
Cardiac Myosin Inhibitors in the Treatment of Hypertrophic Cardiomyopathy: Clinical Trials and Future Challenges



Abstract
1. Introduction
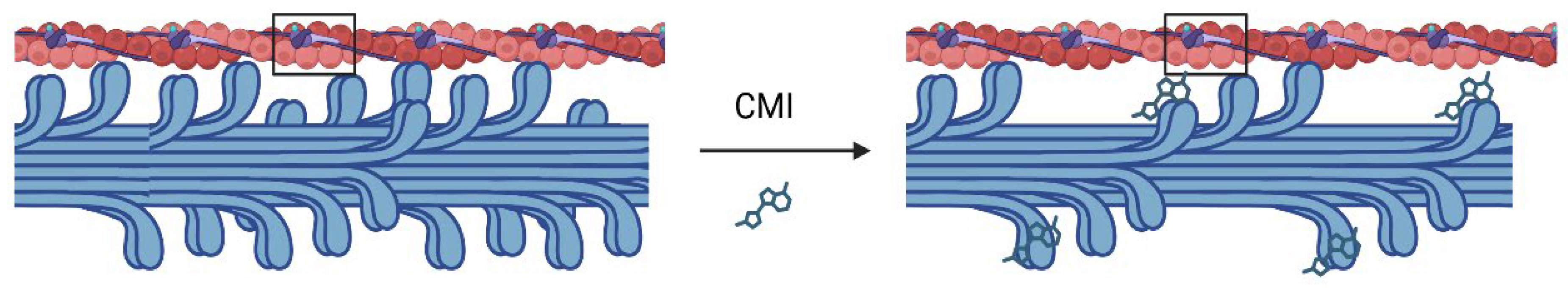
Physiology of Cardiac Muscle Contraction and Pathophysiology of HCM
2. Materials and Methods
2.1. Data Sources
2.2. Search Strategy
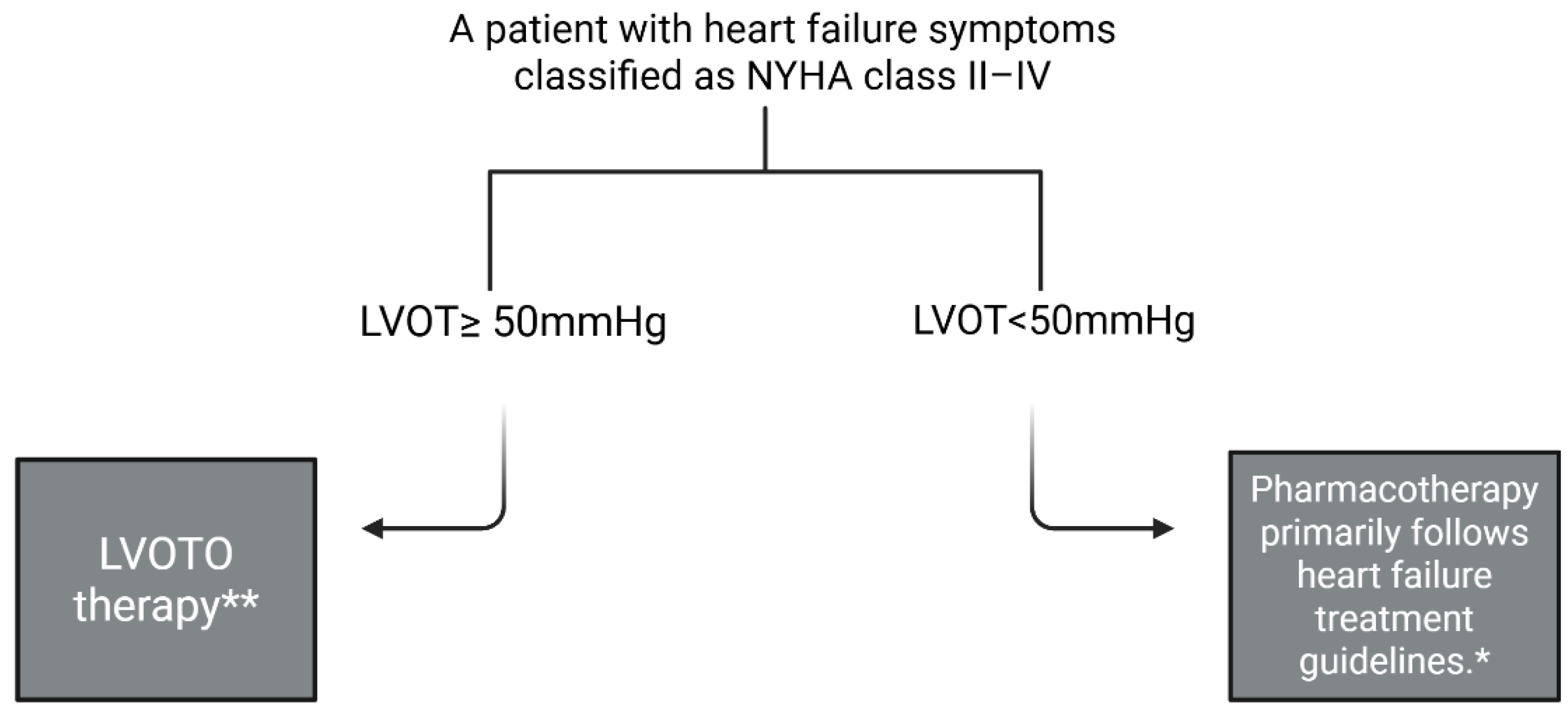
2.3. Inclusion and Exclusion Criteria
2.4. Article Selection and Quality Assessment
2.5. Bias Assessment
2.6. Transparency and Reproducibility
3. Cardiac Myosin Inhibitors
3.1. Mavacamten
3.2. Aficamten
3.3. Experimental and Emerging CMIs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HFpEF | Heart Failure with Preserved Ejection Fraction |
| HCM | Hypertrophic Cardiomyopathy |
| oHCM | Obstructive Hypertrophic Cardiomyopathy |
| nHCM | Non-Obstructive Hypertrophic Cardiomyopathy |
| CMI | Cardiac Myosin Inhibitor |
References
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Mandeş, L.; Roşca, M.; Ciupercă, D.; Popescu, B.A. The role of echocardiography for diagnosis and prognostic stratification in hypertrophic cardiomyopathy. J. Echocardiogr. 2020, 18, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Mizia-Stec, K.; Leszek, P.; Cegłowska, U.; Wiśniewska, A.; Hałgas, K.; Wybraniec, M.; Pachciński, O.; Stec, M.; Cieśla, D.; Gąsior, M.; et al. Incidence and prevalence of cardiomyopathies in Poland and outcomes for patients in the years 2016–2020. Kardiol. Pol. 2024, 82, 217–219. [Google Scholar] [CrossRef]
- Chumakova, O.S.; Baklanova, T.N.; Zateyshchikov, D.A. Clinical Features and Prospective Outcomes of Thin-Filament Hypertrophic Cardiomyopathy: Intrinsic Data and Comparative Insights from Other Cohorts. J. Clin. Med. 2025, 14, 866. [Google Scholar] [CrossRef]
- Hong, Y.; Xi, H.T.; Yang, X.Y.; Su, W.W.; Li, X.P. Pathogenic genes and clinical prognosis in hypertrophic cardiomyopathy. World J. Cardiol. 2025, 17, 99595. [Google Scholar] [CrossRef]
- The Criteria Committee of the New York Heart Association. Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels, 9th ed.; Little, Brown & Co.: Boston, MA, USA, 1994; pp. 253–256. [Google Scholar]
- Vilcant, V.; Hai, O. Left Ventricular Outflow Tract Obstruction. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Huffman, M.D.; Roth, G.A.; Sliwa, K.; Yancy, C.W.; Prabhakaran, D. Heart Failure. In Cardiovascular, Respiratory, and Related Disorders, 3rd ed.; Prabhakaran, D., Anand, S., Gaziano, T.A., Mbanya, J.C., Wu, Y., Nugent, R., Eds.; The International Bank for Reconstruction and Development/The World Bank: Washington, DC, USA, 2017. [Google Scholar]
