
Recent Advances in the Structural Studies of the Proteolytic ClpP/ClpX Molecular Machine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
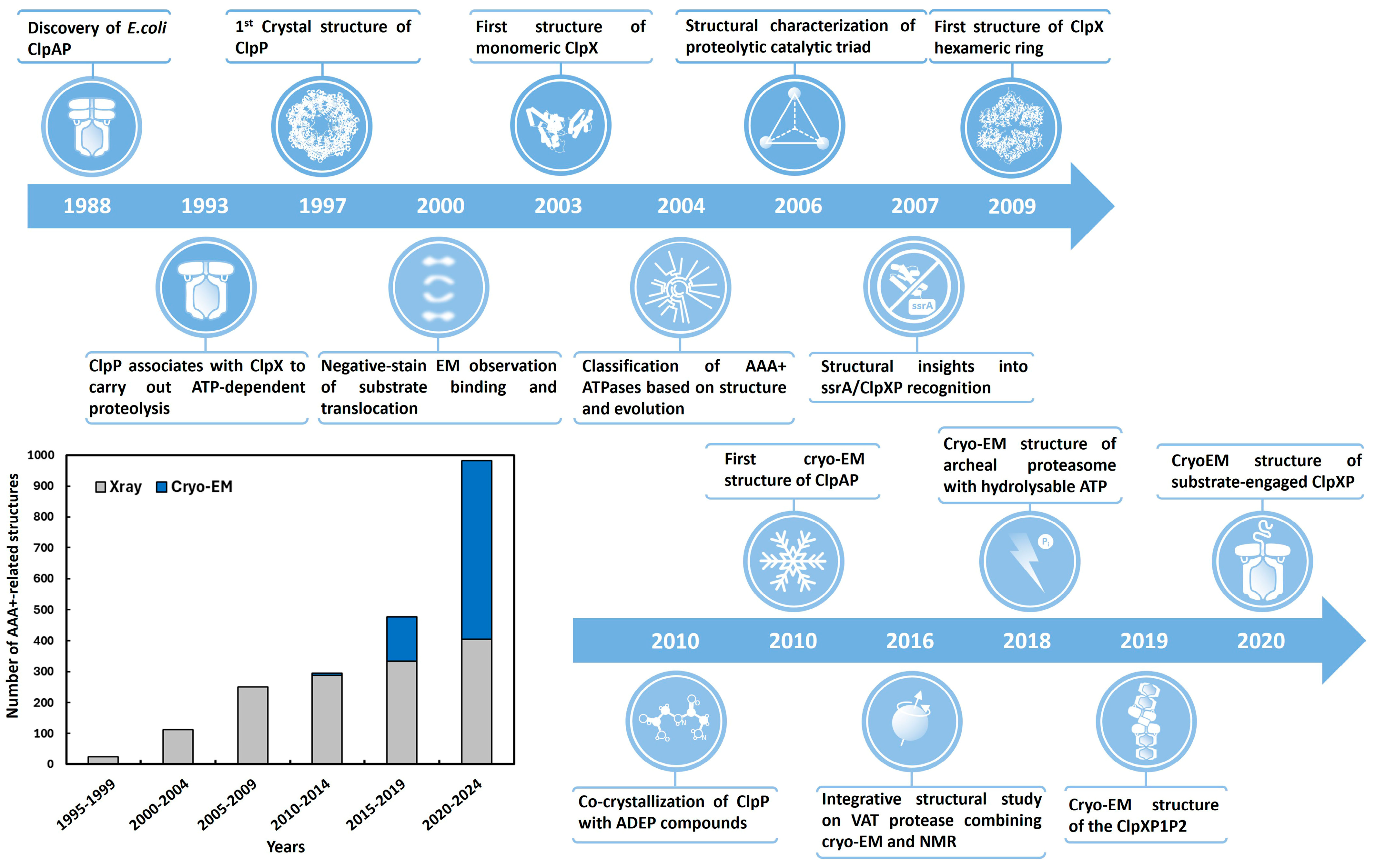
1. Introduction
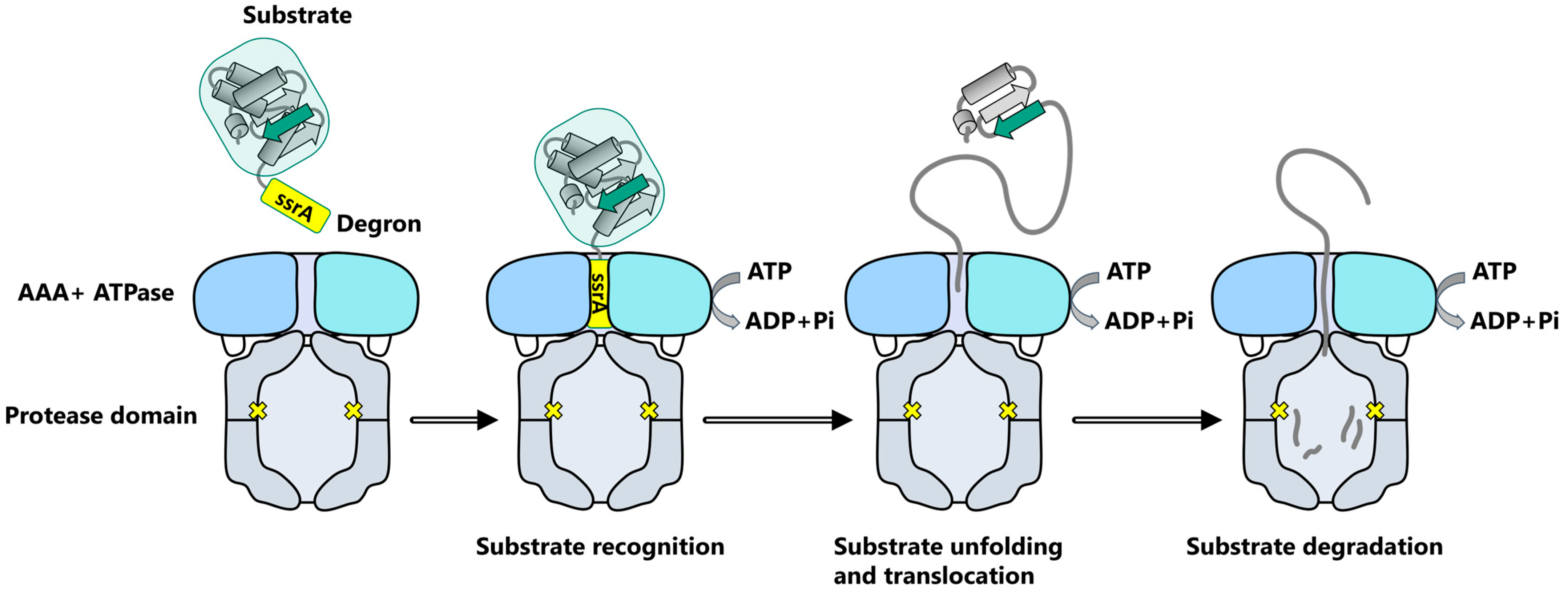
2. ClpXP, a Prototype for the AAA+ Proteolytic Machine Family
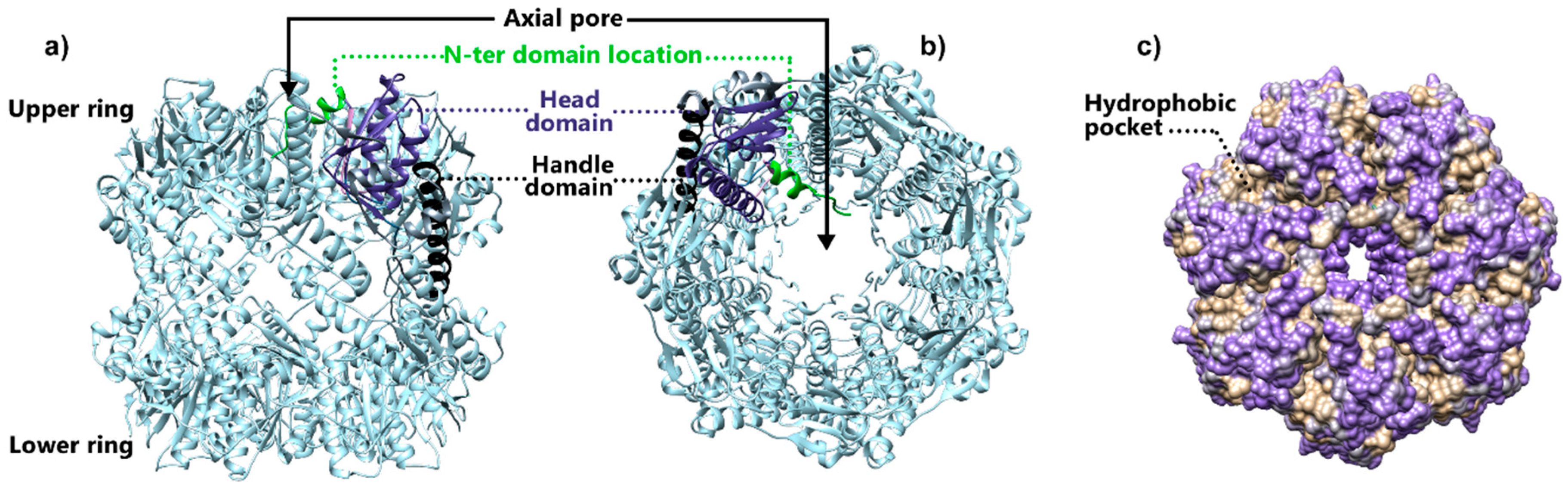
3. Structural Studies of Tetradecameric ClpP Protease
3.1. Structural Insights into ClpP Catalytic Site
3.2. Structural Insights into the Gating Mechanism of the ClpP Axial Pore
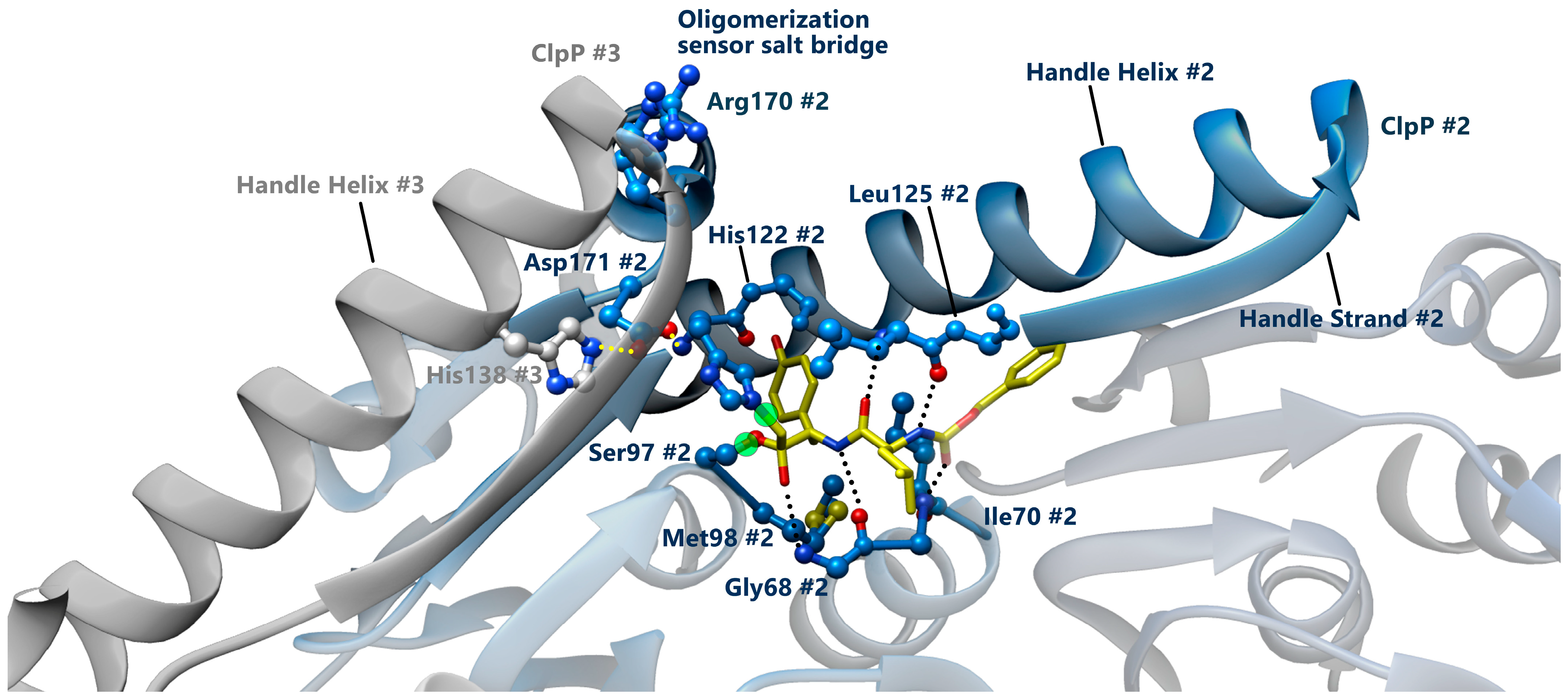
3.3. Conformational Switch of ClpP Handle Domain Controls the Catalytic Activity
4. ClpX Hexameric AAA+ ATPase, a Challenging Target for Structural Biology
4.1. First ClpX Crystal Structures
4.2. Identification of the ClpX Nucleotide Binding Site
5. Cryo-EM Investigations Revealed the High-Resolution Structure of Active ClpXP Machinery
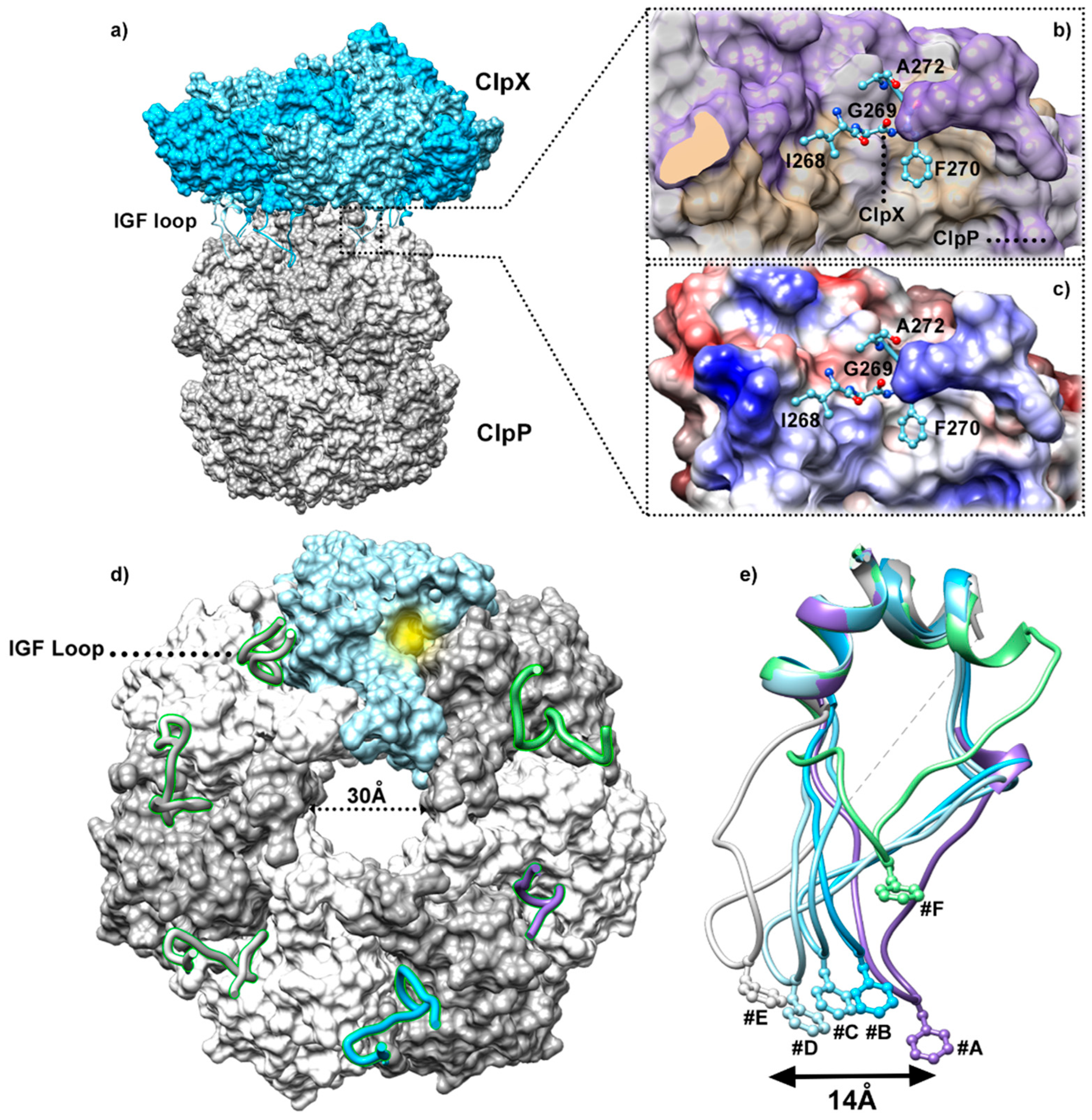
5.1. Insights into the ClpP/ClpX Interaction
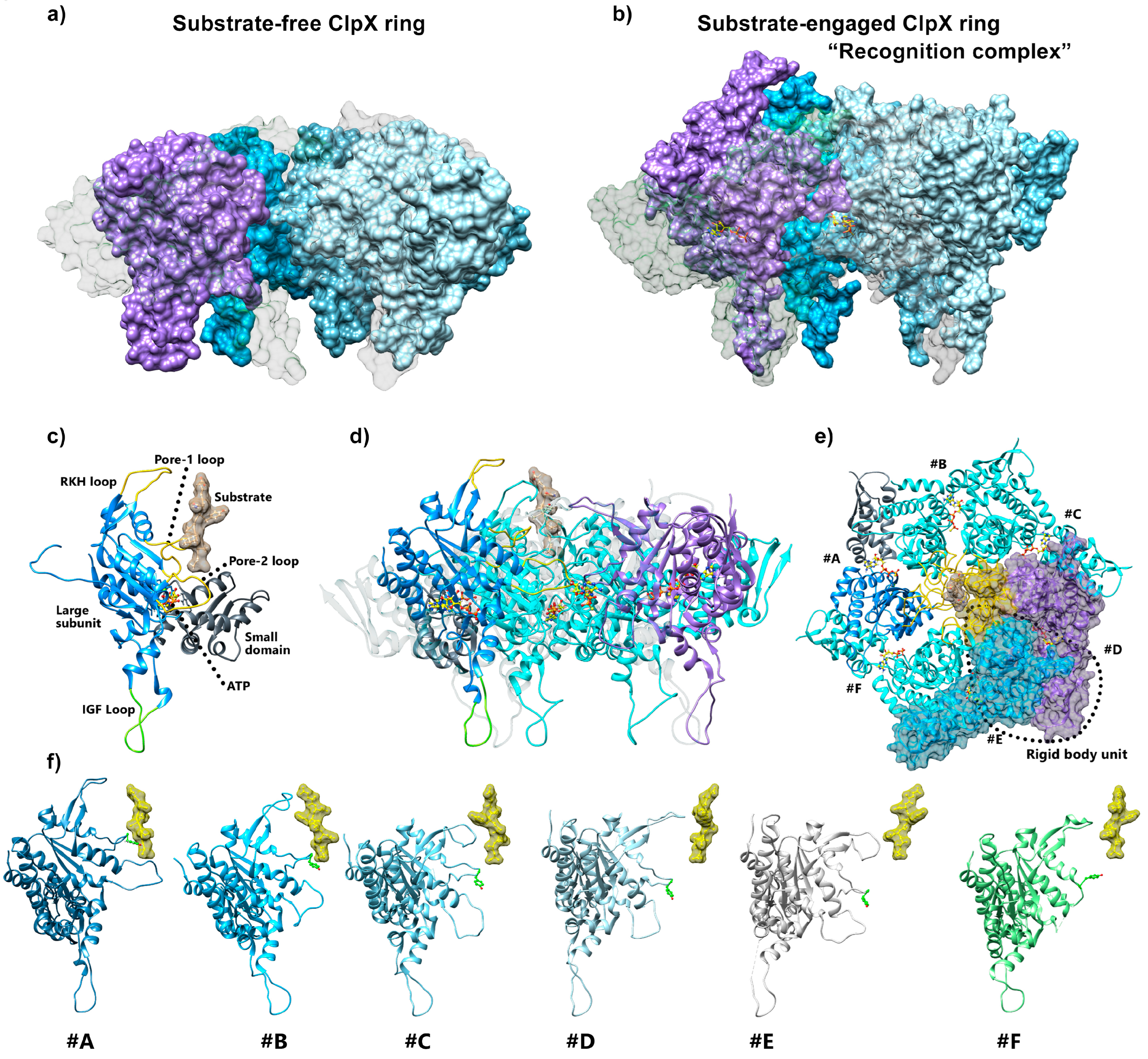
5.2. ClpX Structural Rearrangements upon Substrate Binding
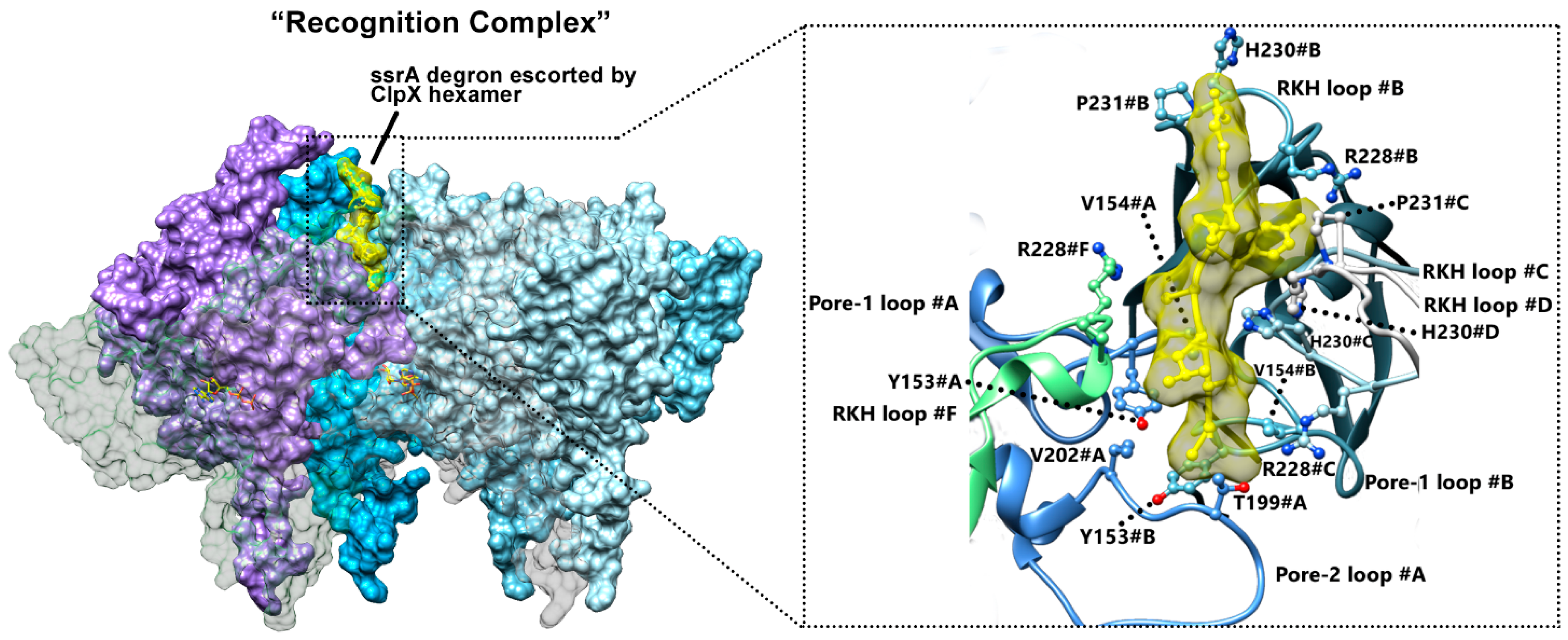
5.3. ClpX Substrate Recognition
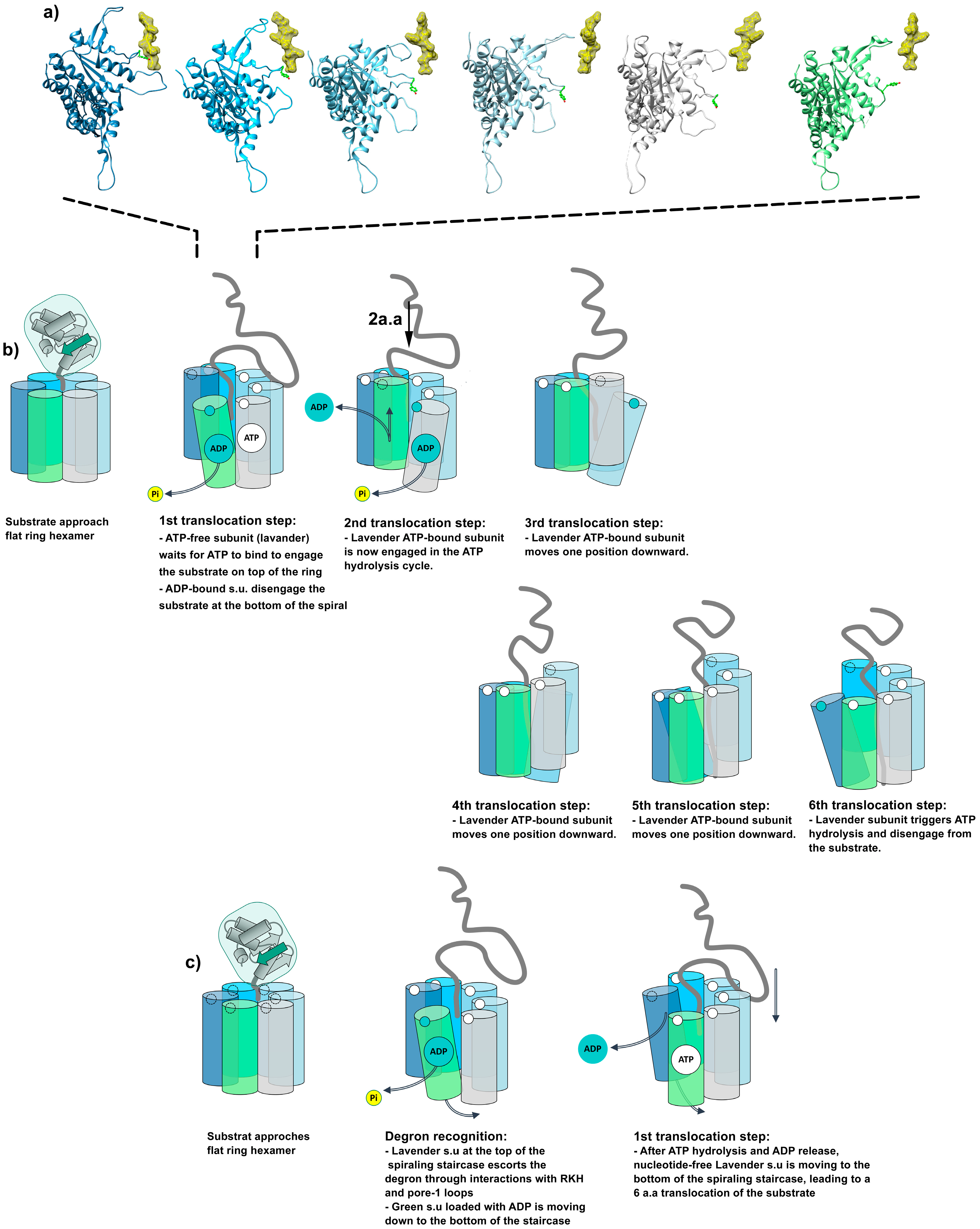
5.4. Intermediate Complex Sheds Light on Substrate Translocation
6. Models for ClpXP Substrate Translocation Mechanism
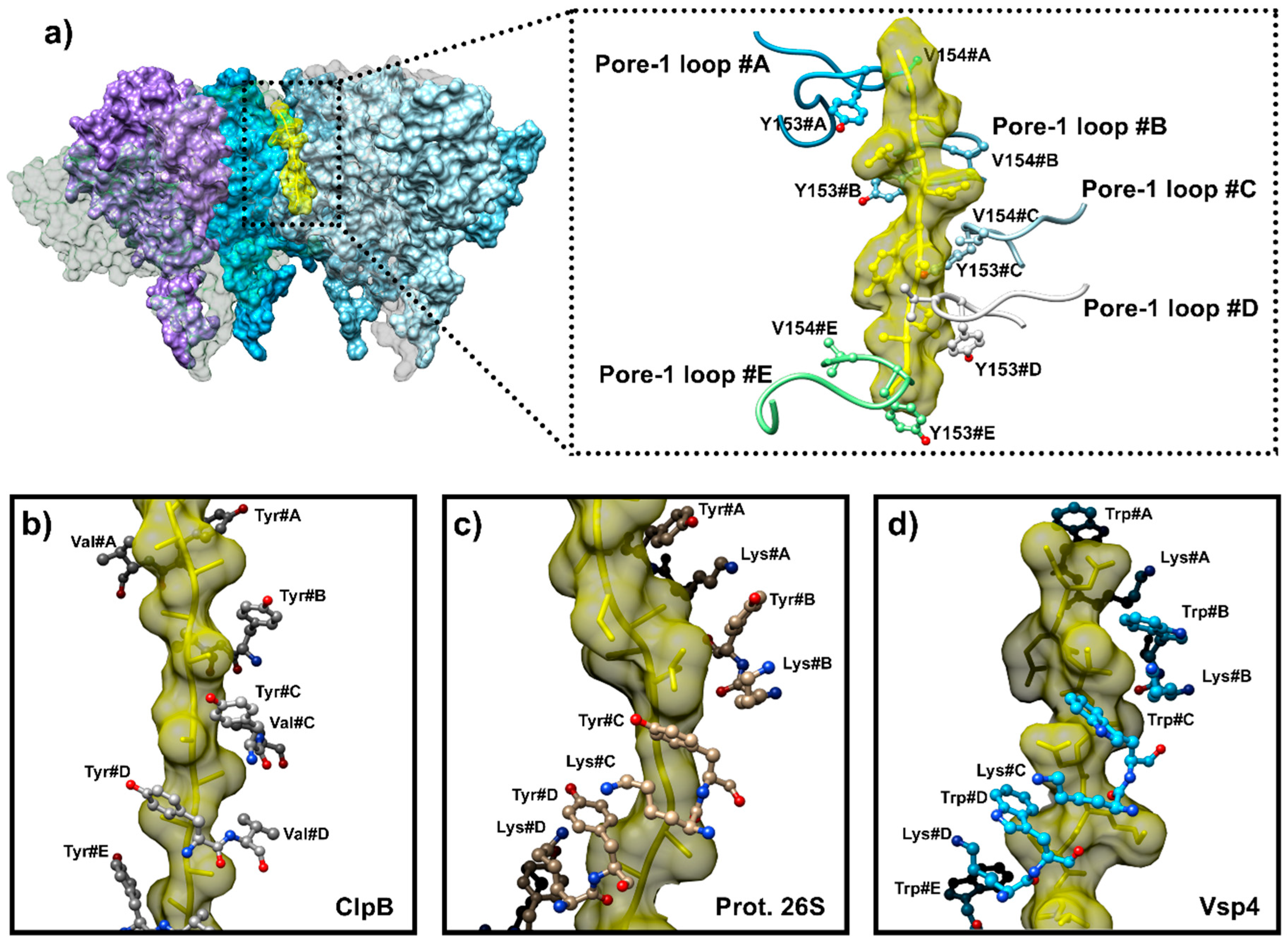
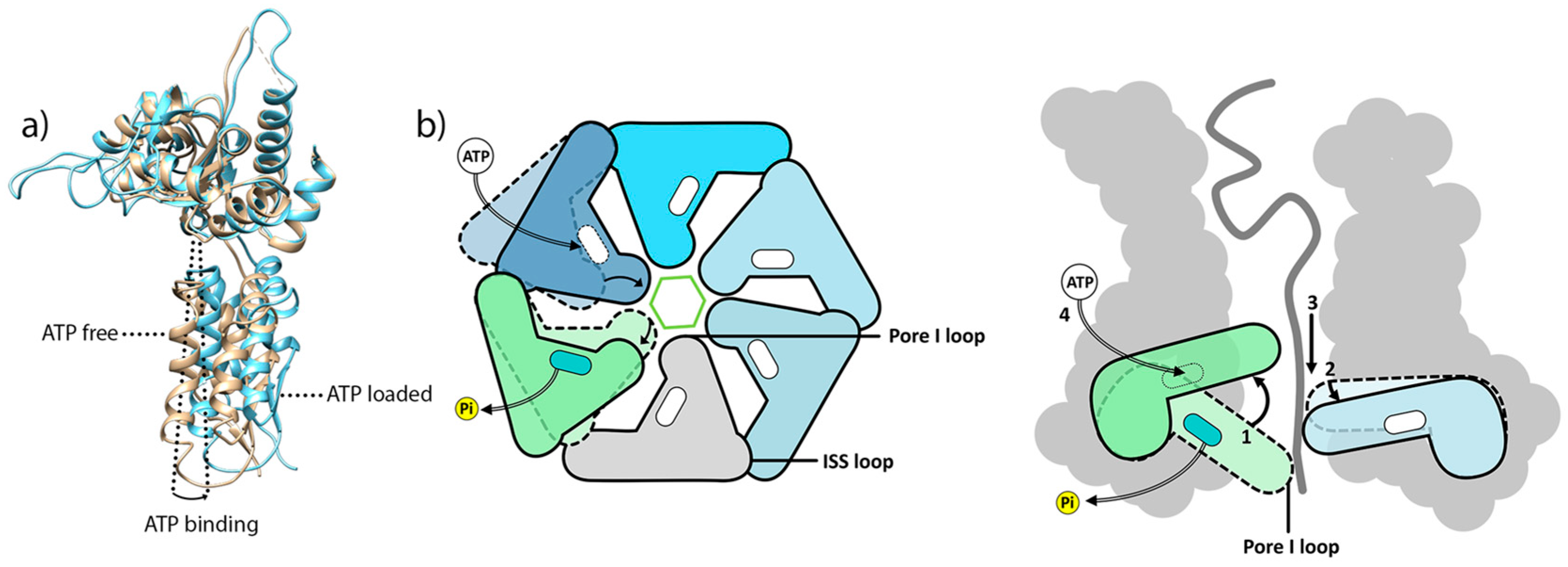
6.1. Coupling ATP Binding to Pore-1 Loop/Substrate Engagement
6.2. A Conserved Mechanism for Substrate Pulling by the AAA+ Subunit
6.3. Allosteric Regulation of Translocation Models
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-Resolution Protein Structure Determination by Cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef]
- Förster, A.; Schulze-Briese, C. A Shared Vision for Macromolecular Crystallography over the next Five Years. Struct. Dyn. 2019, 6, 064302. [Google Scholar] [CrossRef]
- Alderson, T.R.; Kay, L.E. NMR Spectroscopy Captures the Essential Role of Dynamics in Regulating Biomolecular Function. Cell 2021, 184, 577–595. [Google Scholar] [CrossRef]
- Kerfah, R.; Plevin, M.J.; Sounier, R.; Gans, P.; Boisbouvier, J. Methyl-Specific Isotopic Labeling: A Molecular Tool Box for Solution NMR Studies of Large Proteins. Curr. Opin. Struct. Biol. 2015, 32, 113–122. [Google Scholar] [CrossRef]
- Wiesner, S.; Sprangers, R. Methyl Groups as NMR Probes for Biomolecular Interactions. Curr. Opin. Struct. Biol. 2015, 35, 60–67. [Google Scholar] [CrossRef]
- Sprangers, R.; Velyvis, A.; Kay, L.E. Solution NMR of Supramolecular Complexes: Providing New Insights into Function. Nat. Methods 2007, 4, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Kay, L.E. Bringing Dynamic Molecular Machines into Focus by Methyl-TROSY NMR. Annu. Rev. Biochem. 2014, 83, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Geraets, J.A.; Pothula, K.R.; Schröder, G.F. Integrating Cryo-EM and NMR Data. Curr. Opin. Struct. Biol. 2020, 61, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mao, Y. AAA+ ATPases in Protein Degradation: Structures, Functions and Mechanisms. Biomolecules 2020, 10, 629. [Google Scholar] [CrossRef]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-Powered Unfolding and Protein-Degradation Machine. Biochim. Biophys. Acta. 2012, 1823, 15–28. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kim, K.K. Crystal Structure of ClpX Molecular Chaperone from Helicobacter Pylori*. J. Biol. Chem. 2003, 278, 50664–50670. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hartling, J.A.; Flanagan, J.M. The Structure of ClpP at 2.3 A Resolution Suggests a Model for ATP-Dependent Proteolysis. Cell 1997, 91, 447–456. [Google Scholar] [CrossRef]
