Blood 2008, 112, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; Fisher, B.; Surgener, S.P.; Gerhardt, G.A.; Rouault, T. Neurochemical investigations of dopamine neuronal systems in iron-regulatory protein 2 (IRP-2) knockout mice. Mol. Brain Res. 2005, 139, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Grabill, C.; Silva, A.C.; Smith, S.S.; Koretsky, A.P.; Rouault, T.A. MRI detection of ferritin iron overload and associated neuronal pathology in iron regulatory protein-2 knockout mice. Brain Res. 2003, 971, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.; Berger, U.V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004, 23, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Drapier, J.-C. Interplay between NO and [Fe-S] Clusters: Relevance to biological systems. Methods 1997, 11, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Hausladen, A.; Fridovich, I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem. 1994, 269, 29405–29408. [Google Scholar] [PubMed]
- Castro, L.; Rodriguez, M.; Radi, R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J. Biol. Chem. 1994, 269, 29409–29415. [Google Scholar] [PubMed]
- Radi, R. Peroxynitrite, a Stealthy Biological Oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [PubMed]
- Soum, E.; Brazzolotto, X.; Goussias, C.; Bouton, C.; Moulis, J.-M.; Mattioli, T.A.; Drapier, J.-C. Peroxynitrite and nitric oxide differently target the iron-sulfur cluster and amino acid residues of human iron regulatory protein 1. Biochemistry 2003, 42, 7648–7654. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Bergonia, H.A.; Müller, C.; Pitt, B.R.; Watkins, W.D.; Lancaster, J.R. Loss and degradation of enzyme-bound heme induced by cellular nitric oxide synthesis. J. Biol. Chem. 1995, 270, 5710–5713. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Kühn, L.C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Singel, D.J.; Loscalzo, J. Biochemistry of nitric oxide and its redox-activated forms. Science 1992, 258, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.N.; Richardson, D.R. The mechanism of nitrogen monoxide (NO)-mediated iron mobilization from cells. Eur. J. Biochem. 2002, 269, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.N.; Richardson, D.R. Nitrogen monoxide (NO) and glucose: Unexpected links between energy metabolism and NO-mediated iron mobilization from cells. J. Biol. Chem. 2001, 276, 4724–4732. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Arosio, P.; Cozzi, A.; Chasteen, N.D. Identification of the EPR-active iron-nitrosyl complexes in mammalian ferritins. Biochemistry 1994, 33, 3679–3687. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Wing, S.S.; Ponka, P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell. Biol. 2004, 24, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Mikhael, M.; Kim, S.F.; Schranzhofer, M.; Lin, S.S.; Sheftel, A.D.; Mullner, E.W.; Ponka, P. Iron regulatory protein-independent regulation of ferritin synthesis by nitrogen monoxide. FEBS J. 2006, 273, 3828–3836. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Choi, Y.-B.; Pan, Z.-H.; Lei, S.Z.; Chen, H.-S.V.; Sucher, N.J.; Loscalzo, J.; Singel, D.J.; Stamler, J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 1993, 364, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 78, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Vanin, A.F. Dinitrosyl iron complexes and S-nitrosothiols are two possible forms for stabilization and transport of nitric oxide in biological systems. Biochemistry 1998, 63, 782–793. [Google Scholar] [PubMed]
- Boese, M.; Mordvintcev, P.I.; Vanin, A.F.; Busse, R.; Mülsch, A. S-nitrosation of serum albumin by dinitrosyl-iron complex. J. Biol. Chem. 1995, 270, 29244–29249. [Google Scholar] [CrossRef] [PubMed]
- Bosworth, C.A.; Toledo, J.C.; Zmijewski, J.W.; Li, Q.; Lancaster, J.R. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc. Natl. Acad. Sci. USA 2009, 106, 4671–4676. [Google Scholar] [CrossRef] [PubMed]
