

S-Nitrosylation in Cardiovascular Disorders: The State of the Art
 ,
,
Abstract
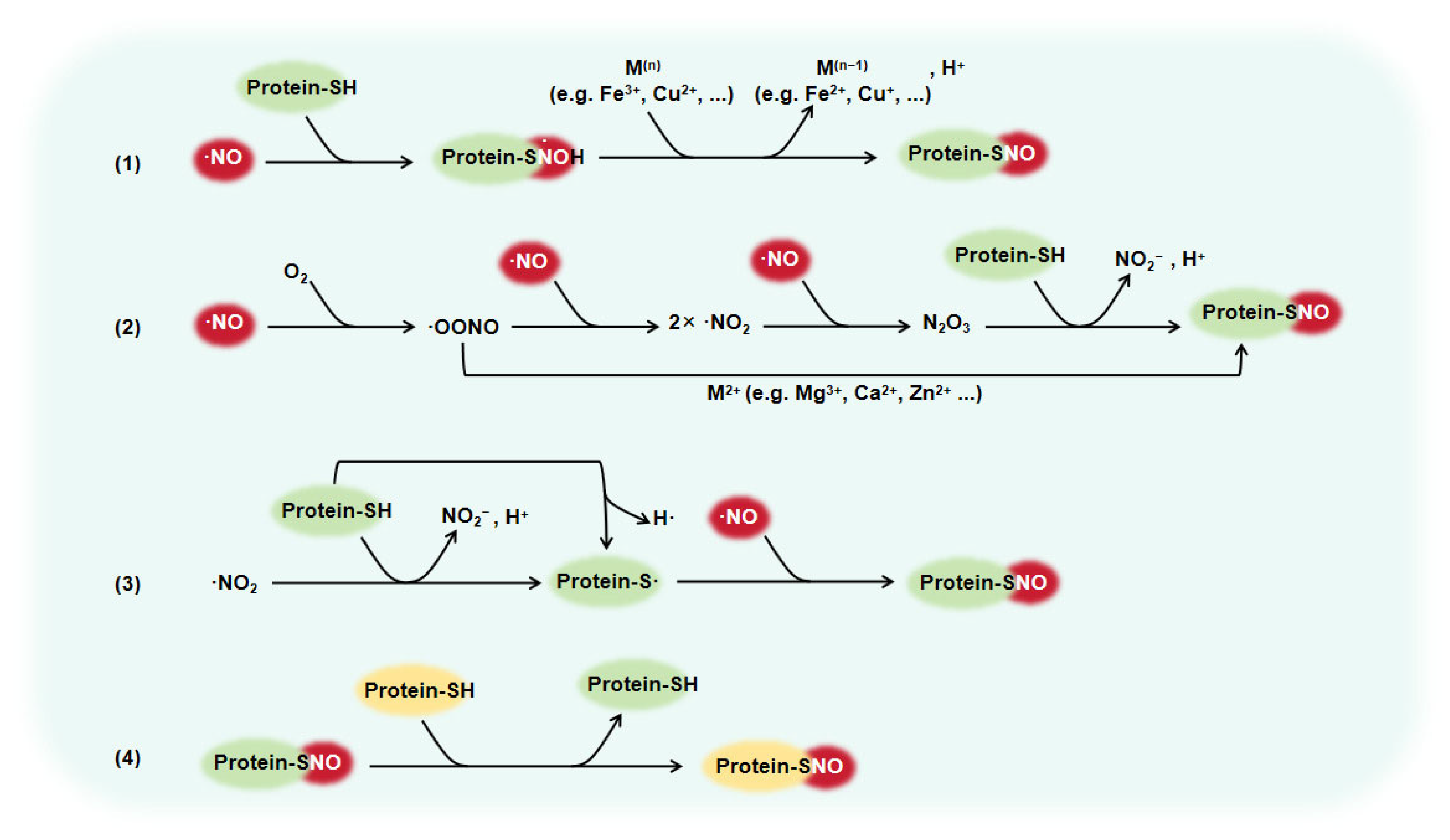
1. Introduction
2. The Role of Protein S-Nitrosylation in Vascular Diseases
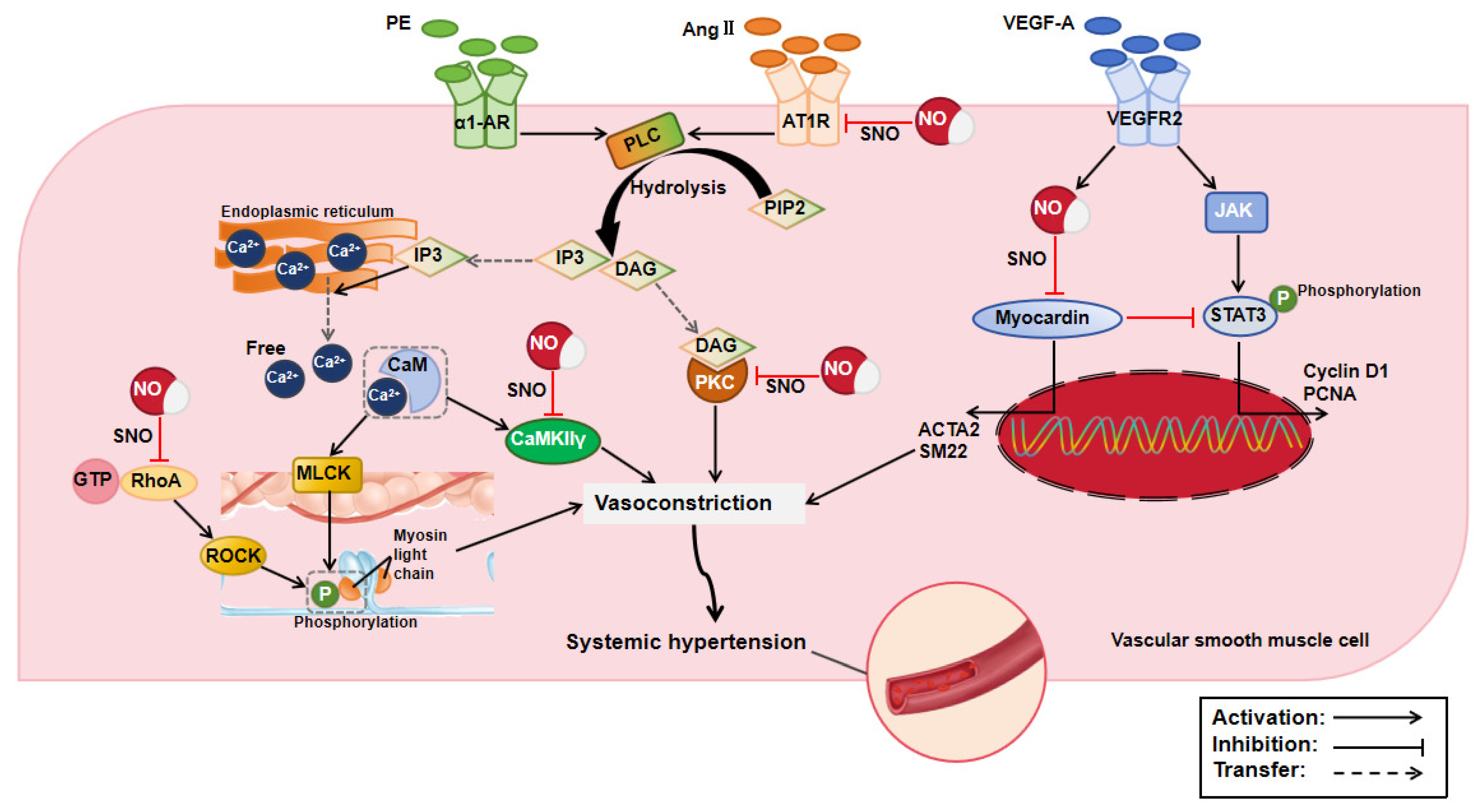
2.1. Effect of Protein S-Nitrosylation in Systemic Hypertension
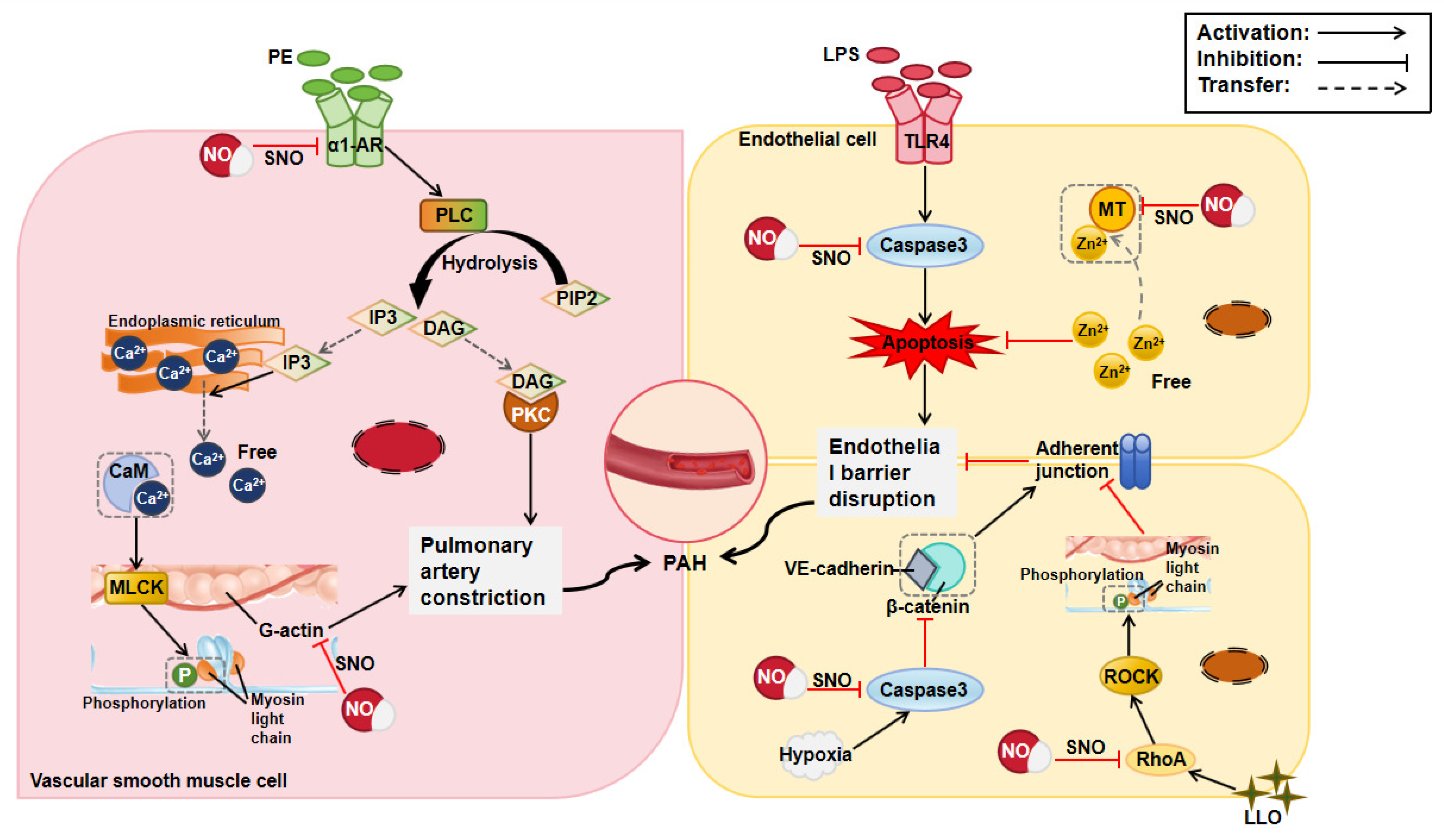
2.2. Effect of Protein S-Nitrosylation in Pulmonary Arterial Hypertension
2.3. Effect of Protein S-Nitrosylation in Atherosclerosis
3. The Role of Protein S-Nitrosylation in Heart Diseases
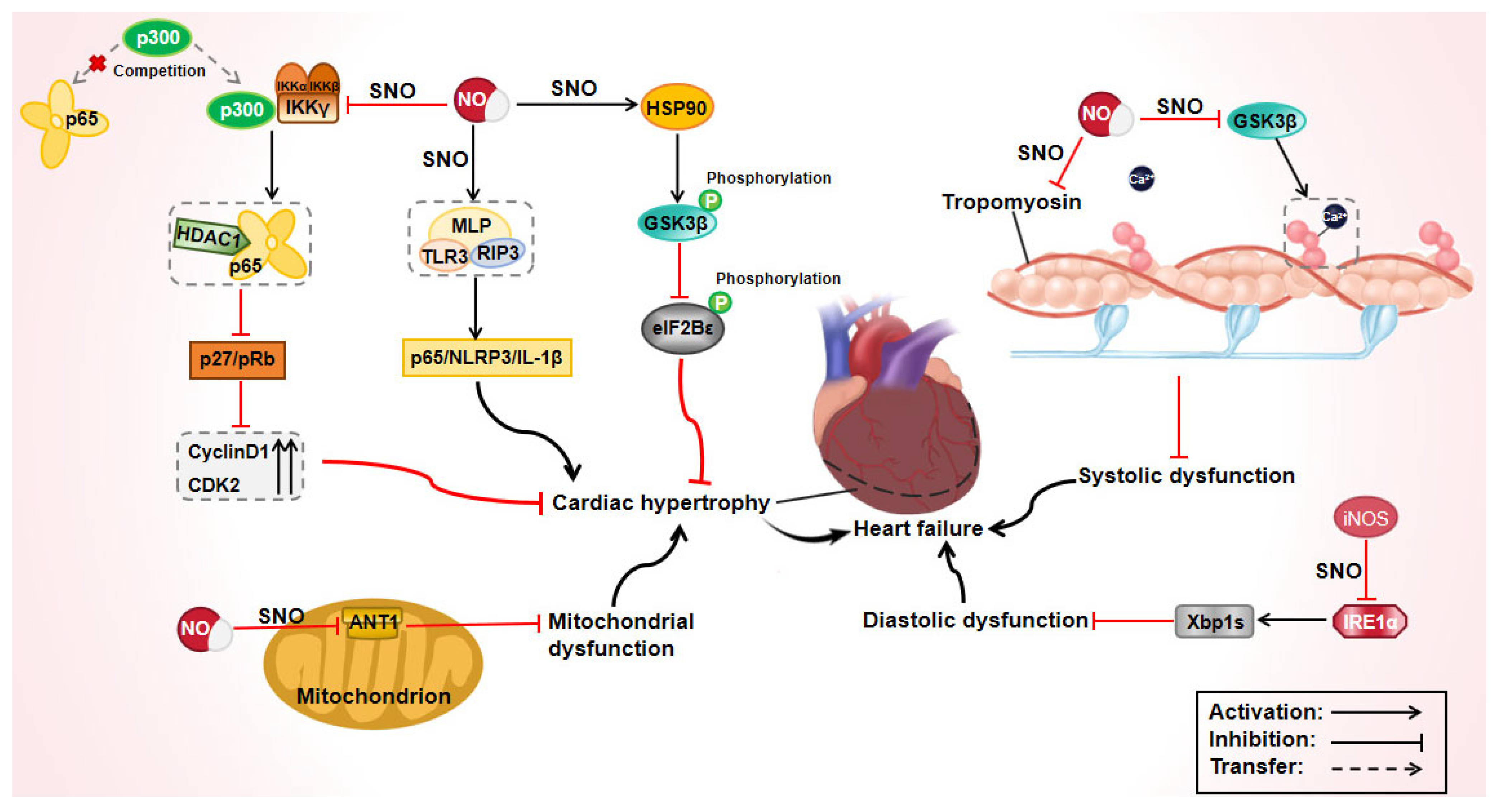
3.1. Effect of Protein S-Nitrosylation in Heart Failure
3.2. Effect of Protein S-Nitrosylation on Myocardial Infarction
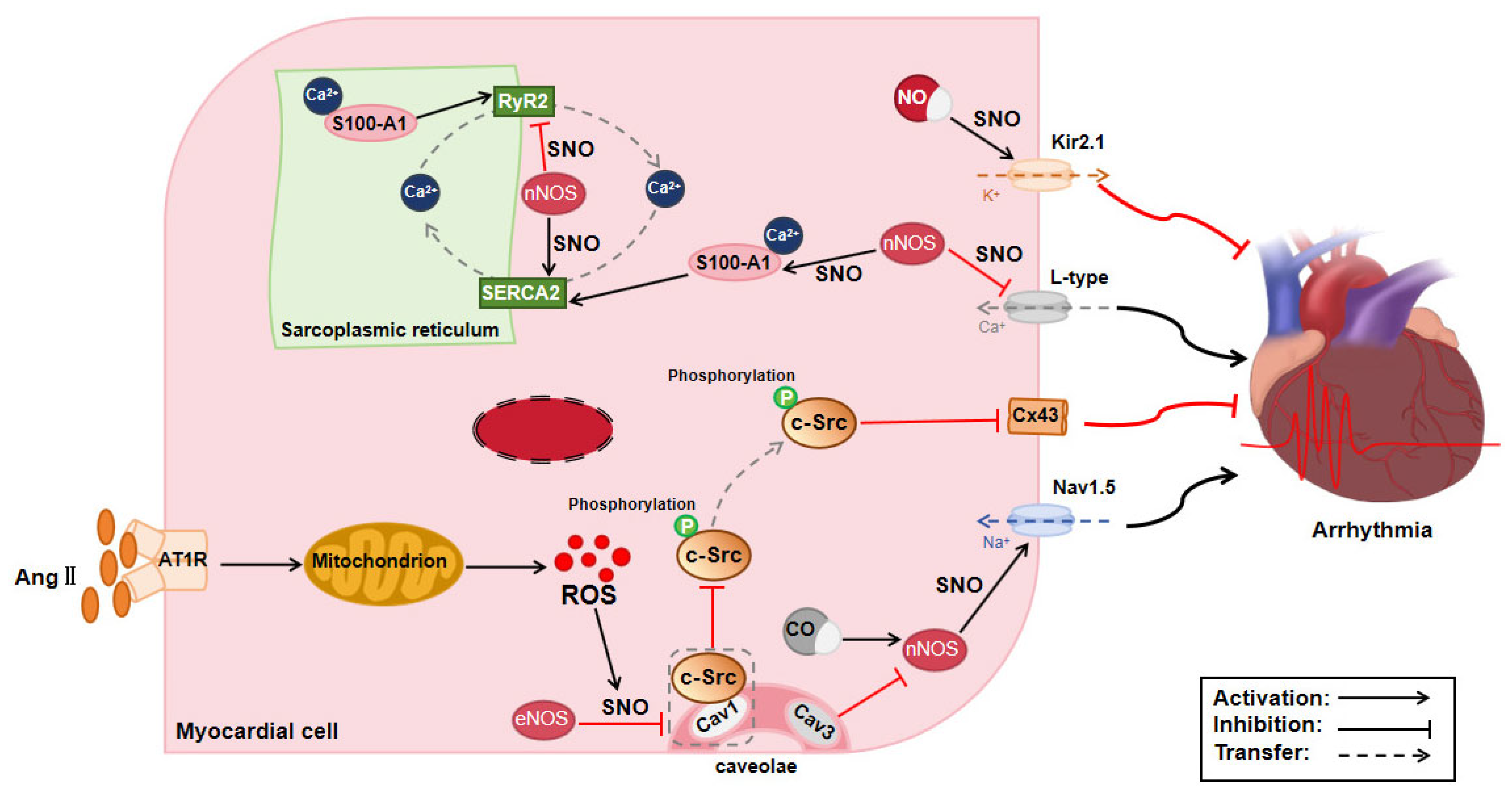
3.3. Effect of Protein S-Nitrosylation on Arrhythmia
3.4. Effect of Protein S-Nitrosylation in Diabetic Cardiomyopathy
4. Conclusions and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADP | adenosine diphosphate |
| AMPK | AMP-activated protein kinase |
| AP-1 | activator protein-1 |
| Caspase3 | cysteine proteinase 3 |
| CDK2 | cyclin-dependent kinase 2 |
| cGMP | cyclic guanosine monophosphate |
| CHC | clathrin heavy chain |
| CO | carbon monoxide |
| CSNO | S-nitrosocysteine |
| DHETs | dihydroxyeicosatrienoic acids |
| Drp1 | dynamin-related protein 1 |
| EETs | epoxyeicosatrienoic acids |
| eIF2Bε | eukaryotic initiation factor 2B epsilon subunit |
| GPX4 | glutathione peroxidase 4 |
| GSSG | oxidized glutathione |
| IL-1β | interleukin-1β |
| L-NAME | Nω-nitro-l-arginine methyl ester |
| L-NNA | NG-nitro-L-arginine |
| MAP3K | mitogen-activated protein kinase kinase kinase |
| MAP4K4 | mitogen-activated protein kinase kinase kinase kinase 4 |
| N2O3 | dinitrogen trioxide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NEMO | NF-κB essential modulator |
| NLRP3 | nod-like receptor protein 3 |
| NSF | N-ethylmaleimide-sensitive factor |
| ODQ | 1H-[1,2,4]oxadiazolo [4,3,-a]quinoxalin-1-one |
| p38MAPK | p38 mitogen-activated protein kinase |
| PAF | platelet activating factor |
| PI3K | phosphatidylinositol 3 kinase |
| PKC | protein kinase C |
| pRb | retinoblastoma protein |
| PTEN | phosphatase and TENsin homolog |
| RIP3 | receptor-interacting protein kinase 3 |
| SERCA2 | sarcoplasmic/endoplasmic reticulum calcium ATPase 2 |
| SNAP | S-nitro-N-acetylpenicillamine |
| SNO-MPG | S-nitroso-2-mercaptopropionyl glycine |
| STAT3 | signal transducer and activator of transcription 3 |
| TGFβ | transforming growth factor-β |
| TLR3 | toll-like receptor 3 |
| VE-cadherin | vascular endothelial-cadherin |
References
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Vassileff, N.; Spiers, J.G.; Bamford, S.E.; Lowe, R.G.T.; Datta, K.K.; Pigram, P.J.; Hill, A.F. Microglial activation induces nitric oxide signalling and alters protein S-nitrosylation patterns in extracellular vesicles. J. Extracell. Vesicles 2024, 13, e12455. [Google Scholar] [CrossRef] [PubMed]
- Wynia-Smith, S.L.; Smith, B.C. Nitrosothiol formation and S-nitrosation signaling through nitric oxide synthases. Nitric Oxide 2017, 63, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Reynolds, J.D.; Zhang, R.; Stamler, J.S. Role of Nitric Oxide Carried by Hemoglobin in Cardiovascular Physiology: Developments on a Three-Gas Respiratory Cycle. Circ. Res. 2020, 126, 129–158. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.W.Z.; Liang, M.C.; Soong, T.W.; Hu, Z. Regulation of cardiovascular calcium channel activity by post-translational modifications or interacting proteins. Pflug. Arch. 2020, 472, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.; Clayton, S.; Wauson, E.; Christian, D.; Tran, Q.K. Promotion of nitric oxide production: Mechanisms, strategies, and possibilities. Front. Physiol. 2025, 16, 1545044. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, O.; Moser, O.; Eckstein, M.L.; Bain, S.C.; Pitt, J.; Bracken, R. Supplementary Nitric Oxide Donors and Exercise as Potential Means to Improve Vascular Health in People with Type 1 Diabetes: Yes to NO? Nutrients 2019, 11, 1571. [Google Scholar] [CrossRef] [PubMed]