- Glynn, S.E.; Martin, A.; Nager, A.R.; Baker, T.A.; Sauer, R.T. Structures of Asymmetric ClpX Hexamers Reveal Nucleotide-Dependent Motions in a AAA+ Protein-Unfolding Machine. Cell 2009, 139, 744–756. [Google Scholar] [CrossRef]
- Bewley, M.C.; Graziano, V.; Griffin, K.; Flanagan, J.M. The Asymmetry in the Mature Amino-Terminus of ClpP Facilitates a Local Symmetry Match in ClpAP and ClpXP Complexes. J. Struct. Biol. 2006, 153, 113–128. [Google Scholar] [CrossRef]
- Gribun, A.; Kimber, M.S.; Ching, R.; Sprangers, R.; Fiebig, K.M.; Houry, W.A. The ClpP Double Ring Tetradecameric Protease Exhibits Plastic Ring-Ring Interactions, and the N Termini of Its Subunits Form Flexible Loops That Are Essential for ClpXP and ClpAP Complex Formation*. J. Biol. Chem. 2005, 280, 16185–16196. [Google Scholar] [CrossRef]
- Kang, S.G.; Maurizi, M.R.; Thompson, M.; Mueser, T.; Ahvazi, B. Crystallography and Mutagenesis Point to an Essential Role for the N-Terminus of Human Mitochondrial ClpP. J. Struct. Biol. 2004, 148, 338–352. [Google Scholar] [CrossRef]
- Szyk, A.; Maurizi, M.R. Crystal Structure at 1.9A of E. coli ClpP with a Peptide Covalently Bound at the Active Site. J. Struct. Biol. 2006, 156, 165–174. [Google Scholar] [CrossRef]
- Gatsogiannis, C.; Balogh, D.; Merino, F.; Sieber, S.A.; Raunser, S. Cryo-EM Structure of the ClpXP Protein Degradation Machinery. Nat. Struct. Mol. Biol. 2019, 26, 946–954. [Google Scholar] [CrossRef]
- Fei, X.; Bell, T.A.; Barkow, S.R.; Baker, T.A.; Sauer, R.T. Structural Basis of ClpXP Recognition and Unfolding of ssrA-Tagged Substrates. eLife 2020, 9, e61496. [Google Scholar] [CrossRef] [PubMed]
- Striebel, F.; Kress, W.; Weber-Ban, E. Controlled Destruction: AAA+ ATPases in Protein Degradation from Bacteria to Eukaryotes. Curr. Opin. Struct. Biol. 2009, 19, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.S.; Chung, Y.S.; Gloyd, M.; Joseph, E.; Ghirlando, R.; Wright, G.D.; Cheng, Y.-Q.; Maurizi, M.R.; Guarné, A.; Ortega, J. Acyldepsipeptide Antibiotics Induce the Formation of a Structured Axial Channel in ClpP: A Model for the ClpX/ClpA-Bound State of ClpP. Chem. Biol. 2010, 17, 959–969. [Google Scholar] [CrossRef]
- Park, E.Y.; Lee, B.-G.; Hong, S.-B.; Kim, H.-W.; Jeon, H.; Song, H.K. Structural Basis of SspB-Tail Recognition by the Zinc Binding Domain of ClpX. J. Mol. Biol. 2007, 367, 514–526. [Google Scholar] [CrossRef]
- Effantin, G.; Ishikawa, T.; De Donatis, G.M.; Maurizi, M.R.; Steven, A.C. Local and Global Mobility in the ClpA AAA+ Chaperone Detected by Cryo-Electron Microscopy: Functional Connotations. Structure 2010, 18, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Ripstein, Z.A.; Augustyniak, R.; Lazniewski, M.; Ginalski, K.; Kay, L.E.; Rubinstein, J.L. Unfolding the Mechanism of the AAA+ Unfoldase VAT by a Combined Cryo-EM, Solution NMR Study. Proc. Natl. Acad. Sci. USA 2016, 113, E4190–E4199. [Google Scholar] [CrossRef]