- Elliott, P.M.; Gimeno, J.R.; Tomé, M.T.; Shah, J.; Ward, D.; Thaman, R.; Mogensen, J.; McKenna, W.J. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2006, 27, 1933–1941. [Google Scholar] [CrossRef]
- Kalinski, J.K.; Xu, B.; Boyd, R.; Tasseff, N.; Rutkowski, K.; Ospina, S.; Smedira, N.; Thamilarasan, M.; Popovic, Z.B.; Desai, M.Y. Novel Cardiac Myosin Inhibitor Therapy for Hypertrophic Cardiomyopathy in Adults: A Contemporary Review. Am. J. Cardiovasc. Drugs 2024, 24, 591–602. [Google Scholar] [CrossRef]
- Sherrod Iv, C.F.; Saberi, S.; Nassif, M.E.; Claggett, B.L.; Coats, C.J.; Garcia-Pavia, P.; Januzzi, J.L.; Lewis, G.D.; Ma, C.; Maron, M.S.; et al. Effect of Aficamten on Health Status Outcomes in Obstructive Hypertrophic Cardiomyopathy: Results From SEQUOIA-HCM. J. Am. Coll. Cardiol. 2024, 84, 1773–1785. [Google Scholar] [CrossRef]
- Desai, M.Y.; Owens, A.; Wolski, K.; Geske, J.B.; Saberi, S.; Wang, A.; Sherrid, M.; Cremer, P.C.; Lakdawala, N.K.; Tower-Rader, A.; et al. Mavacamten in Patients With Hypertrophic Cardiomyopathy Referred for Septal Reduction: Week 56 Results From the VALOR-HCM Randomized Clinical Trial. JAMA Cardiol. 2023, 8, 968–977. [Google Scholar] [CrossRef]
- Ommen, S.R.; Ho, C.Y.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1239–e1311. [Google Scholar] [CrossRef] [PubMed]
- U.S. Foods and Drugs Administration (FDA). Available online: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-new-drug-improve-heart-function-adults-rare-heart-condition (accessed on 15 May 2025).
- European Medicine Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/camzyos (accessed on 15 May 2025).
- Clinical Trails. Available online: https://clinicaltrials.gov/study/NCT05186818 (accessed on 15 May 2025).
- Clinical Trails. Available online: https://clinicaltrials.eu/inn/bms-986435/ (accessed on 15 May 2025).
- Zhang, H.; Yu, C.; Cheng, Y.; Chen, Z.; Chen, M.; He, W.; Jin, Z.; Cai, S.; Yu, L. Clinical Trials in Hypertrophic Cardiomyopathy Therapy: A Comprehensive Analysis of Trials Registered in Global Clinical Databases. Drug Des. Devel. Ther. 2023, 17, 1863–1877. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef] [PubMed]
- Squire, J. Special Issue: The Actin-Myosin Interaction in Muscle: Background and Overview. Int. J. Mol. Sci. 2019, 20, 5715. [Google Scholar] [CrossRef] [PubMed]