- Gaston, B.; Reilly, J.; Drazen, J.M.; Fackler, J.; Ramdev, P.; Arnelle, D.; Mullins, M.E.; Sugarbaker, D.J.; Chee, C.; Singel, D.J. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc. Natl. Acad. Sci. USA 1993, 90, 10957–10961. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.J.; Buerk, D.G.; Ischiropoulos, H. A novel reaction mechanism for the formation of S-nitrosothiol in vivo. J. Biol. Chem. 1997, 272, 2841–2845. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N.; Singh, R.J.; Kalyanaraman, B. The role of glutathione in the transport and catabolism of nitric oxide. FEBS Lett. 1996, 382, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Keszler, A.; Zhang, Y.; Hogg, N. Reaction between nitric oxide, glutathione, and oxygen in the presence and absence of protein: How are S-nitrosothiols formed? Free Radic. Biol. Med. 2010, 48, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Nims, R.W.; Darbyshire, J.F.; Christodoulou, D.; Hanbauer, I.; Cox, G.W.; Laval, F.; Laval, J.; Cook, J.A. Reaction kinetics for nitrosation of cysteine and glutathione in aerobic nitric oxide solutions at neutral pH. Insights into the fate and physiological effects of intermediates generated in the NO/O2 reaction. Chem. Res. Toxicol. 1994, 7, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ruiz, A.; Lamas, S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: Convergences and divergences. Cardiovasc. Res. 2007, 75, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Rauhala, P.; Lin, A.M.-Y.; Chiueh, C.C. Neuroprotection by S-nitrosoglutathione of brain dopamine neurons from oxidative stress. FASEB J. 1998, 12, 165–173. [Google Scholar] [PubMed]
- Rauhala, P.; Parameswarannay Mohanakumar, K.; Sziraki, I.; Lin, A.M.Y.; Chiueh, C.C. S-nitrosothiols and nitric oxide, but not sodium nitroprusside, protect nigrostriatal dopamine neurons against iron-induced oxidative stress in vivo. Synapse 1996, 23, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Struck, A.T.; Hogg, N.; Thomas, J.P.; Kalyanaraman, B. Nitric oxide donor compounds inhibit the toxicity of oxidized low-density lipoprotein to endothelial cells. FEBS Lett. 1995, 361, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Cook, J.A.; Pacelli, R.; DeGraff, W.; Gamson, J.; Liebmann, J.; Krishna, M.C.; Mitchell, J.B. The effect of various nitric oxide-donor agents on hydrogen peroxide-mediated toxicity: A direct correlation between nitric oxide formation and protection. Arch. Biochem. Biophys. 1996, 331, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Chiueh, C.C.; Rauhala, P. The redox pathway of S-nitrosoglutathione, glutathione and nitric oxide in cell to neuron communications. Free Radic. Res. 1999, 31, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Chiueh, C.C. Neuroprotective Properties of Nitric Oxide. Ann. NY Acad. Sci. 1999, 890, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Meloche, B.A.; O'Brien, P.J. S-nitrosyl glutathione-mediated hepatocyte cytotoxicity. Xenobiotica 1993, 23, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ponka, P. Effects of interferon-γ and lipopolysaccharide on macrophage iron metabolism are mediated by nitric oxide-induced degradation of iron regulatory protein 2. J. Biol. Chem. 2000, 275, 6220–6226. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ponka, P. Control of transferrin receptor expression via nitric oxide-mediated modulation of iron-regulatory protein 2. J. Biol. Chem. 1999, 274, 33035–33042. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.C.; Antholine, W.E.; Beinert, H. An EPR Investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J. Biol. Chem. 1997, 272, 20340–20347. [Google Scholar] [CrossRef] [PubMed]
- Bouton, C.; Raveau, M.; Drapier, J.-C. Modulation of Iron regulatory protein functions. J. Biol. Chem. 1996, 271, 2300–2306. [Google Scholar] [CrossRef] [PubMed]
- Drapier, J.C.; Hirling, H.; Wietzerbin, J.; Kaldy, P.; Kuhn, L.C. Biosynthesis of nitric oxide activates iron regulatory factor in macrophages. EMBO J. 1993, 12, 3643–3649. [Google Scholar] [PubMed]
- Recalcati, S.; Taramelli, D.; Conte, D.; Cairo, G. Nitric oxide-mediated induction of ferritin synthesis in j774 macrophages by inflammatory cytokines: Role of selective iron regulatory protein-2 downregulation. Blood 1998, 91, 1059–1066. [Google Scholar] [PubMed]
- Pantopoulos, K. Iron Metabolism and the IRE/IRP regulatory system: An update. Ann. NY Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Weiss, G.; Hentze, M.W. Nitric oxide and oxidative stress (H2O2) control mammalian iron metabolism by different pathways. Mol. Cell. Biol. 1996, 16, 3781–3788. [Google Scholar] [PubMed]
- Mulero, V.; Brock, J.H. Regulation of Iron Metabolism in Murine J774 Macrophages: Role of nitric oxide-dependent and -independent pathways following activation with gamma interferon and lipopolysaccharide. Blood 1999, 94, 2383–2389. [Google Scholar] [PubMed]