- de la Peña, A.H.; Goodall, E.A.; Gates, S.N.; Lander, G.C.; Martin, A. Substrate-Engaged 26S Proteasome Structures Reveal Mechanisms for ATP-Hydrolysis–Driven Translocation. Science 2018, 362, eaav0725. [Google Scholar] [CrossRef]
- Katayama-Fujimura, Y.; Gottesman, S.; Maurizi, M.R. A Multiple-Component, ATP-Dependent Protease from Escherichia Coli. J. Biol. Chem. 1987, 262, 4477–4485. [Google Scholar] [CrossRef]
- Wojtkowiak, D.; Georgopoulos, C.; Zylicz, M. Isolation and Characterization of ClpX, a New ATP-Dependent Specificity Component of the Clp Protease of Escherichia Coli. J. Biol. Chem. 1993, 268, 22609–22617. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.; Singh, S.K.; Ishikawa, T.; Maurizi, M.R.; Steven, A.C. Visualization of Substrate Binding and Translocation by the ATP-Dependent Protease, ClpXP. Mol. Cell 2000, 6, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary History and Higher Order Classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef]
- Sakai, J.; Zhang, S.; Chen, H.; Atsumi, F.; Matsui, T.; Shiono, H.; Sanada, S.; Okada, T. Primary Structure of a Thrombin-like Serine Protease, Kangshuanmei, from the Venom of Agkistrodon Halys Brevicaudus Stejneger. Toxicon 2006, 48, 313–322. [Google Scholar] [CrossRef]
- Alexopoulos, J.; Ahsan, B.; Homchaudhuri, L.; Husain, N.; Cheng, Y.-Q.; Ortega, J. Structural Determinants Stabilizing the Axial Channel of ClpP for Substrate Translocation. Mol. Microbiol. 2013, 90, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Ghanbarpour, A.; Sauer, R.T.; Davis, J.H. A Proteolytic AAA+ Machine Poised to Unfold Protein Substrates. Nat. Commun. 2024, 15, 9681. [Google Scholar] [CrossRef]
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-Fueled ClpXP Proteolytic Machine Bound to Protein Substrate. eLife 2020, 9, e52774. [Google Scholar] [CrossRef] [PubMed]
- Ghanbarpour, A.; Cohen, S.E.; Fei, X.; Kinman, L.F.; Bell, T.A.; Zhang, J.J.; Baker, T.A.; Davis, J.H.; Sauer, R.T. A Closed Translocation Channel in the Substrate-Free AAA+ ClpXP Protease Diminishes Rogue Degradation. Nat. Commun. 2023, 14, 7281. [Google Scholar] [CrossRef]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A Class of Chaperone-Like ATPases Associated with the Assembly, Operation, and Disassembly of Protein Complexes. Genome Res. 1999, 9, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Wilkinson, A.J. AAA+ Superfamily ATPases: Common Structure–Diverse Function. Genes. Cells 2001, 6, 575–597. [Google Scholar] [CrossRef]
- Hanson, P.I.; Whiteheart, S.W. AAA+ Proteins: Have Engine, Will Work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef]
- Sauer, R.T.; Baker, T.A. AAA+ Proteases: ATP-Fueled Machines of Protein Destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanistic Insights into Bacterial AAA+ Proteases and Protein-Remodelling Machines. Nat. Rev. Microbiol. 2016, 14, 33–44. [Google Scholar] [CrossRef]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanical Protein Unfolding and Degradation. Annu. Rev. Physiol. 2018, 80, 413–429. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Puchades, C.; Sandate, C.R.; Lander, G.C. The Molecular Principles Governing the Activity and Functional Diversity of AAA+ Proteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 43–58. [Google Scholar] [CrossRef]