- Jani, V.P.; Song, T.; Gao, C.; Gong, H.; Sadayappan, S.; Kass, D.A.; Irving, T.C.; Ma, W. The structural OFF and ON states of myosin can be decoupled from the biochemical super- and disordered-relaxed states. PNAS Nexus 2024, 3, 39. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Fang, T.; Huang, J.; Guo, Y.; Alam, M.; Qian, H. Hypertrophic cardiomyopathy: From phenotype and pathogenesis to treatment. Front. Cardiovasc. Med. 2021, 8, 722340. [Google Scholar] [CrossRef]
- Trivedi, D.V.; Adhikari, A.S.; Sarkar, S.S.; Ruppel, K.M.; Spudich, J.A. Hypertrophic cardiomyopathy and the myosin mesa: Viewing an old disease in a new light. Biophys. Rev. 2018, 10, 27–48. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, H.K.; Hwang, I.C.; Lee, S.P.; Park, E.A.; Lee, W.; Kim, Y.J.; Park, J.H.; Sohn, D.W. Myocardial scarring on cardiovascular magnetic resonance in asymptomatic or minimally symptomatic patients with “pure” apical hypertrophic cardiomyopathy. J. Cardiovasc. Magn. Reson. 2012, 14, 52. [Google Scholar] [CrossRef]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef]
- Turer, A.T.; Wang, A. Cardiac Myosin Inhibitors: Unlocking Potential to Improve Treatment in Hypertrophic Cardiomyopathy. Circulation 2023, 147, 700–702. [Google Scholar] [CrossRef] [PubMed]
- Aman, A.; Akram, A.; Akram, B.; Maham, M.; Bokhari, M.Z.; Akram, A.; Akram, S.; Yaqub, F. Efficacy of cardiac myosin inhibitors mavacamten and aficamten in hypertrophic cardiomyopathy: A systematic review and meta-analysis of randomised controlled trials. Open Heart 2025, 12, e003215. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Saberi, S.; Abraham, T.P.; Elliott, P.M.; Olivotto, I. Mavacamten: A first-in-class myosin inhibitor for obstructive hypertrophic cardiomyopathy. Eur. Heart J. 2023, 44, 4622–4633. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, C.L.; Aprajita, Y.; Ferguson, B.S.; Christopher, Z.; Trisha, S.; Frank, R.; John, S.; Lee-Jae, G.; Allison, H.; Julie, G.; et al. Abstract 14585: Chronic Treatment with a Mavacamten-Like Myosin-Modulator (MYK-581) Blunts Disease Progression in a Mini-Pig Genetic Model of Non-Obstructed Hypertrophic Cardiomyopathy: In Vivo Evidence for Improved Relaxation and Functional Reserve. Circulation 2019, 140, A14585. [Google Scholar]
- Swift, L.M.; Asfour, H.; Posnack, N.G.; Arutunyan, A.; Kay, M.W.; Sarvazyan, N. Properties of blebbistatin for cardiac optical mapping and other imaging applications. Pflug. Arch. 2012, 464, 503–512. [Google Scholar] [CrossRef]