- Timby, N.; Domellöf, M.; Hernell, O.; Lönnerdal, B.; Nihlen, C.; Johanssson, I.; Weitzberg, E. Effects of age, sex and diet on salivary nitrate and nitrite in infants. Nitric Oxide 2020, 94, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.H.; Xiang, Y.; Li, H.; Zheng, L.; Xu, Y.; Xi Yu, C.; Li, J.P.; Zhang, X.Y.; Xing, W.B.; Cao, D.S.; et al. VEGF-A Stimulates STAT3 Activity via Nitrosylation of Myocardin to Regulate the Expression of Vascular Smooth Muscle Cell Differentiation Markers. Sci. Rep. 2017, 7, 2660. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.C.; Oliveira-Paula, G.H.; Ferreira, G.C.; Dal-Cin de Paula, T.; Duarte, D.A.; Costa-Neto, C.M.; Tanus-Santos, J.E. Oral nitrite treatment increases S-nitrosylation of vascular protein kinase C and attenuates the responses to angiotensin II. Redox Biol. 2021, 38, 101769. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, P.C.; Lanctot, P.M.; Auger-Messier, M.; Escher, E.; Leduc, R.; Guillemette, G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. Br. J. Pharmacol. 2006, 148, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhang, B.; Lim, L.J.Y.; Loh, W.Z.K.; Yu, D.; Tan, B.W.Q.; Liang, M.C.; Huang, Z.; Leo, C.H.; Huang, H.; et al. S-Nitrosylation-Mediated Reduction of CaV1.2 Surface Expression and Open Probability Underlies Attenuated Vasoconstriction Induced by Nitric Oxide. Hypertension 2022, 79, 2854–2866. [Google Scholar] [CrossRef] [PubMed]
- Novello, M.J.; Zhu, J.; Zhang, M.; Feng, Q.; Stathopulos, P.B. Synergistic stabilization by nitrosoglutathione-induced thiol modifications in the stromal interaction molecule-2 luminal domain suppresses basal and store operated calcium entry. Sci. Rep. 2020, 10, 10177. [Google Scholar] [CrossRef] [PubMed]
- Brunner, F.; Stessel, H.; Kukovetz, W.R. Novel guanylyl cyclase inhibitor, ODQ reveals role of nitric oxide, but not of cyclic GMP in endothelin-1 secretion. FEBS Lett. 1995, 376, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Paula, G.H.; Batista, R.I.M.; Stransky, S.; Tella, S.C.; Ferreira, G.C.; Portella, R.L.; Pinheiro, L.C.; Damacena-Angelis, C.; Riascos-Bernal, D.F.; Sidoli, S.; et al. Orally administered sodium nitrite prevents the increased α-1 adrenergic vasoconstriction induced by hypertension and promotes the S-nitrosylation of calcium/calmodulin-dependent protein kinase II. Biochem. Pharmacol. 2023, 212, 115571. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Whalen, E.J.; Nelson, C.D.; Mu, Y.; Hess, D.T.; Lefkowitz, R.J.; Stamler, J.S. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol. Cell. 2008, 31, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Xu, C.; Carraway, M.S.; Piantadosi, C.A.; Whorton, A.R.; Li, S. RhoA inactivation by S-nitrosylation regulates vascular smooth muscle contractive signaling. Nitric Oxide 2018, 74, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Milzani, A.; Giustarini, D.; Di Simplicio, P.; Colombo, R.; Rossi, R. S-NO-actin: S-nitrosylation kinetics and the effect on isolated vascular smooth muscle. J. Muscle Res. Cell Motil. 2000, 21, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.L.; Wasserloos, K.J.; Liu, X.; Stitt, M.S.; Reynolds, I.J.; Pitt, B.R.; St Croix, C.M. Nitric oxide decreases the sensitivity of pulmonary endothelial cells to LPS-induced apoptosis in a zinc-dependent fashion. Mol. Cell. Biochem. 2002, 234–235, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, Y.; Rafikov, R.; Haigh, S.; Zhi, W.B.; Kumar, S.; Doulias, P.T.; Rafikova, O.; Pillich, H.; Chakraborty, T.; et al. RhoA S-nitrosylation as a regulatory mechanism influencing endothelial barrier function in response to G+-bacterial toxins. Biochem. Pharmacol. 2017, 127, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Pan, K.T.; Chang, G.F.; Hsu, C.H.; Khoo, K.H.; Hung, C.H.; Jiang, Y.J.; Ho, F.M.; Meng, T.C. Nitrite-mediated S-nitrosylation of caspase-3 prevents hypoxia-induced endothelial barrier dysfunction. Circ. Res. 2011, 109, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.A.; Gaston, B.; Johns, R.A. Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: Redox-dependent effect of nitrogen oxides. Mol. Pharmacol. 2000, 58, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Zhang, Y.; Lü, S.; Miao, Y.; Yang, J.; Huang, S.; Ma, X.; Han, L.; Deng, J.; et al. GSNOR modulates hyperhomocysteinemia-induced T cell activation and atherosclerosis by switching Akt S-nitrosylation to phosphorylation. Redox Biol. 2018, 17, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Khatib, S.; Artoul, F.; Gershko, M.; Markman, G.; Vaya, J. The synthesis and analysis of S-nitorsylated paraoxonase 1. Biochem. Biophys. Res. Commun. 2014, 444, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Hajouj, H.; Khattib, A.; Atrahimovich, D.; Musa, S.; Khatib, S. S-Nitrosylation of Paraxonase 1 (PON1) Elevates Its Hydrolytic and Antioxidant Activities. Biomolecules 2022, 12, 414. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Tang, X.; Miao, Z.; Chen, Y.; Cao, J.; Song, T.; You, D.; Zhong, Y.; Lin, Z.; Wang, D.; et al. Hsp90 S-nitrosylation at Cys521, as a conformational switch, modulates cycling of Hsp90-AHA1-CDC37 chaperone machine to aggravate atherosclerosis. Redox Biol. 2022, 52, 102290. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Cui, Q.; Liu, J.; Xie, X.; Jiang, T.; Wang, H.; Zhao, Z.; Zhao, W.; Du, X.; Lai, B.; et al. Free fatty acids stabilize integrin β1via S-nitrosylation to promote monocyte-endothelial adhesion. J. Biol. Chem. 2023, 299, 102765. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.S.; Hausladen, A.; Slaughter, T.F.; Eu, J.P.; Stamler, J.S.; Greenberg, C.S. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry 2001, 40, 4904–4910. [Google Scholar] [CrossRef] [PubMed]