- Jessop, M.; Felix, J.; Gutsche, I. AAA+ ATPases: Structural Insertions under the Magnifying Glass. Curr. Opin. Struct. Biol. 2021, 66, 119–128. [Google Scholar] [CrossRef]
- Mabanglo, M.F.; Houry, W.A. Recent Structural Insights into the Mechanism of ClpP Protease Regulation by AAA+ Chaperones and Small Molecules. J. Biol. Chem. 2022, 298, 101781. [Google Scholar] [CrossRef]
- Ishikawa, F.; Homma, M.; Tanabe, G.; Uchihashi, T. Protein Degradation by a Component of the Chaperonin-Linked Protease ClpP. Genes Cells 2024, 29, 695–709. [Google Scholar] [CrossRef]
- Lo, H.-H.; Liao, C.-T.; Li, C.-E.; Chiang, Y.-C.; Hsiao, Y.-M. The clpX Gene Plays an Important Role in Bacterial Attachment, Stress Tolerance, and Virulence in Xanthomonas Campestris Pv. Campestris. Arch. Microbiol. 2020, 202, 597–607. [Google Scholar] [CrossRef]
- Aljghami, M.E.; Barghash, M.M.; Majaesic, E.; Bhandari, V.; Houry, W.A. Cellular Functions of the ClpP Protease Impacting Bacterial Virulence. Front. Mol. Biosci. 2022, 9, 1054408. [Google Scholar] [CrossRef]
- Nouri, K.; Feng, Y.; Schimmer, A.D. Mitochondrial ClpP Serine Protease-Biological Function and Emerging Target for Cancer Therapy. Cell Death Dis. 2020, 11, 841. [Google Scholar] [CrossRef]
- Camberg, J.L.; Hoskins, J.R.; Wickner, S. ClpXP Protease Degrades the Cytoskeletal Protein, FtsZ, and Modulates FtsZ Polymer Dynamics. Proc. Natl. Acad. Sci. USA 2009, 106, 10614–10619. [Google Scholar] [CrossRef]
- Weart, R.B.; Nakano, S.; Lane, B.E.; Zuber, P.; Levin, P.A. The ClpX Chaperone Modulates Assembly of the Tubulin-like Protein FtsZ. Mol. Microbiol. 2005, 57, 238–249. [Google Scholar] [CrossRef]
- Fischer, F.; Langer, J.; Osiewacz, H. Identification of Potential Mitochondrial CLPXP Protease Interactors and Substrates Suggests Its Central Role in Energy Metabolism. Sci. Rep. 2015, 5, 18375. [Google Scholar] [CrossRef]
- Feng, Y.; Nouri, K.; Schimmer, A.D. Mitochondrial ATP-Dependent Proteases—Biological Function and Potential Anti-Cancer Targets. Cancers 2021, 13, 2020. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Kukat, A.; Szczepanowska, K.; Hermans, S.; Senft, K.; Brandscheid, C.P.; Maiti, P.; Trifunovic, A. CLPP Deficiency Protects against Metabolic Syndrome but Hinders Adaptive Thermogenesis. EMBO Rep. 2018, 19, e45126. [Google Scholar] [CrossRef]
- Guo, C.; Xiao, Y.; Gu, J.; Zhao, P.; Hu, Z.; Zheng, J.; Hua, R.; Hai, Z.; Su, J.; Zhang, J.V.; et al. ClpP/ClpX Deficiency Impairs Mitochondrial Functions and mTORC1 Signaling during Spermatogenesis. Commun. Biol. 2023, 6, 1012. [Google Scholar] [CrossRef]
- Ripstein, Z.A.; Vahidi, S.; Houry, W.A.; Rubinstein, J.L.; Kay, L.E. A Processive Rotary Mechanism Couples Substrate Unfolding and Proteolysis in the ClpXP Degradation Machinery. eLife 2020, 9, e52158. [Google Scholar] [CrossRef]
- Ahlawat, S.; Morrison, D.A. ClpXP Degrades SsrA-Tagged Proteins in Streptococcus Pneumoniae. J. Bacteriol. 2009, 191, 2894–2898. [Google Scholar] [CrossRef]