- Lehman, S.J.; Crocini, C.; Leinwand, L.A. Targeting the sarcomere in inherited cardiomyopathies. Nat. Rev. Cardiol. 2022, 19, 353–363. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 117761397, Mavacamten. PubChem Identifier. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Mavacamten#section=2D-Structure&fullscreen=true (accessed on 21 May 2025).
- Keam, S.J. Mavacamten: First Approval. Drugs 2022, 82, 1127–1135, Erratum in Drugs 2022, 82, 1235. [Google Scholar] [CrossRef]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214998s000lbl.pdf (accessed on 15 May 2025).
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten Treatment for Obstructive Hypertrophic Cardiomyopathy: A Clinical Trial. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef]
- Masri, A.; Lester, S.J.; Stendahl, J.C.; Hegde, S.M.; Sehnert, A.J.; Balaratnam, G.; Shah, A.; Fox, S.; Wang, A. Long-Term Safety and Efficacy of Mavacamten in Symptomatic Obstructive Hypertrophic Cardiomyopathy: Interim Results of the PIONEER-OLE Study. J. Am. Heart Assoc. 2024, 13, e030607. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769, Erratum in Lancet 2020, 396, 758. [Google Scholar] [CrossRef]
- Kitaoka, H.; Ieda, M.; Ebato, M.; Kozuma, K.; Takayama, M.; Tanno, K.; Komiyama, N.; Sakata, Y.; Maekawa, Y.; Minami, Y.; et al. Phase 3 Open-Label Study Evaluating the Efficacy and Safety of Mavacamten in Japanese Adults with Obstructive Hypertrophic Cardiomyopathy—The HORIZON-HCM Study. Circ. J. 2024, 89, 130–138. [Google Scholar] [CrossRef]
- Rader, F.; Oręziak, A.; Choudhury, L.; Saberi, S.; Fermin, D.; Wheeler, M.T.; Abraham, T.P.; Garcia-Pavia, P.; Zwas, D.R.; Masri, A.; et al. Mavacamten Treatment for Symptomatic Obstructive Hypertrophic Cardiomyopathy: Interim Results From the MAVA-LTE Study, EXPLORER-LTE Cohort. JACC Heart Fail. 2024, 12, 164–177. [Google Scholar] [CrossRef]
- Inestroza, K.; Mijares-Rojas, I.; Matute-Martínez, C.; Ergui, L.; Albosta, M.; Vergara-Sanchez, C.; Dangl, M.; Hernandez, R.J.; Ebner, B.; Vincent, L.T.; et al. In-hospital outcomes of septal myectomy vs. alcohol septal ablation for hypertrophic cardiomyopathy with outflow tract obstruction: An update and insights from the national inpatient sample from 2011 to 2019. J. Investig. Med. 2024, 72, 262–269. [Google Scholar] [CrossRef]