- Bekendam, R.H.; Iyu, D.; Passam, F.; Stopa, J.D.; De Ceunynck, K.; Muse, O.; Bendapudi, P.K.; Garnier, C.L.; Gopal, S.; Crescence, L.; et al. Protein disulfide isomerase regulation by nitric oxide maintains vascular quiescence and controls thrombus formation. J. Thromb. Haemost. 2018, 16, 2322–2335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, X.; Sheng, C.; Chen, X.; Chen, Y.; Zhu, D.; Gao, P. Macrophages activate iNOS signaling in adventitial fibroblasts and contribute to adventitia fibrosis. Nitric Oxide 2016, 61, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Whalen, E.J.; Stamler, J.S.; McMahon, T.J.; Chitano, P.; Piantadosi, C.A. S-nitrosoglutathione inhibits alpha1-adrenergic receptor-mediated vasoconstriction and ligand binding in pulmonary artery. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L136–L143. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; McMahon, T.J.; Huang, Y.C.; Dieterle, C.S.; Stamler, J.S.; Piantadosi, C.A. Pulmonary vasoconstriction by serotonin is inhibited by S-nitrosoglutathione. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1057–L1065. [Google Scholar] [CrossRef] [PubMed]
- Ceneviva, G.D.; Tzeng, E.; Hoyt, D.G.; Yee, E.; Gallagher, A.; Engelhardt, J.F.; Kim, Y.M.; Billiar, T.R.; Watkins, S.A.; Pitt, B.R. Nitric oxide inhibits lipopolysaccharide-induced apoptosis in pulmonary artery endothelial cells. Am. J. Physiol. 1998, 275, L717–L728. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.B.; Lee, J.E. Protein trafficking dysfunctions: Role in the pathogenesis of pulmonary arterial hypertension. Pulm. Circ. 2011, 1, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Lee, J.; Sehgal, P.B. Depletion of the ATPase NSF from Golgi membranes with hypo-S-nitrosylation of vasorelevant proteins in endothelial cells exposed to monocrotaline pyrrole. Am. J. physiol. Heart Circ. Physiol. 2008, 295, H1943–H1955. [Google Scholar] [CrossRef] [PubMed]
- McMahon, T.J.; Ahearn, G.S.; Moya, M.P.; Gow, A.J.; Huang, Y.C.; Luchsinger, B.P.; Nudelman, R.; Yan, Y.; Krichman, A.D.; Bashore, T.M.; et al. A nitric oxide processing defect of red blood cells created by hypoxia: Deficiency of S-nitrosohemoglobin in pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2005, 102, 14801–14806. [Google Scholar] [CrossRef] [PubMed]
- Grau, M.; Lauten, A.; Hoeppener, S.; Goebel, B.; Brenig, J.; Jung, C.; Bloch, W.; Suhr, F. Regulation of red blood cell deformability is independent of red blood cell-nitric oxide synthase under hypoxia. Clin. Hemorheol. Microcirc. 2016, 63, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Hausladen, A.; Qian, Z.; Zhang, R.; Premont, R.T.; Stamler, J.S. Optimized S-nitrosohemoglobin Synthesis in Red Blood Cells to Preserve Hypoxic Vasodilation Via βCys93. J. Pharmacol. Exp. Ther. 2022, 382, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fenk, S.; Melnikova, E.V.; Anashkina, A.A.; Poluektov, Y.M.; Zaripov, P.I.; Mitkevich, V.A.; Tkachev, Y.V.; Kaestner, L.; Minetti, G.; Mairbäurl, H.; et al. Hemoglobin is an oxygen-dependent glutathione buffer adapting the intracellular reduced glutathione levels to oxygen availability. Redox Biol. 2022, 58, 102535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hausladen, A.; Qian, Z.; Liao, X.; Premont, R.T.; Stamler, J.S. Hypoxic vasodilatory defect and pulmonary hypertension in mice lacking hemoglobin β-cysteine93 S-nitrosylation. JCI Insight 2022, 7, e155234. [Google Scholar] [CrossRef] [PubMed]
- Gaston, B.; May, W.J.; Sullivan, S.; Yemen, S.; Marozkina, N.V.; Palmer, L.A.; Bates, J.N.; Lewis, S.J. Essential role of hemoglobin beta-93-cysteine in posthypoxia facilitation of breathing in conscious mice. J. Appl. Physiol. (1985) 2014, 116, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Carver, D.J.; Gaston, B.; Deronde, K.; Palmer, L.A. Akt-mediated activation of HIF-1 in pulmonary vascular endothelial cells by S-nitrosoglutathione. Am. J. Respir. Cell Mol. Biol. 2007, 37, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Huang, B.; Liu, Y.C.; Shyu, K.G.; Lin, P.Y.; Wang, D.L. Acute hypoxia enhances proteins’ S-nitrosylation in endothelial cells. Biochem. Biophys. Res. Commun. 2008, 377, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Gomberg-Maitland, M.; Zhao, L.; Grimminger, F. Mechanisms and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 2025, 22, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Chu, J.; Lin, H.; Zhu, G.; Qian, J.; Yu, Y.; Yao, T.; Ping, F.; Chen, F.; Liu, X. Mechanism of homocysteine-mediated endothelial injury and its consequences for atherosclerosis. Front. Cardiovasc. Med. 2023, 9, 1109445. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, R.; Zhang, G.; Yu, Q.; Jia, M.; Zheng, C.; Wang, Y.; Xu, C.; Zhang, Y.; Liu, E. Hypercysteinemia promotes atherosclerosis by reducing protein S-nitrosylation. Biomed. Pharmacother. 2015, 70, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, S.; Huang, B.; Wang, Y.; Li, Y.; Waqar, A.B.; Liu, R.; Bai, L.; Fan, J.; Liu, E. Probucol and cilostazol exert a combinatorial anti-atherogenic effect in cholesterol-fed rabbits. Thromb. Res. 2013, 132, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Lekontseva, O.; Peters, A.; Davidge, S.T. 17beta-Estradiol induces protein S-nitrosylation in the endothelium. Cardiovasc. Res. 2010, 85, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, C.; Zhu, N.; Chen, Y.; Yu, Q.; Liu, E.; Wang, R. Sex differences in the formation of atherosclerosis lesion in apoE-/-mice and the effect of 17β-estrodiol on protein S-nitrosylation. Biomed. Pharmacother. 2018, 99, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Boichenko, V.; Noakes, V.M.; Reilly-O’Donnell, B.; Luciani, G.B.; Emanueli, C.; Martelli, F.; Gorelik, J. Circulating Non-Coding RNAs as Indicators of Fibrosis and Heart Failure Severity. Cells 2025, 14, 553. [Google Scholar] [CrossRef] [PubMed]
- Bernier, T.D.; Buckley, L.F. Cardiac Myosin Activation for the Treatment of Systolic Heart Failure. J. Cardiovasc. Pharmacol. 2021, 77, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Menazza, S.; Sheeran, F.L.; Polverino de Laureto, P.; Di Lisa, F.; Pepe, S. Oxidation of myofibrillar proteins in human heart failure. J. Am. Coll. Cardiol. 2011, 57, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.A.; Holewinski, R.J.; Kooij, V.; Agnetti, G.; Tunin, R.S.; Witayavanitkul, N.; de Tombe, P.P.; Gao, W.D.; Van Eyk, J.; Kass, D.A. Cardiac resynchronization sensitizes the sarcomere to calcium by reactivating GSK-3β. J. Clin. Investig. 2014, 124, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Venkatraman, V.; Crowgey, E.L.; Liu, T.; Fu, Z.; Holewinski, R.; Ranek, M.; Kass, D.A.; O’Rourke, B.; Van Eyk, J.E. Protein S-Nitrosylation Controls Glycogen Synthase Kinase 3β Function Independent of Its Phosphorylation State. Circ. Res. 2018, 122, 1517–1531. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wen, J.; He, A.; Qu, C.; Peng, Y.; Luo, S.; Wang, X. iNOS contributes to heart failure with preserved ejection fraction through mitochondrial dysfunction and Akt S-nitrosylation. J. Adv. Res. 2023, 43, 175–186. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Gou, Q.; Dong, M.; Chang, F.; Xiu, J. Exploring the role of iNOS in HFpEF-Related myocardial fibrosis: Involvement of PTEN-PI3K/AKT signaling pathway. Biochem. Biophys. Res. Commun. 2024, 734, 150589. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.R.; Treuer, A.V.; Castellanos, J.; Dulce, R.A.; Hare, J.M. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J. Biol. Chem. 2010, 285, 28938–28945. [Google Scholar] [CrossRef] [PubMed]
- Vielma, A.Z.; Boric, M.P.; Gonzalez, D.R. Apocynin Treatment Prevents Cardiac Connexin 43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice. Int. J. Mol. Sci. 2020, 21, 5415. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Hess, D.T.; Zhang, R.; Sugi, K.; Gao, H.; Tan, B.L.; Bowles, D.E.; Milano, C.A.; Jain, M.K.; Koch, W.J.; et al. S-Nitrosylation of β-Arrestins Biases Receptor Signaling and Confers Ligand Independence. Mol. Cell. 2018, 70, 473–487.e6. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Duan, X.; Chen, J.; Zhang, D.; Xu, J.; Zhuang, J.; Wang, S. Metabolic adaptations in pressure overload hypertrophic heart. Heart Fail. Rev. 2024, 29, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Makarewich, C.A.; Munir, A.Z.; Schiattarella, G.G.; Bezprozvannaya, S.; Raguimova, O.N.; Cho, E.E.; Vidal, A.H.; Robia, S.L.; Bassel-Duby, R.; Olson, E.N. The DWORF micropeptide enhances contractility and prevents heart failure in a mouse model of dilated cardiomyopathy. eLife 2018, 7, e38319. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.; Frank, D.; Dierck, F.; Oehl, U.; Krebs, J.; Will, R.; Lehmann, L.H.; Backs, J.; Katus, H.A.; Frey, N. Cardiac remodeling is not modulated by overexpression of muscle LIM protein (MLP). Basic Res. Cardiol. 2012, 107, 262. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Pan, L.; Zhao, S.; Dai, F.; Chao, M.; Jiang, H.; Li, X.; Lin, Z.; Huang, Z.; Meng, G.; et al. SNO-MLP (S-Nitrosylation of Muscle LIM Protein) Facilitates Myocardial Hypertrophy Through TLR3 (Toll-Like Receptor 3)-Mediated RIP3 (Receptor-Interacting Protein Kinase 3) and NLRP3 (NOD-Like Receptor Pyrin Domain Containing 3) Inflammasome Activation. Circulation 2020, 141, 984–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Song, T.Y.; Wang, Z.Y.; Gao, J.; Cao, J.W.; Hu, L.L.; Huang, Z.R.; Xie, L.P.; Ji, Y. S-nitrosylation of Hsp90 promotes cardiac hypertrophy in mice through GSK3β signaling. Acta Pharmacol. Sin. 2022, 43, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; LaPenna, K.B.; Gehred, N.D.; Yu, X.; Tang, W.H.W.; Doiron, J.E.; Xia, H.; Chen, J.; Driver, I.H.; Sachse, F.B.; et al. Dysregulation of Nitrosylation Dynamics Promotes Nitrosative Stress and Contributes to Cardiometabolic Heart Failure with Preserved Ejection Fraction. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liu, X.; Sha, X.; Zhang, Y.; Zu, Y.; Fan, Q.; Hu, L.; Sun, S.; Zhang, Z.; Chen, F.; et al. NEDD4-Mediated GSNOR Degradation Aggravates Cardiac Hypertrophy and Dysfunction. Circ. Res. 2025, 136, 422–438. [Google Scholar] [CrossRef] [PubMed]
- Sips, P.Y.; Irie, T.; Zou, L.; Shinozaki, S.; Sakai, M.; Shimizu, N.; Nguyen, R.; Stamler, J.S.; Chao, W.; Kaneki, M.; et al. Reduction of cardiomyocyte S-nitrosylation by S-nitrosoglutathione reductase protects against sepsis-induced myocardial depression. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1134–H1146. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhao, S.; Liu, J.; Liu, X.; Sha, X.; Huang, C.; Hu, L.; Sun, S.; Gao, Y.; Chen, H.; et al. Mitochondrial GSNOR Alleviates Cardiac Dysfunction via ANT1 Denitrosylation. Circ. Res. 2023, 133, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Datta Chaudhuri, R.; Datta, R.; Rana, S.; Kar, A.; Vinh Nguyen Lam, P.; Mazumder, R.; Mohanty, S.; Sarkar, S. Cardiomyocyte-specific regression of nitrosative stress-mediated S-Nitrosylation of IKKγ alleviates pathological cardiac hypertrophy. Cell. Signal. 2022, 98, 110403. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Gao, E.; Bryan, N.S.; Qu, Y.; Liu, H.R.; Hu, A.; Christopher, T.A.; Lopez, B.L.; Yodoi, J.; Koch, W.J.; et al. Cardioprotective effects of thioredoxin in myocardial ischemia and reperfusion: Role of S-nitrosation [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 11471–11476. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Bibli, S.I.; Papapetropoulos, A.; Iliodromitis, E.K.; Daiber, A.; Randriamboavonjy, V.; Steven, S.; Brouckaert, P.; Chatzianastasiou, A.; Kypreos, K.E.; Hausenloy, D.J.; et al. Nitroglycerine limits infarct size through S-nitrosation of cyclophilin D: A novel mechanism for an old drug. Cardiovasc. Res. 2019, 115, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, Y.; Zhang, X.; He, J.; Lu, D.; Fang, X.; Wang, Y.; Wang, J.; Zhang, Y.; Qiao, X.; et al. Soluble epoxide hydrolase activation by S-nitrosation contributes to cardiac ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2017, 110, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Baskaran, P.; Ma, X.; van den Akker, F.; Beuve, A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc. Natl. Acad. Sci. USA 2007, 104, 12312–12317. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Zhang, H.M.; An, G.P.; Liu, M.Y.; Han, S.F.; Jin, Q.; Song, Y.; Lin, Y.M.; Dong, B.; Wang, S.X.; et al. S-Nitrosylation of Akt by organic nitrate delays revascularization and the recovery of cardiac function in mice following myocardial infarction. J. Cell. Mol. Med. 2021, 25, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.M.; Gao, E.; Fonseca, F.V.; Hayashi, H.; Shang, X.; Hoffman, N.E.; Chuprun, J.K.; Tian, X.; Tilley, D.G.; Madesh, M.; et al. Convergence of G protein-coupled receptor and S-nitrosylation signaling determines the outcome to cardiac ischemic injury. Sci. Signal. 2013, 6, ra95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Miao, Q.; Shi, Z.; Hu, L.; Liu, S.; Gao, J.; Zhao, S.; Chen, H.; Huang, Z.; et al. Inhibition of HSP90 S-nitrosylation alleviates cardiac fibrosis via TGFβ/SMAD3 signalling pathway. Br. J. Pharmacol. 2021, 178, 4608–4625. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Chen, J.Y.; Chao, M.L.; Zhang, C.; Shi, Z.G.; Zhou, X.C.; Xie, L.P.; Sun, S.X.; Huang, Z.R.; Luo, S.S.; et al. S-nitrosylation of c-Jun N-terminal kinase mediates pressure overload-induced cardiac dysfunction and fibrosis. Acta Pharmacol. Sin. 2022, 43, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.E.; Lu, X.; Lei, M.; Xiang, F.L.; Hammoud, L.; Jiang, M.; Wang, H.; Jones, D.L.; Sims, S.M.; Feng, Q. Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation 2009, 120, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Živković, M.L.; Zaręba-Kozioł, M.; Zhukova, L.; Poznański, J.; Zhukov, I.; Wysłouch-Cieszyńska, A. Post-translational S-nitrosylation is an endogenous factor fine tuning the properties of human S100A1 protein. J. Biol. Chem. 2012, 287, 40457–40470. [Google Scholar] [CrossRef] [PubMed]