- Thompson, M.W.; Singh, S.K.; Maurizi, M.R. Processive Degradation of Proteins by the ATP-Dependent Clp Protease from Escherichia Coli. Requirement for the Multiple Array of Active Sites in ClpP but Not ATP Hydrolysis. J. Biol. Chem. 1994, 269, 18209–18215. [Google Scholar] [CrossRef]
- Lee, M.E.; Baker, T.A.; Sauer, R.T. Control of Substrate Gating and Translocation into ClpP by Channel Residues and ClpX Binding. J. Mol. Biol. 2010, 399, 707–718. [Google Scholar] [CrossRef]
- Wawrzynow, A.; Wojtkowiak, D.; Marszalek, J.; Banecki, B.; Jonsen, M.; Graves, B.; Georgopoulos, C.; Zylicz, M. The ClpX Heat-Shock Protein of Escherichia Coli, the ATP-Dependent Substrate Specificity Component of the ClpP-ClpX Protease, Is a Novel Molecular Chaperone. EMBO J. 1995, 14, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Krüger, E.; Witt, E.; Ohlmeier, S.; Hanschke, R.; Hecker, M. The Clp Proteases of Bacillus Subtilis Are Directly Involved in Degradation of Misfolded Proteins. J. Bacteriol. 2000, 182, 3259–3265. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, M.R.; Clark, W.P.; Kim, S.H.; Gottesman, S. Clp P Represents a Unique Family of Serine Proteases. J. Biol. Chem. 1990, 265, 12546–12552. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Powers, J.C.; Odake, S.; Oleksyszyn, J.; Hori, H.; Ueda, T.; Boduszek, B.; Kam, C. Proteases--Structures, Mechanism and Inhibitors. Agents Actions Suppl. 1993, 42, 3–18. [Google Scholar]
- Illigmann, A.; Vielberg, M.-T.; Lakemeyer, M.; Wolf, F.; Dema, T.; Stange, P.; Kuttenlochner, W.; Liebhart, E.; Kulik, A.; Staudt, N.D.; et al. Structure of Staphylococcus Aureus ClpP Bound to the Covalent Active-Site Inhibitor Cystargolide A. Angew. Chem. Int. Ed. 2024, 63, e202314028. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cinos, C.; Sassetti, E.; Salado, I.G.; Witt, G.; Benramdane, S.; Reinhardt, L.; Cruz, C.D.; Joossens, J.; Van der Veken, P.; Brötz-Oesterhelt, H.; et al. α-Amino Diphenyl Phosphonates as Novel Inhibitors of Escherichia Coli ClpP Protease. J. Med. Chem. 2019, 62, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, E.; Durante Cruz, C.; Tammela, P.; Winterhalter, M.; Augustyns, K.; Gribbon, P.; Windshügel, B. Identification and Characterization of Approved Drugs and Drug-Like Compounds as Covalent Escherichia Coli ClpP Inhibitors. Int. J. Mol. Sci. 2019, 20, 2686. [Google Scholar] [CrossRef] [PubMed]
- Mabanglo, M.F.; Leung, E.; Vahidi, S.; Seraphim, T.V.; Eger, B.T.; Bryson, S.; Bhandari, V.; Zhou, J.L.; Mao, Y.-Q.; Rizzolo, K.; et al. ClpP Protease Activation Results from the Reorganization of the Electrostatic Interaction Networks at the Entrance Pores. Commun. Biol. 2019, 2, 410. [Google Scholar] [CrossRef]
- Alexopoulos, J.A.; Guarné, A.; Ortega, J. ClpP: A Structurally Dynamic Protease Regulated by AAA+ Proteins. J. Struct. Biol. 2012, 179, 202–210. [Google Scholar] [CrossRef]
- Liu, K.; Ologbenla, A.; Houry, W.A. Dynamics of the ClpP Serine Protease: A Model for Self-Compartmentalized Proteases. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 400–412. [Google Scholar] [CrossRef]
- Religa, T.L.; Ruschak, A.M.; Rosenzweig, R.; Kay, L.E. Site-Directed Methyl Group Labeling as an NMR Probe of Structure and Dynamics in Supramolecular Protein Systems: Applications to the Proteasome and to the ClpP Protease. J. Am. Chem. Soc. 2011, 133, 9063–9068. [Google Scholar] [CrossRef]