- Ramonfaur, D.; Gasperetti, A.; Blake, V.E.; Rivers, B.; Kassamali, A.A.; Kasper, E.K.; Barouch, L.A.; Wu, K.C.; Madrazo, J.A.; Carrick, R.T. Eighteen-Month Real-World Experience Using Mavacamten for Treatment of Obstructive Hypertrophic Cardiomyopathy in a Racially Diverse Population. J. Am. Heart Assoc. 2024, 13, e034069. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Cho, J.Y.; Kwak, S.; Park, C.S.; Park, J.; Choi, H.M.; Cho, G.Y.; Choi, G.H.; Kim, J.; Na, J.O. Real-World Experience of Mavacamten for Patients With Obstructive Hypertrophic Cardiomyopathy in South Korea: A Prospective Multi-Center Observational Study. Korean Circ. J. 2025, 55, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Scholtz, S.; Coppée, C.; Mohemed, K.; Potratz, M.; Sequeira, V.; Rudolph, V.; Scholtz, W.; Reil, J.C. Mavacamten maintenance dose determination: Insights into individualised therapy for hypertrophic cardiomyopathy. Open Heart 2025, 12, e003192. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, N.; Hsu, P.; Sun, J.; Li, W.T.; Wang, Q.; Samira, M.; Wei, Q.; Yu, J.C.; Cao, G.Y.; et al. Pharmacokinetics and safety of mavacamten in healthy Chinese participants with different CYP2C19 phenotypes. Clin. Transl. Sci. 2024, 17, e13877. [Google Scholar] [CrossRef]
- Ricci, F.; Molinari, L.V.; Mansour, D.; Galanti, K.; Vagnarelli, F.; Renda, G.; Gallina, S.; Owens, A.; Luzum, J.A.; Olivotto, I.; et al. Managing drug-drug interactions with mavacamten: A focus on combined use of antiarrhythmic drugs and anticoagulants. Heart Rhythm 2025, 22, 510–525. [Google Scholar] [CrossRef]
- Ammirati, E.; Contri, R.; Coppini, R.; Cecchi, F.; Frigerio, M.; Olivotto, I. Pharmacological treatment of hypertrophic cardiomyopathy: Current practice and novel perspec-tives. Eur. J. Heart Fail. 2016, 18, 1106–1118. [Google Scholar] [CrossRef]
- Badr, A.; Roehl, K.; Suppah, M.; Abdullah, H.A.; Arsanjani, R.; Siontis, K.C.; Geske, J.B.; Ommen, S.R.; Giudicessi, J.R.; Alsidawi, R. Temporal Patterns of Holter-Detected Arrhythmias in Hypertrophic Cardiomyopathy Patients Treated with Mavacamten. Biomedicines 2025, 13, 1005. [Google Scholar] [CrossRef]
- Cytokinetics. Available online: https://pnr-files.pro1.gus.wdc.dianum.io/globenewswire/articles/2989688/en/cytokinetics-announces-fda-acceptance-of-new-drug.pdf (accessed on 15 May 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 139331495, Aficamten. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Aficamten (accessed on 21 May 2025).