- Seitz, A.; Busch, M.; Kroemer, J.; Schneider, A.; Simon, S.; Jungmann, A.; Katus, H.A.; Most, P.; Ritterhoff, J. S100A1’s single cysteine is an indispensable redox switch for the protection against diastolic calcium waves in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2024, 327, H275–H286. [Google Scholar] [CrossRef] [PubMed]
- Power, A.S.; Asamudo, E.U.; Worthington, L.P.I.; Alim, C.C.; Parackal, R.E.; Wallace, R.S.; Ebenebe, O.V.; Heller Brown, J.; Kohr, M.J.; Bers, D.M.; et al. Nitric Oxide Modulates Ca2+ Leak and Arrhythmias via S-Nitrosylation of CaMKII. Circ. Res. 2023, 133, 1040–1055. [Google Scholar] [CrossRef] [PubMed]
- Gómez, R.; Caballero, R.; Barana, A.; Amorós, I.; Calvo, E.; López, J.A.; Klein, H.; Vaquero, M.; Osuna, L.; Atienza, F.; et al. Nitric oxide increases cardiac IK1 by nitrosylation of cysteine 76 of Kir2.1 channels. Circ. Res. 2009, 105, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Dallas, M.L.; Yang, Z.; Boyle, J.P.; Boycott, H.E.; Scragg, J.L.; Milligan, C.J.; Elies, J.; Duke, A.; Thireau, J.; Reboul, C.; et al. Carbon monoxide induces cardiac arrhythmia via induction of the late Na+ current. Am. J. Respir. Crit. Care Med. 2012, 186, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Valdivia, C.R.; Vaidyanathan, R.; Balijepalli, R.C.; Ackerman, M.J.; Makielski, J.C. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J. Mol. Cell. Cardiol. 2013, 61, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.C.; Rutledge, C.A.; Mao, M.; Bakhshi, F.R.; Xie, A.; Liu, H.; Bonini, M.G.; Patel, H.H.; Minshall, R.D.; Dudley, S.C., Jr. Caveolin-1 modulates cardiac gap junction homeostasis and arrhythmogenecity by regulating cSrc tyrosine kinase. Circ. Arrhythm. Electrophysiol. 2014, 7, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Guo, T.; Wang, H.; Li, B.; Sun, Q.; Wu, W.; Zhang, J.; Zhou, J.; Luo, J.; Zhu, M.; et al. S-nitrosylation of AMPKγ impairs coronary collateral circulation and disrupts VSMC reprogramming. EMBO Rep. 2024, 25, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Otani, H.; Shimazu, T.; Yoshioka, K.; Fujita, M.; Katano, T.; Ito, S.; Iwasaka, T. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. J. Mol. Cell. Cardiol. 2011, 50, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Xia, M.L.; Wang, J.; Zhou, X.R.; Lou, Y.Y.; Tang, L.H.; Zhang, F.J.; Yang, J.T.; Qian, L.B. Luteolin Attenuates Cardiac Ischemia/Reperfusion Injury in Diabetic Rats by Modulating Nrf2 Antioxidative Function. Oxid. Med. Cell. Longev. 2019, 2019, 2719252. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, S.; Guan, B.; Yan, X.; Huang, C.; Du, Y.; Yang, F.; Zhang, N.; Li, Y.; Lu, J.; et al. MAP4K4 exacerbates cardiac microvascular injury in diabetes by facilitating S-nitrosylation modification of Drp1. Cardiovasc. Diabetol. 2024, 23, 164. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Casin, K.M.; Sinha, P.; Sun, J.; Ma, H.; Boylston, J.; Noguchi, A.; Liu, C.; Wang, N.; Zhou, G.; et al. A knock-in mutation at cysteine 144 of TRIM72 is cardioprotective and reduces myocardial TRIM72 release. J. Mol. Cell. Cardiol. 2019, 136, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Ghanta, S.N.; Kattamuri, L.P.V.; Odueke, A.; Mehta, J.L. Molecular Insights into Ischemia-Reperfusion Injury in Coronary Artery Disease: Mechanisms and Therapeutic Implications: A Comprehensive Review. Antioxidants 2025, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, D.; Zhao, L.; Zhou, D.; Rong, J.; Zhang, L.; Xia, Z. Myocardial ischemia/reperfusion injury: Mechanisms of injury and implications for management (Review). Exp. Ther. Med. 2022, 23, 430. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Zou, L.; Peana, M.; Chasapis, C.T.; Hangan, T.; Lu, J.; Maes, M. The Role of the Thioredoxin System in Brain Diseases. Antioxidants 2022, 11, 2161. [Google Scholar] [CrossRef] [PubMed]
- Al-Kandari, N.; Fadel, F.; Al-Saleh, F.; Khashab, F.; Al-Maghrebi, M. The Thioredoxin System is Regulated by the ASK-1/JNK/p38/Survivin Pathway During Germ Cell Apoptosis. Molecules 2019, 24, 3333. [Google Scholar] [CrossRef] [PubMed]
- Obsilova, V.; Honzejkova, K.; Obsil, T. Structural Insights Support Targeting ASK1 Kinase for Therapeutic Interventions. Int. J. Mol. Sci. 2021, 22, 13395. [Google Scholar] [CrossRef] [PubMed]