- Weiss, G.; Goossen, B.; Doppler, W.; Fuchs, D.; Pantopoulos, K.; Werner-Felmayer, G.; Wachter, H.; Hentze, M.W. Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J. 1993, 12, 3651–3657. [Google Scholar] [PubMed]
- Wang, J.; Chen, G.; Pantopoulos, K. Nitric oxide inhibits the degradation of IRP2. Mol. Cell. Biol. 2005, 25, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Werner-Felmayer, G.; Werner, E.R.; Grünewald, K.; Wachter, H.; Hentze, M.W. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J. Exp. Med. 1994, 180, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, J.B., Jr.; Taintor, R.R.; Vavrin, Z. Iron depletion: Possible cause of tumor cell cytotoxicity induced by activated macrophages. Biochem. Biophys. Res. Commun. 1984, 123, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Ponka, P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta 1997, 1331, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Ghosh, M.C.; Zhang, D.-L.; Jeong, S.Y.; Kovtunovych, G.; Ollivierre-Wilson, H.; Noguchi, A.; Tu, T.; Senecal, T.; Robinson, G.; Crooks, D.R.; et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational de-repression of HIF2α. Cell Metab. 2013, 17, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef]
- Liu, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Invest. 2012, 122, 4635–4644. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Mathieu, J.R.R.; Delga, S.; Mayeux, P.; Vaulont, S.; Peyssonnaux, C. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica 2012, 97, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2α mRNA translation. Blood 2013, 122, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Qu, A.; Anderson, E.R.; Matsubara, T.; Martin, A.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-Inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 2011, 140, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2α, but not HIF-1α, promotes iron absorption in mice. J. Clin. Invest. 2009, 119, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Matsubara, T.; Ito, S.; Yim, S.-H.; Gonzalez, F.J. Intestinal hypoxia inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009, 9, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar]
- Li, Z.; Rich, J. Hypoxia and Hypoxia Inducible Factors in Cancer Stem Cell Maintenance. In Diverse Effects of Hypoxia on Tumor Progression; Simon, M.C., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 345, pp. 21–30. [Google Scholar]
- Wang, W.; Deng, Z.; Hatcher, H.; Miller, L.D.; Di, X.; Tesfay, L.; Sui, G.; D’Agostino, R.B.; Torti, F.M.; Torti, S.V. IRP2 regulates breast tumor growth. Cancer Res. 2014, 74, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-J.; Polack, A.; Dalla-Favera, R. Coordinated regulation of iron-controlling genes, H-Ferritin and IRP2, by c-MYC. Science 1999, 283, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Templeton, D.M.; Liu, Y. Genetic regulation of cell function in response to iron overload or chelation. Biochim. Biophys. Acta Gen. Subj. 2003, 1619, 113–124. [Google Scholar] [CrossRef]
- Wesselius, L.J.; Nelson, M.E.; Skikne, B.S. Increased release of ferritin and iron by iron-loaded alveolar macrophages in cigarette smokers. Am. J. Respir. Crit. Care Med. 1994, 150, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Ohlenschlager, I.; Wacht, N.; Afazel, S.; Ladurner, G.; Eckl, P.M. Ferritin and FasL (CD95L) mediate density dependent apoptosis in primary rat hepatocytes. J. Cell. Physiol. 2008, 217, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Rolinek, R.; Hochleitner, E.; Lottspeich, F.; Eckl, P.M. Induction of apoptosis by a hepatocyte conditioned medium. J. Cell. Physiol. 2004, 198, 452–460. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bresgen, N.; Eckl, P.M. Oxidative Stress and the Homeodynamics of Iron Metabolism. Biomolecules 2015, 5, 808-847. https://doi.org/10.3390/biom5020808
Bresgen N, Eckl PM. Oxidative Stress and the Homeodynamics of Iron Metabolism. Biomolecules. 2015; 5(2):808-847. https://doi.org/10.3390/biom5020808
Chicago/Turabian StyleBresgen, Nikolaus, and Peter M. Eckl. 2015. "Oxidative Stress and the Homeodynamics of Iron Metabolism" Biomolecules 5, no. 2: 808-847. https://doi.org/10.3390/biom5020808
APA StyleBresgen, N., & Eckl, P. M. (2015). Oxidative Stress and the Homeodynamics of Iron Metabolism. Biomolecules, 5(2), 808-847. https://doi.org/10.3390/biom5020808