- Hartman, J.J.; Hwee, D.T.; Robert-Paganin, J.; Chuang, C.; Chin, E.R.; Edell, S.; Lee, K.H.; Madhvani, R.; Paliwal, P.; Pernier, J.; et al. Aficamten is a small-molecule cardiac myosin inhibitor designed to treat hypertrophic cardiomyopathy. Nat. Cardiovasc. Res. 2024, 3, 1003–1016. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Coats, C.J.; Düngen, H.D.; et al. Aficamten for Symptomatic Obstructive Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2024, 390, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.; Collibee, S.; Ashcraft, L.; Wang, W.; Wal, M.V.; Wang, X.L.; Hwee, D.T.; Wu, Y.S.; Wang, J.Y.; Chin, E.R.; et al. Discovery of Aficamten (CK-274), a Next-Generation Cardiac Myosin Inhibitor for the Treatment of Hypertrophic Cardiomyopathy. J. Med. Chem. 2021, 64, 14142–14152. [Google Scholar] [CrossRef] [PubMed]
- Malik, F.I.; Robertson, L.A.; Armas, D.R.; Robbie, E.P.; Osmukhina, A.; Xu, D.; Li, H.; Solomon, S.D. A Phase 1 Dose-Escalation Study of the Cardiac Myosin Inhibitor Aficamten in Healthy Participants. JACC Basic Transl. Sci. 2022, 7, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Divanji, P.; Griffith, A.; Sukhun, R.; Cheplo, K.; Li, J.; German, P. Pharmacokinetics, disposition, and biotransformation of the cardiac myosin inhibitor aficamten in humans. Pharmacol. Res. Perspect. 2024, 12, e70006. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Abraham, T.P.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Coats, C.J.; et al. Impact of Aficamten on Disease and Symptom Burden in Obstructive Hypertrophic Cardiomyopathy: Results From SEQUOIA-HCM. J. Am. Coll. Cardiol. 2024, 84, 1821–1831. [Google Scholar] [CrossRef]
- Schulze, C.; Abraham, T.P.; Barriales-Villa, R.; Claggett, B.; Coats, C.; García-Pavía, P.; Hagège, A.; Januzzi, J.L.; Kulac, I.J.; Kwong, R.Y.; et al. Abstract 4140102: Changes in EQ-5D-5L with Aficamten in Obstructive Hypertrophic Cardiomyopathy (oHCM): The SEQUOIA-HCM Trial. Circulation 2024, 150, Suppl_1. [Google Scholar] [CrossRef]
- Saberi, S.; Abraham, T.P.; Choudhury, L.; Owens, A.T.; Tower-Rader, A.; Rader, F.; Pavia, P.G.; Olivotto, I.; Coats, C.; Fifer, M.A.; et al. Efficacy and safety of Aficamten in the first cohort of patients with symptomatic obstructive hypertrophic cardiomyopathy completing 1-year follow-up: Findings from the FOREST-HCM study. J. Am. Coll. Cardiol. 2024, 83, 356. [Google Scholar] [CrossRef]
- Owens, A.T.; Masri, A.; Abraham, T.P.; Choudhury, L.; Rader, F.; Symanski, J.D.; Turer, A.T.; Wong, T.C.; Tower-Rader, A.; Coats, C.J.; et al. Aficamten for Drug-Refractory Severe Obstructive Hypertrophic Cardiomyopathy in Patients Receiving Disopyramide: REDWOOD-HCM Cohort 3. J. Card. Fail. 2023, 29, 1576–1582. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Choudhury, L.; Olivotto, I.; Saberi, S.; Wang, A.; Garcia-Pavia, P.; Lakdawala, N.K.; Nagueh, S.F.; Rader, F.; et al. Phase 2 Study of Aficamten in Patients With Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 34–45. [Google Scholar] [CrossRef]
- Coats, C.J.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Düngen, H.; Garcia-Pavia, P.; et al. Dosing and Safety Profile of Aficamten in Symptomatic Obstructive Hypertrophic Cardiomyopathy: Results From SEQUOIA-HCM. J. Am. Heart Assoc. 2024, 13, e035993. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J.; Volk, H.; Nguyen, O.; Kamna, D.; Chen, H.; Barriales-Villa, R.; Garcia-Pavia, P.; Olivotto, I.; Owens, A.T.; Coats, C.J.; et al. Safety and Efficacy of Mavacamten and Aficamten in Patients With Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2025, 14, e038758. [Google Scholar] [CrossRef] [PubMed]
- Maharao, N.; Xu, D.; Simkins, T.J.; Bowles, O.; Liu, G.; Benattia, Y.; Griffith, A.; Heitner, S.B.; Kupfer, S.; German, P. Clinical Evaluation of the Effect of Aficamten on QT/QTc Interval in Healthy Participants. Clin. Transl. Sci. 2025, 18, e70218. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lutz, J.D.; Divanji, P.; Li, J.; Benattia, Y.; Griffith, A.; Heitner, S.B.; Kupfer, S.; German, P. Effect of Hepatic Impairment or Renal Impairment on the Pharmacokinetics of Aficamten. Clin. Pharmacokinet. 2025, 64, 397–406. [Google Scholar] [CrossRef]
- Cytokinetics. Available online: https://pnr-files.pro1.gus.wdc.dianum.io/globenewswire/articles/3001161/en/cytokinetics-announces-european-medicines-agency.pdf (accessed on 15 May 2025).