- Leary, P.J.; Rajasekaran, S.; Morrison, R.R.; Tuomanen, E.I.; Chin, T.K.; Hofmann, P.A. A cardioprotective role for platelet-activating factor through NOS-dependent S-nitrosylation. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2775–H2784. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Picht, E.; Ginsburg, K.S.; Bers, D.M.; Steenbergen, C.; Murphy, E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ. Res. 2006, 98, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Yu, Y.; Yuan, H.; Li, Y.; Xue, Y. Mitochondrial dysfunction in AMI: Mechanisms and therapeutic perspectives. J. Transl. Med. 2025, 23, 418. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.M.; Burwell, L.S.; Ingraham, C.A.; Spencer, C.M.; Friedman, A.E.; Pinkert, C.A.; Brookes, P.S. In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. J. Mol. Cell. Cardiol. 2009, 46, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Kohr, M.J.; Sun, J.; Aponte, A.; Wang, G.; Gucek, M.; Murphy, E.; Steenbergen, C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ. Res. 2011, 108, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ruan, Y.; Li, C.; Zheng, H.; Tang, Y.; Chen, Y.; He, F.; Liu, Y.; Wu, G.; Li, Z.; et al. Hypoxia Microenvironment Preconditioning Attenuated Myocardial Ischemia-Reperfusion Injury via Stc1-Mediating Cardiomyocyte Self-Protection and Neutrophil Polarization. Adv. Sci. 2025, 12, e2411880. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Kim, D.D.; Fioramonti, X.; Iwahashi, T.; Durán, W.N.; Beuve, A. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ. Res. 2008, 103, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M.; Suvorava, T.; Freudenberger, T.; Dao, V.T.; Fischer, J.W.; Weber, M.; Kojda, G. Regulation of vascular guanylyl cyclase by endothelial nitric oxide-dependent posttranslational modification. Basic Res. Cardiol. 2011, 106, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Treuer, A.V.; Gonzalez, D.R. NOS1AP modulates intracellular Ca2+ in cardiac myocytes and is up-regulated in dystrophic cardiomyopathy. Int. J. Physiol. Pathophysiol. Pharmacol. 2014, 6, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Puebla, M.; Muñoz, M.F.; Lillo, M.A.; Contreras, J.E.; Figueroa, X.F. Control of astrocytic Ca2+ signaling by nitric oxide-dependent S-nitrosylation of Ca2+ homeostasis modulator 1 channels. Biol. Res. 2024, 57, 19. [Google Scholar] [CrossRef] [PubMed]
- Rohde, D.; Ritterhoff, J.; Voelkers, M.; Katus, H.A.; Parker, T.G.; Most, P. S100A1: A multifaceted therapeutic target in cardiovascular disease. J. Cardiovasc. Transl. Res. 2010, 3, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Dhamoon, A.S.; Jalife, J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm. 2005, 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, N.; Le Bouter, S.; Szuts, V.; Varro, A.; Escande, D.; Nattel, S.; Demolombe, S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J. Physiol. 2007, 582, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Valdivia, C.; Medeiros-Domingo, A.; Tester, D.J.; Vatta, M.; Farrugia, G.; Ackerman, M.J.; Makielski, J.C. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. USA 2008, 105, 9355–9360. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, A.; Battault, S.; Belaidi, E.; Tanguy, S.; Rosselin, M.; Boulghobra, D.; Meyer, G.; Gayrard, S.; Walther, G.; Geny, B.; et al. Exercise does not activate the β3 adrenergic receptor-eNOS pathway, but reduces inducible NOS expression to protect the heart of obese diabetic mice. Basic Res. Cardiol. 2016, 111, 40. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; O’Connor, M.; Qiu, H. Valosin-containing protein acts as a target and mediator of S-nitrosylation in the heart through distinct mechanisms. Redox Biol. 2024, 72, 103166. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Lu, C.T.; Su, M.G.; Huang, K.Y.; Ching, W.C.; Yang, H.H.; Liao, Y.C.; Chen, Y.J.; Lee, T.Y. dbSNO 2.0: A resource for exploring structural environment, functional and disease association and regulatory network of protein S-nitrosylation. Nucleic. Acids Res. 2015, 43, D503–D511. [Google Scholar] [CrossRef] [PubMed]
- Aboalroub, A.A.; Al Azzam, K.M. Protein S-Nitrosylation: A Chemical Modification with Ubiquitous Biological Activities. Protein J. 2024, 43, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Qiu, H. Post-Translational S-Nitrosylation of Proteins in Regulating Cardiac Oxidative Stress. Antioxidants 2020, 9, 1051. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Gupta, S.; Matilal, A.; Sarkar, S. Tissue engineering with targeted delivery of nanotized S-nitrosyl mutant of NEMO ameliorates myocardial infarction. Nanomedicine 2025, 20, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Choi, W.; Hwang, P.T.J.; Oh, Y.; Jun, T.; Ryu, D.Y.; Kim, N.K.; Jang, E.H.; Shin, Y.R.; Youn, Y.N.; et al. Engineered silk fibroin bio-hybrid artificial graft with releasing biological gas for enhanced circulatory stability and surgical performance. Int. J. Biol. Macromol. 2025, 309, 142760. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Protein | Site | Effect | Reference |
|---|---|---|---|---|
| Systemic hypertension | Myocardin | C596 | Reduced the expression of contractile markers | [10] |
