
Engineered Glibenclamide-Loaded Nanovectors Hamper Inflammasome Activation in an Ex Vivo Alzheimer’s Disease Model—A Novel Potential Therapy for Neuroinflammation: A Pilot Study

 , , , , , , , ,
, , , , , , , ,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Controls
2.2. Blood Sample Collection, DNA Extraction, and ApoE and NLRP3 Genotyping
2.3. Peripheral Blood Mononuclear Cell Processing
2.4. Preparation and Characterization of GNVs
2.5. Intracellular ASC Protein Staining and Imaging Flow Cytometry by FlowSight AMNIS
2.6. Cytokines Production and Caspase-1(p20) Release
2.7. RNA Extraction, and Analysis of mRNA and miRNA Expression by ddPCR
2.8. Statistical Analysis
3. Results
3.1. Clinical Characteristics of AD and HC Subjects Included in the Study
3.2. ApoE and NLRP3 Genotypic Characterization
3.3. Characterization of GNV
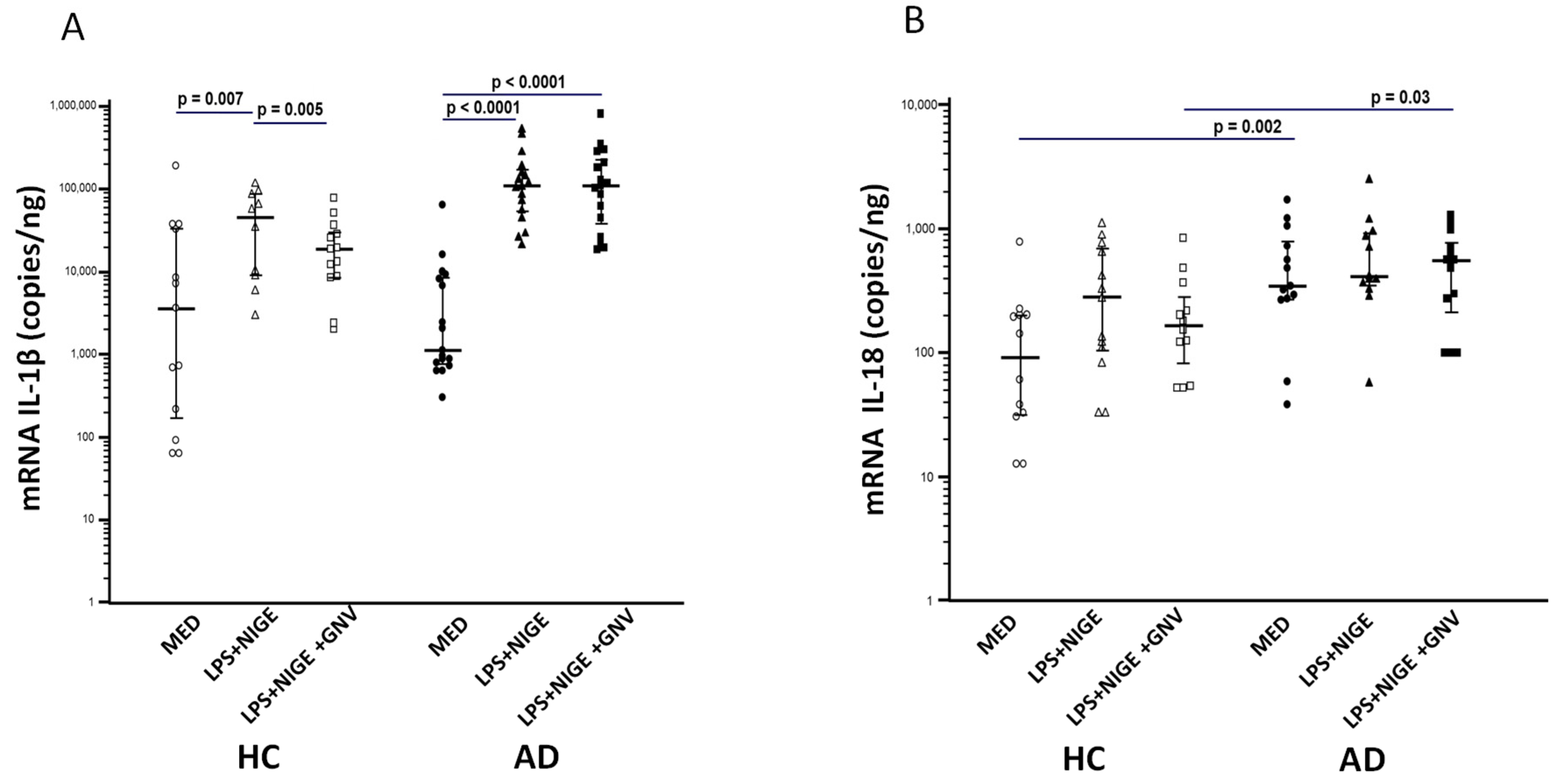