- Vahidi, S.; Ripstein, Z.A.; Bonomi, M.; Yuwen, T.; Mabanglo, M.F.; Juravsky, J.B.; Rizzolo, K.; Velyvis, A.; Houry, W.A.; Vendruscolo, M.; et al. Reversible Inhibition of the ClpP Protease via an N-Terminal Conformational Switch. Proc. Natl. Acad. Sci. USA 2018, 115, E6447–E6456. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, F.; Lan, L.; Jiang, H.; Luo, C.; Yang, C.-G. Structural Switching of Staphylococcus Aureus Clp Protease. J. Biol. Chem. 2011, 286, 37590–37601. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhang, J.; Liu, H.; Hilgenfeld, R.; Zhang, R.; Kong, X.; Li, L.; Lu, J.; Zhang, X.; Li, D.; et al. Helix Unfolding/Refolding Characterizes the Functional Dynamics of Staphylococcus Aureus Clp Protease. J. Biol. Chem. 2013, 288, 17643–17653. [Google Scholar] [CrossRef] [PubMed]
- Geiger, S.R.; Böttcher, T.; Sieber, S.A.; Cramer, P. A Conformational Switch Underlies ClpP Protease Function. Angew. Chem. Int. Ed. 2011, 50, 5749–5752. [Google Scholar] [CrossRef] [PubMed]
- Vahidi, S.; Ripstein, Z.A.; Juravsky, J.B.; Rennella, E.; Goldberg, A.L.; Mittermaier, A.K.; Rubinstein, J.L.; Kay, L.E. An Allosteric Switch Regulates Mycobacterium Tuberculosis ClpP1P2 Protease Function as Established by Cryo-EM and Methyl-TROSY NMR. Proc. Natl. Acad. Sci. USA 2020, 117, 5895–5906. [Google Scholar] [CrossRef] [PubMed]
- Ripstein, Z.A.; Vahidi, S.; Rubinstein, J.L.; Kay, L.E. A pH-Dependent Conformational Switch Controls, N. Meningitidis ClpP Protease Function. J. Am. Chem. Soc. 2020, 142, 20519–20523. [Google Scholar] [CrossRef]
- Sprangers, R.; Gribun, A.; Hwang, P.M.; Houry, W.A.; Kay, L.E. Quantitative NMR Spectroscopy of Supramolecular Complexes: Dynamic Side Pores in ClpP Are Important for Product Release. Proc. Natl. Acad. Sci. USA 2005, 102, 16678–16683. [Google Scholar] [CrossRef]
- Díaz-Sáez, L.; Pankov, G.; Hunter, W.N. Open and Compressed Conformations of Francisella Tularensis ClpP. Proteins 2017, 85, 188–194. [Google Scholar] [CrossRef]
- Kim, L.; Lee, B.; Kim, M.; Kim, M.K.; Kwon, D.H.; Kim, H.; Brötz-Oesterhelt, H.; Roh, S.; Song, H.K. Structural Insights into ClpP Protease Side Exit Pore-opening by a pH Drop Coupled with Substrate Hydrolysis. EMBO J. 2022, 41, e109755. [Google Scholar] [CrossRef]
- Singh, S.K.; Rozycki, J.; Ortega, J.; Ishikawa, T.; Lo, J.; Steven, A.C.; Maurizi, M.R. Functional Domains of the ClpA and ClpX Molecular Chaperones Identified by Limited Proteolysis and Deletion Analysis*. J. Biol. Chem. 2001, 276, 29420–29429. [Google Scholar] [CrossRef]
- Wojtyra, U.A.; Thibault, G.; Tuite, A.; Houry, W.A. The N-Terminal Zinc Binding Domain of ClpX Is a Dimerization Domain That Modulates the Chaperone Function*. J. Biol. Chem. 2003, 278, 48981–48990. [Google Scholar] [CrossRef]
- Glynn, S.E.; Nager, A.R.; Baker, T.A.; Sauer, R.T. Dynamic and Static Components Power Unfolding in Topologically Closed Rings of a AAA+ Proteolytic Machine. Nat. Struct. Mol. Biol. 2012, 19, 616–622. [Google Scholar] [CrossRef]
- Martin, A.; Baker, T.A.; Sauer, R.T. Rebuilt AAA + Motors Reveal Operating Principles for ATP-Fuelled Machines. Nature 2005, 437, 1115–1120. [Google Scholar] [CrossRef]