| PKC | - | Inhibited vasoconstriction | [11] | |
| AT1R | C289 | Decreased AT1R’s binding affinity for AngII | [12] | |
| Cav1.2 channel | C1180, C1280 | Limited Ca2+ influx and vasoconstriction | [13] | |
| STIM2 | C15, C53, C60 | Reduced intracellular Ca2+ concentration and vasoconstriction | [14] | |
| CaMKIIγ | - | Inhibited α1-adrenergic vasoconstriction | [16] | |
| β-arrestin 2 | C410 | Accelerated β-adrenergic receptor endocytosis | [17] | |
| RhoA | C16, C20 | Inhibited vasoconstriction | [18] | |
| Pulmonary arterial hypertension | Actin | C374 | Promoted vasodilation | [19] |
| MT | - | Reduced pulmonary artery endothelial cell apoptosis | [20] | |
| RhoA | C16, C20, C159 | Reduced pulmonary vascular endothelial barrier destruction | [21] | |
| Caspase3 | C163 | Maintained adherent junctions of pulmonary vascular endothelial cells | [22] | |
| HIF-1 | - | Promoted angiogenesis and improved oxygenation | [23] | |
| Atherosclerosis | Akt | C224 | Reduced T cell inflammation and atherosclerosis | [24] |
| PON1 | C284 | Inhibited LDL oxidation and atherosclerosis process | [25,26] | |
| HSP90 | C521 | Aggravated atherosclerosis | [27] | |
| Integrin β1 | C555 | Promoted vascular inflammation and atherosclerosis progression | [28] | |
| tTG | C277 | Inhibited platelet aggregation and thrombosis | [29] | |
| PDI | C397, C400 | Reduced platelet aggregation and thrombosis | [30] |
| Disease | Protein | Site | Effect | Reference |
|---|---|---|---|---|
| Heart failure | Tropomyosin | - | Induced systolic dysfunction | [55] |
| GSK-3β | C76, C178, C199, C245, C317, C335 | Induced systolic dysfunction | [57] | |
| IRE1α | - | Induced diastolic dysfunction | [58] | |
| Akt | C224 | Decreased myocardial glucose uptake | [59] | |
| RyR2 | - | Improved diastolic SR Ca2+ leakage and decreased contractility | [62] | |
| Cx43 | - | Induced cardiomyocyte apoptosis and accelerated heart failure | [63] | |
| β-arrestin 2 | C253 | Maintained β-adrenergic response | [64] | |
| MLP | C79 | Myocardial hypertrophy occurred and developed | [68] | |
| HSP90 | C589 | Accelerated cardiac hypertrophy | [69] | |
| ANT1 | C160 | Myocardial cell mitochondrial dysfunction occurred | [73] | |
| IKKγ | C410 | Induced cardiomyocyte apoptosis and cardiac hypertrophy | [74] | |
| Myocardial infarction | Trx | C69 | Inhibited ASK-1 proapoptotic effect and reduced MIRI | [75] |
| Complex I ND3 subunit | C39 | Reduced ROS production and alleviated MIRI | [76] | |
| CypD | - | Inhibited mPTP opening and limited myocardial infarction area | [77] | |
| sEH | C141 | Aggravated MIRI | [78] | |
| sGC | C122, C243 | Decreased the response of sGC to NO | [79] | |
| Akt | C296, C344 | Impaired angiogenesis | [80] | |
| GRK2 | C340 | Reduced ischemic heart damage | [81] | |
| HSP90 | C589 | Promoted cardiac fibrosis | [82] | |
| JNK | C116, C163 | Induced myocardial fibroblast differentiation | [83] | |
| Arrhythmia | L-type Ca2+ channel RyR2 SERCA2 | - | Reduced ventricular arrhythmias after myocardial infarction | [84] |
| S100-A1 | C85 | Induced positive inotropic action and anti-arrhythmia | [85,86] | |
| CaMKIIδ | C273, C290 | Limited or worsened β-adrenergic receptor-induced arrhythmias | [87] | |
| Kir2.1 | C76 | Increased Kir2.1 channel opening and reduced arrhythmias | [88] | |
| Nav1.5 | - | Promoted late INa and arrhythmia | [89,90] | |
| Caveolin-1 | C156 | Increased risk of ventricular arrhythmia | [91] | |
| Diabetic cardiomyopathy | AMPKγ1 | C130 | Disrupted coronary collateral circulation | [92] |
| BH4 | - | Reduced iNOS-derived superoxide and improved left ventricular function | [93] | |
| Keap1 | - | Inhibited oxidative stress and reduced diabetic MIRI | [94] | |
| Drp1 | C650 | Promoted endothelial dysfunction in diabetic cardiomyopathy | [95] | |
| TRIM72 | C144 | Reduced degradation of TRIM72 and protected the heart | [96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, C.; Zhao, J.; Cheng, N.; Xu, Z.; Ma, H.; Song, Y.; Sun, X. S-Nitrosylation in Cardiovascular Disorders: The State of the Art. Biomolecules 2025, 15, 1073. https://doi.org/10.3390/biom15081073
Mao C, Zhao J, Cheng N, Xu Z, Ma H, Song Y, Sun X. S-Nitrosylation in Cardiovascular Disorders: The State of the Art. Biomolecules. 2025; 15(8):1073. https://doi.org/10.3390/biom15081073
Chicago/Turabian StyleMao, Caiyun, Jieyou Zhao, Nana Cheng, Zihang Xu, Haoming Ma, Yunjia Song, and Xutao Sun. 2025. "S-Nitrosylation in Cardiovascular Disorders: The State of the Art" Biomolecules 15, no. 8: 1073. https://doi.org/10.3390/biom15081073
APA StyleMao, C., Zhao, J., Cheng, N., Xu, Z., Ma, H., Song, Y., & Sun, X. (2025). S-Nitrosylation in Cardiovascular Disorders: The State of the Art. Biomolecules, 15(8), 1073. https://doi.org/10.3390/biom15081073