- Rauscher, A.Á.; Gyimesi, M.; Kovács, M.; Málnási-Csizmadia, A. Targeting Myosin by Blebbistatin Derivatives: Optimization and Pharmacological Potential. Trends. Biochem. Sci. 2018, 43, 700–713. [Google Scholar] [CrossRef]
- Roman, B.I.; Verhasselt, S.; Stevens, C.V. Medicinal Chemistry and Use of Myosin II Inhibitor (S)-Blebbistatin and Its Derivatives. J. Med. Chem. 2018, 61, 9410–9428. [Google Scholar] [CrossRef]
- Gyimesi, M.; Rauscher, A.Á.; Suthar, S.K.; Hamow, K.Á.; Oravecz, K.; Lőrincz, I.; Borhegyi, Z.; Déri, M.T.; Kiss, Á.F.; Monostory, K.; et al. Improved Inhibitory and Absorption, Distribution, Metabolism, Excretion, and Toxicology (ADMET) Properties of Blebbistatin Derivatives Indicate That Blebbistatin Scaffold Is Ideal for drug Development Targeting Myosin-2. J. Pharmacol. Exp. Ther. 2021, 376, 358–373. [Google Scholar] [CrossRef]
- Alsulami, K.; Marston, S. Small Molecules acting on Myofilaments as Treatments for Heart and Skeletal Muscle Diseases. Int. J. Mol. Sci. 2020, 21, 9599. [Google Scholar] [CrossRef]
- AdisInsight. Available online: https://adisinsight.springer.com/drugs/800056091 (accessed on 15 May 2025).
- Clinical Trails. Available online: https://clinicaltrials.gov/study/NCT06122779 (accessed on 15 May 2025).
- Shah, S.J.; Rigolli, M.; Javidialsaadi, A.; Patel, R.B.; Khadra, S.; Goyal, P.; Little, S.; Wever-Pinzon, O.; Owens, A.T.; Skali, H.; et al. Cardiac Myosin Inhibition in Heart Failure with Normal and Supranormal Ejection Fraction: Primary Results of the EMBARK-HFpEF Trial. JAMA Cardiol. 2025, 10, 170–175. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Bilen, O.; Burroughs, M.; Costabel, J.P.; Correia, E.d.B.; Dybro, A.M.; Elliott, P.; Lakdawala, N.K.; Mann, A.; Nair, A.; et al. Aficamten vs Metoprolol for Obstructive Hypertrophic Cardiomyopathy: MAPLE-HCM Rationale, Study Design, and Baseline Characteristics. JACC Heart Fail. 2025, 13, 346–357. [Google Scholar] [CrossRef]
- Wheeler, M.T.; Jacoby, D.; Elliott, P.M.; Saberi, S.; Hegde, S.M.; Lakdawala, N.K.; Myers, J.; Sehnert, A.J.; Edelberg, J.M.; Li, W.; et al. Effect of beta-blocker therapy on the response to mavacamten in patients with symptomatic obstructive hypertrophic cardiomyopathy. Eur. J. Heart Fail. 2023, 25, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trails. Available online: https://clinicaltrials.gov/study/NCT06081894 (accessed on 15 May 2025).