- Donaldson, L.W.; Wojtyra, U.; Houry, W.A. Solution Structure of the Dimeric Zinc Binding Domain of the Chaperone ClpX*. J. Biol. Chem. 2003, 278, 48991–48996. [Google Scholar] [CrossRef]
- Stinson, B.M.; Nager, A.R.; Glynn, S.E.; Schmitz, K.R.; Baker, T.A.; Sauer, R.T. Nucleotide Binding and Conformational Switching in the Hexameric Ring of a AAA+ Machine. Cell 2013, 153, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Levchenko, I.; Fraczkowska, K.; Woodruff, R.V.; Sauer, R.T.; Baker, T.A. Molecular Determinants of Complex Formation between Clp/Hsp100 ATPases and the ClpP Peptidase. Nat. Struct. Biol. 2001, 8, 230–233. [Google Scholar] [CrossRef]
- Shein, M.; Hitzenberger, M.; Cheng, T.C.; Rout, S.R.; Leitl, K.D.; Sato, Y.; Zacharias, M.; Sakata, E.; Schütz, A.K. Characterizing ATP Processing by the AAA+ Protein P97 at the Atomic Level. Nat. Chem. 2024, 16, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Carroni, M.; Saibil, H.R. Cryo Electron Microscopy to Determine the Structure of Macromolecular Complexes. Methods 2016, 95, 78–85. [Google Scholar] [CrossRef]
- Han, H.; Monroe, N.; Sundquist, W.I.; Shen, P.S.; Hill, C.P. The AAA ATPase Vps4 Binds ESCRT-III Substrates through a Repeating Array of Dipeptide-Binding Pockets. eLife 2017, 6, e31324. [Google Scholar] [CrossRef]
- Deville, C.; Franke, K.; Mogk, A.; Bukau, B.; Saibil, H.R. Two-Step Activation Mechanism of the ClpB Disaggregase for Sequential Substrate Threading by the Main ATPase Motor. Cell Rep. 2019, 27, 3433–3446.e4. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM Structures and Dynamics of Substrate-Engaged Human 26S Proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef]
- Fritze, J.; Zhang, M.; Luo, Q.; Lu, X. An Overview of the Bacterial SsrA System Modulating Intracellular Protein Levels and Activities. Appl. Microbiol. Biotechnol. 2020, 104, 5229–5241. [Google Scholar] [CrossRef] [PubMed]
- Keiler, K.C.; Waller, P.R.H.; Sauer, R.T. Role of a Peptide Tagging System in Degradation of Proteins Synthesized from Damaged Messenger RNA. Science 1996, 271, 990–993. [Google Scholar] [CrossRef]
- Gottesman, S.; Roche, E.; Zhou, Y.; Sauer, R.T. The ClpXP and ClpAP Proteases Degrade Proteins with Carboxy-Terminal Peptide Tails Added by the SsrA-Tagging System. Genes. Dev. 1998, 12, 1338–1347. [Google Scholar] [CrossRef]
- Janssen, B.D.; Hayes, C.S. The tmRNA Ribosome Rescue System. Adv. Protein Chem. Struct. Biol. 2012, 86, 151–191. [Google Scholar] [CrossRef] [PubMed]
- Puchades, C.; Rampello, A.J.; Shin, M.; Giuliano, C.J.; Wiseman, R.L.; Glynn, S.E.; Lander, G.C. Structure of the Mitochondrial Inner Membrane AAA+ Protease YME1 Gives Insight into Substrate Processing. Science 2017, 358, eaao0464. [Google Scholar] [CrossRef] [PubMed]
- Botos, I.; Lountos, G.T.; Wu, W.; Cherry, S.; Ghirlando, R.; Kudzhaev, A.M.; Rotanova, T.V.; de Val, N.; Tropea, J.E.; Gustchina, A.; et al. Cryo-EM Structure of Substrate-Free E. coli Lon Protease Provides Insights into the Dynamics of Lon Machinery. Curr. Res. Struct. Biol. 2019, 1, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Schmitz, K.A.; Schenck, N.; Balasopoulos, D.; Topitsch, A.; Maier, T.; Abrahams, J.P. Catalytic Cycling of Human Mitochondrial Lon Protease. Structure 2022, 30, 1254–1268.e7. [Google Scholar] [CrossRef] [PubMed]