- Masri, A.; Maron, M.S.; Abraham, T.P.; Nassif, M.E.; Barriales-Villa, R.; Bilen, O.; Coats, C.J.; Elliott, P.; Garcia-Pavia, P.; Massera, D.; et al. Concomitant Aficamten and Disopyramide in Symptomatic Obstructive Hypertrophic Cardiomyopathy. JACC Heart Fail. 2025. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C. Appropriate and inappropriate polypharmacy-Choosing the right strategy. Br. J. Clin. Pharmacol. 2021, 87, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Delara, M.; Murray, L.; Jafari, B.; Bahji, A.; Goodarzi, Z.; Kirkham, J.; Chowdhury, M.; Seitz, D.P. Prevalence and factors associated with polypharmacy: A systematic review and Meta-analysis. BMC Geriatr. 2022, 22, 601, Erratum in BMC Geriatr. 2022, 22, 742. [Google Scholar] [CrossRef]
- Ho, C.Y.; Mealiffe, M.E.; Bach, R.G.; Bhattacharya, M.; Choudhury, L.; Edelberg, J.M.; Hegde, S.M.; Jacoby, D.; Lakdawala, N.K.; Lester, S.J.; et al. Evaluation of Mavacamten in Symptomatic Patients With Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2649–2660. [Google Scholar] [CrossRef]
- Desai, M.Y.; Nissen, S.E.; Abraham, T.; Olivotto, I.; Garcia-Pavia, P.; Lopes, R.D.; Verheyen, N.; Wever-Pinzon, O.; Wolski, K.; Jaber, W.; et al. Mavacamten in Symptomatic Nonobstructive Hypertrophic Cardiomyopathy: Design, Rationale, and Baseline Characteristics of ODYSSEY-HCM. JACC Heart Fail. 2025, 13, 358–370. [Google Scholar] [CrossRef]
- Bristol Myers Squibb. Available online: https://news.bms.com/news/details/2025/Bristol-Myers-Squibb-Provides-Update-on-Phase-3-ODYSSEY-HCM-Trial/ (accessed on 15 May 2025).
- Amr, A.; Kayvanpour, E.; Reich, C.; Koelemen, J.; Asokan, S.; Frey, N.; Meder, B.; Sedaghat-Hamedani, F. Assessing the Applicability of Cardiac Myosin Inhibitors for Hypertrophic Cardiomyopathy Management in a Large Single Center Cohort. Rev. Cardiovasc. Med. 2024, 25, 225. [Google Scholar] [CrossRef]
- Clinical Trails. Available online: https://clinicaltrials.gov/study/NCT06412666 (accessed on 15 May 2025).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | FDA Approval | EMA Approval | Clinical Development Stage |
|---|---|---|---|
| Mavacamten | Approved (2022) | Approved (2023) | Available on the market |
| Aficamten | Pending approval | Pending approval | Phase III completed |
| MYK-224 | Not approved | Not approved | Phase II |
| MYK-581 | Not approved | Not approved | Preclinical phase |
| Blebbistatin | Not approved | Not approved | Research use only |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kukowka, A.; Droździk, M. Cardiac Myosin Inhibitors in the Treatment of Hypertrophic Cardiomyopathy: Clinical Trials and Future Challenges. Biomolecules 2025, 15, 1098. https://doi.org/10.3390/biom15081098
Kukowka A, Droździk M. Cardiac Myosin Inhibitors in the Treatment of Hypertrophic Cardiomyopathy: Clinical Trials and Future Challenges. Biomolecules. 2025; 15(8):1098. https://doi.org/10.3390/biom15081098
Chicago/Turabian StyleKukowka, Arnold, and Marek Droździk. 2025. "Cardiac Myosin Inhibitors in the Treatment of Hypertrophic Cardiomyopathy: Clinical Trials and Future Challenges" Biomolecules 15, no. 8: 1098. https://doi.org/10.3390/biom15081098
APA StyleKukowka, A., & Droździk, M. (2025). Cardiac Myosin Inhibitors in the Treatment of Hypertrophic Cardiomyopathy: Clinical Trials and Future Challenges. Biomolecules, 15(8), 1098. https://doi.org/10.3390/biom15081098