- Ripstein, Z.A.; Huang, R.; Augustyniak, R.; Kay, L.E.; Rubinstein, J.L. Structure of a AAA+ Unfoldase in the Process of Unfolding Substrate. eLife 2017, 6, e25754. [Google Scholar] [CrossRef]
- Monroe, N.; Han, H.; Shen, P.S.; Sundquist, W.I.; Hill, C.P. Structural Basis of Protein Translocation by the Vps4-Vta1 AAA ATPase. eLife 2017, 6, e24487. [Google Scholar] [CrossRef]
- Yu, H.; Lupoli, T.J.; Kovach, A.; Meng, X.; Zhao, G.; Nathan, C.F.; Li, H. ATP Hydrolysis-Coupled Peptide Translocation Mechanism of Mycobacterium Tuberculosis ClpB. Proc. Natl. Acad. Sci. USA 2018, 115, E9560–E9569. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Baker, T.A.; Sauer, R.T. Protein Unfolding by a AAA+ Protease Is Dependent on ATP-Hydrolysis Rates and Substrate Energy Landscapes. Nat. Struct. Mol. Biol. 2008, 15, 139–145. [Google Scholar] [CrossRef]
- Aubin-Tam, M.-E.; Olivares, A.O.; Sauer, R.T.; Baker, T.A.; Lang, M.J. Single-Molecule Protein Unfolding and Translocation by an ATP-Fueled Proteolytic Machine. Cell 2011, 145, 257–267. [Google Scholar] [CrossRef]
- Maillard, R.A.; Chistol, G.; Sen, M.; Righini, M.; Tan, J.; Kaiser, C.M.; Hodges, C.; Martin, A.; Bustamante, C. ClpX(P) Generates Mechanical Force to Unfold and Translocate Its Protein Substrates. Cell 2011, 145, 459–469. [Google Scholar] [CrossRef]
- Sen, M.; Maillard, R.A.; Nyquist, K.; Rodriguez-Aliaga, P.; Pressé, S.; Martin, A.; Bustamante, C. The ClpXP Protease Functions as a Motor with Constant “Rpm” but Different “Gears”. Cell 2013, 155, 636–646. [Google Scholar] [CrossRef]
- Cordova, J.C.; Olivares, A.O.; Shin, Y.; Stinson, B.M.; Calmat, S.; Schmitz, K.R.; Aubin-Tam, M.-E.; Baker, T.A.; Lang, M.J.; Sauer, R.T. Stochastic but Highly Coordinated Protein Unfolding and Translocation by the ClpXP Proteolytic Machine. Cell 2014, 158, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Fei, X.; Bell, T.A.; Baker, T.A. Structure and Function of ClpXP, a AAA+ Proteolytic Machine Powered by Probabilistic ATP Hydrolysis. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 188–204. [Google Scholar] [CrossRef] [PubMed]
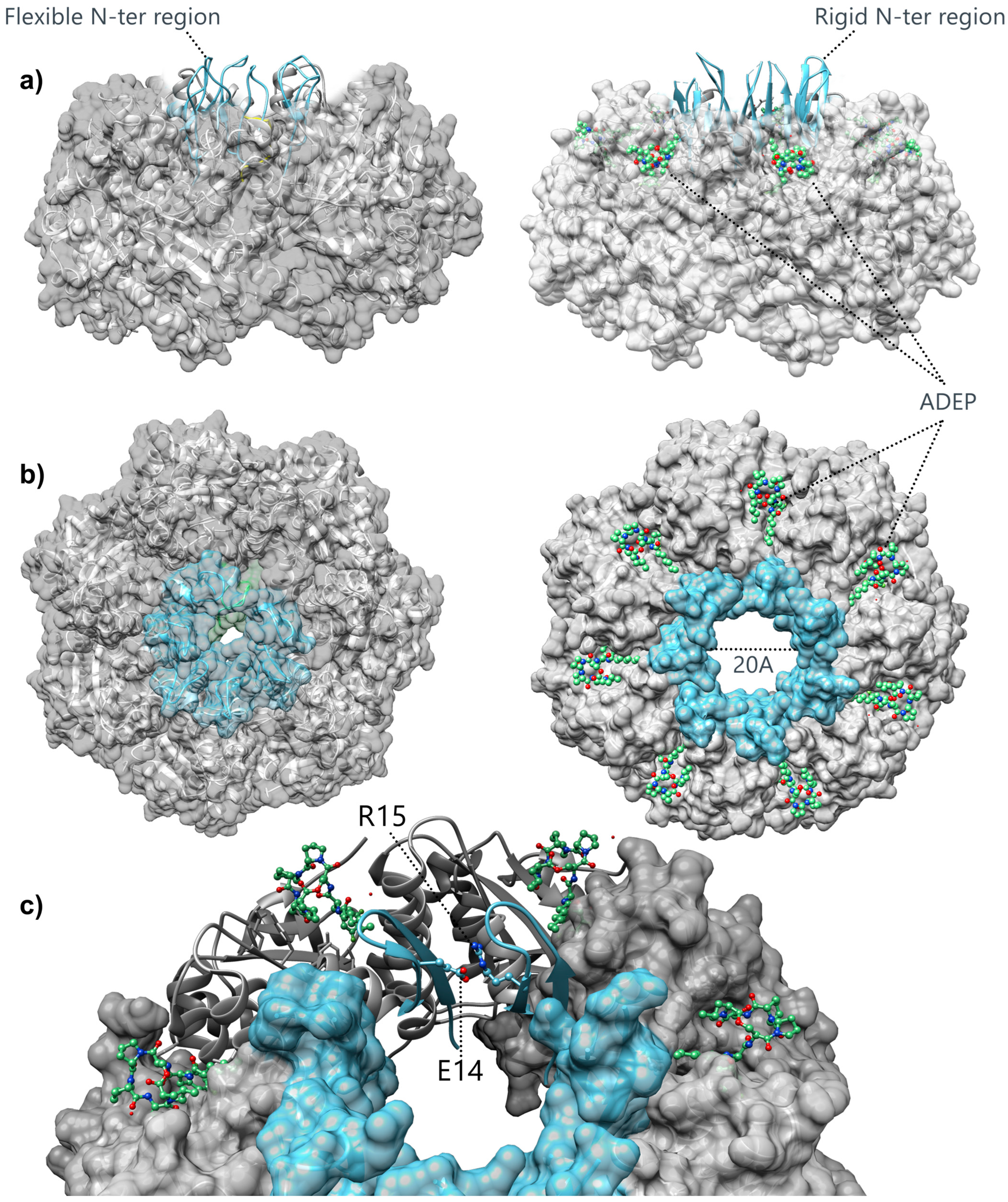
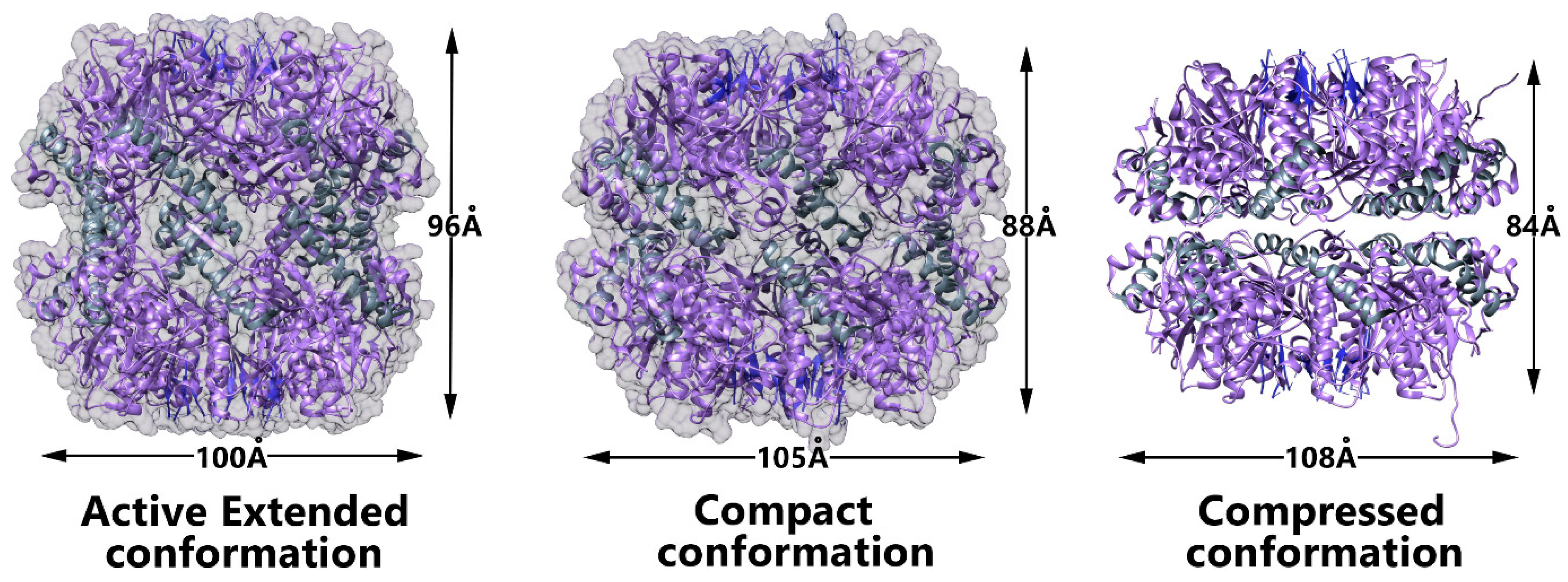
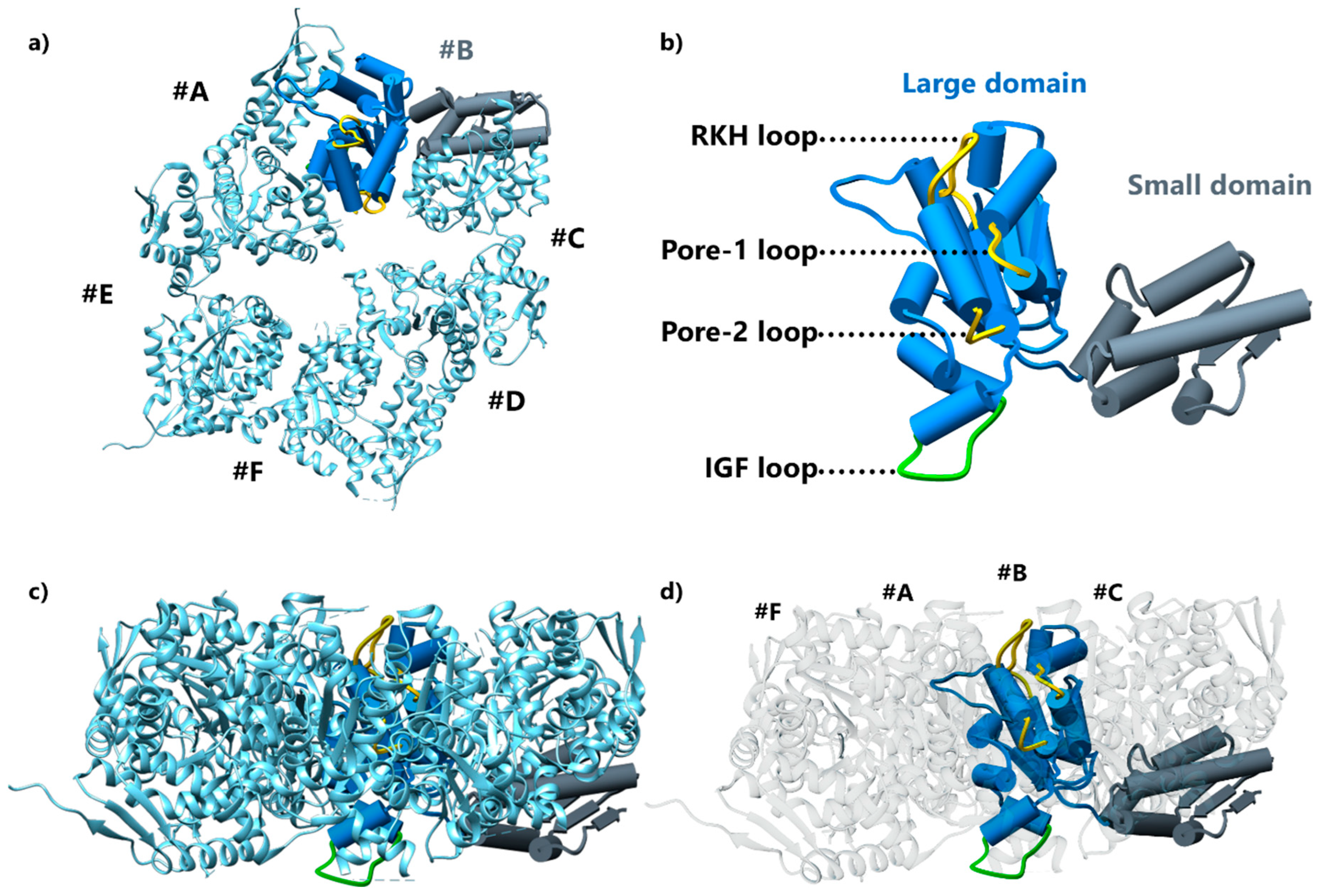
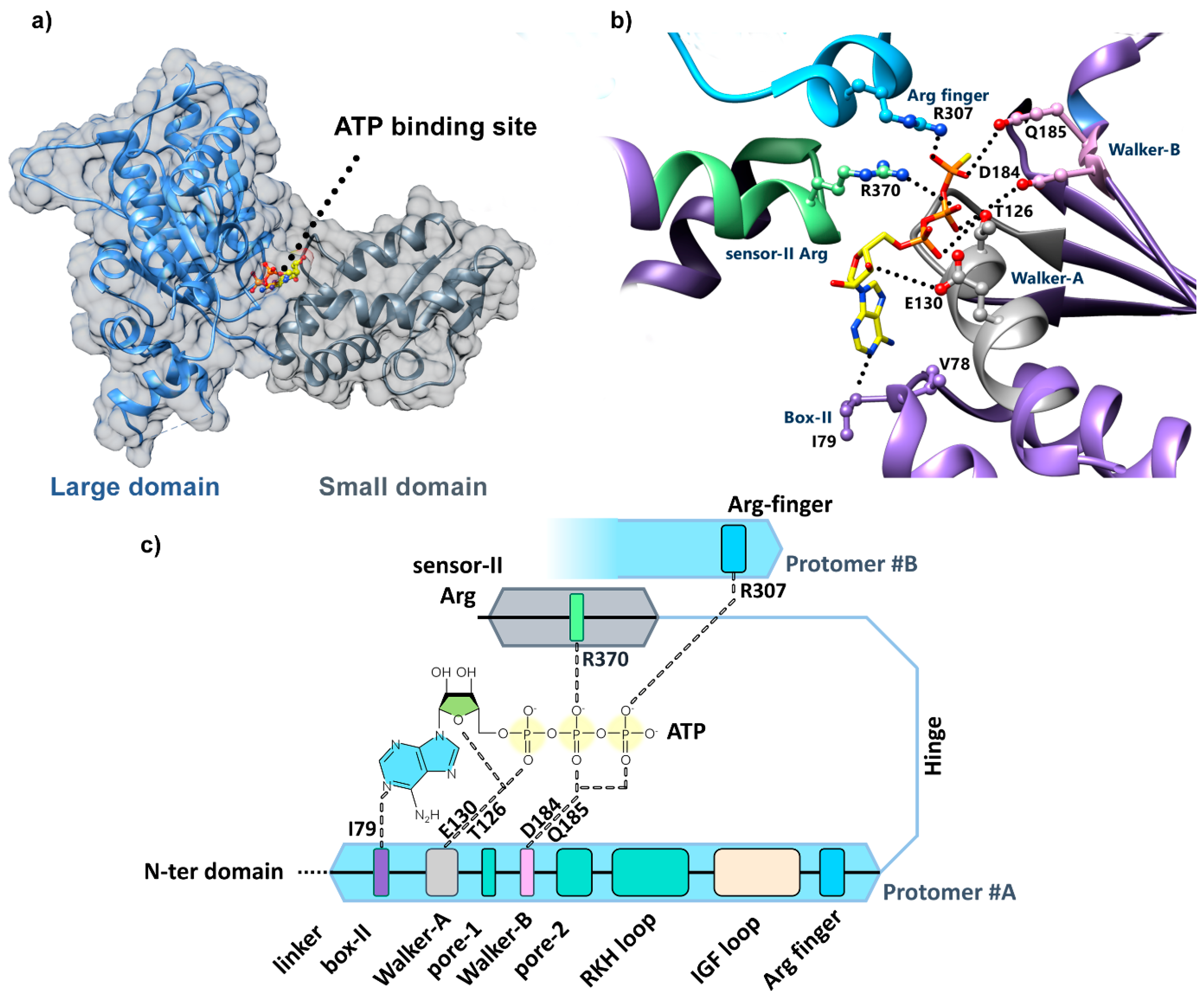

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Audibert, A.; Boisbouvier, J.; Vermot, A. Recent Advances in the Structural Studies of the Proteolytic ClpP/ClpX Molecular Machine. Biomolecules 2025, 15, 1097. https://doi.org/10.3390/biom15081097
Audibert A, Boisbouvier J, Vermot A. Recent Advances in the Structural Studies of the Proteolytic ClpP/ClpX Molecular Machine. Biomolecules. 2025; 15(8):1097. https://doi.org/10.3390/biom15081097
Chicago/Turabian StyleAudibert, Astrid, Jerome Boisbouvier, and Annelise Vermot. 2025. "Recent Advances in the Structural Studies of the Proteolytic ClpP/ClpX Molecular Machine" Biomolecules 15, no. 8: 1097. https://doi.org/10.3390/biom15081097
APA StyleAudibert, A., Boisbouvier, J., & Vermot, A. (2025). Recent Advances in the Structural Studies of the Proteolytic ClpP/ClpX Molecular Machine. Biomolecules, 15(8), 1097. https://doi.org/10.3390/biom15081097