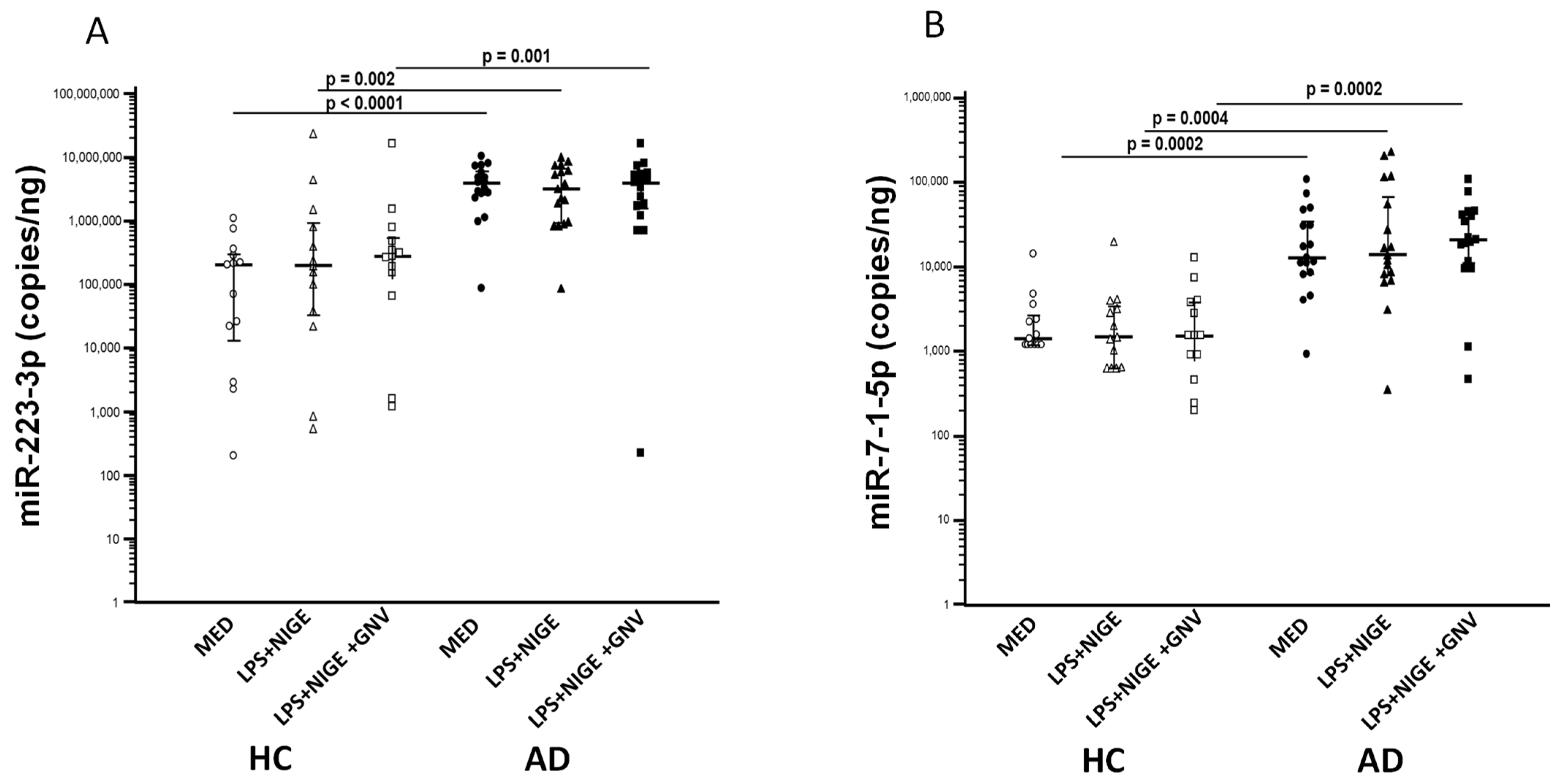
3.4. GNV-Effects on Inflammasome Gene and miRNAs Expression in LPS-Primed and Nigericin-Stimulated Monocytes
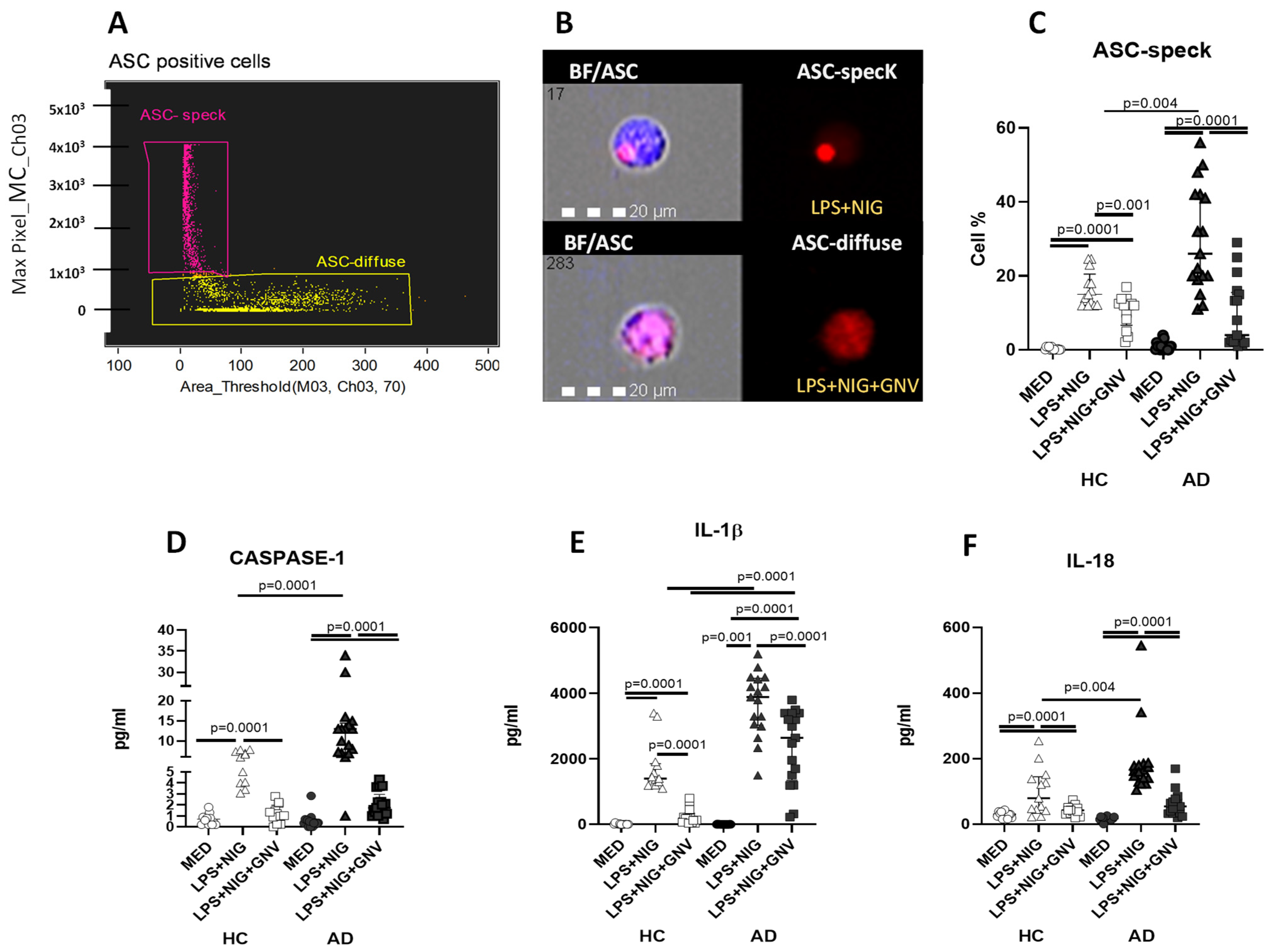
3.5. GNV-Effects on Inflammasome and Downstream Protein Activation in Monocytes
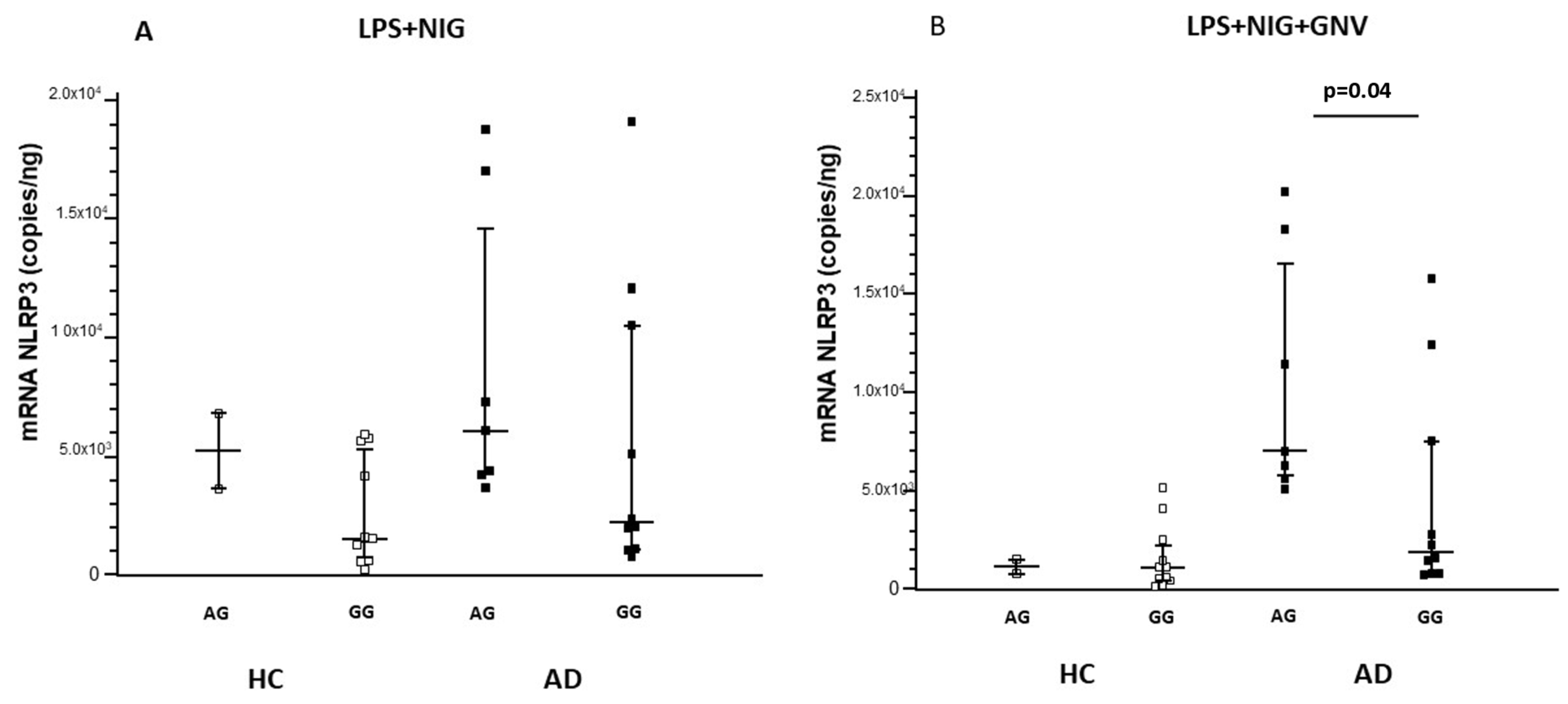
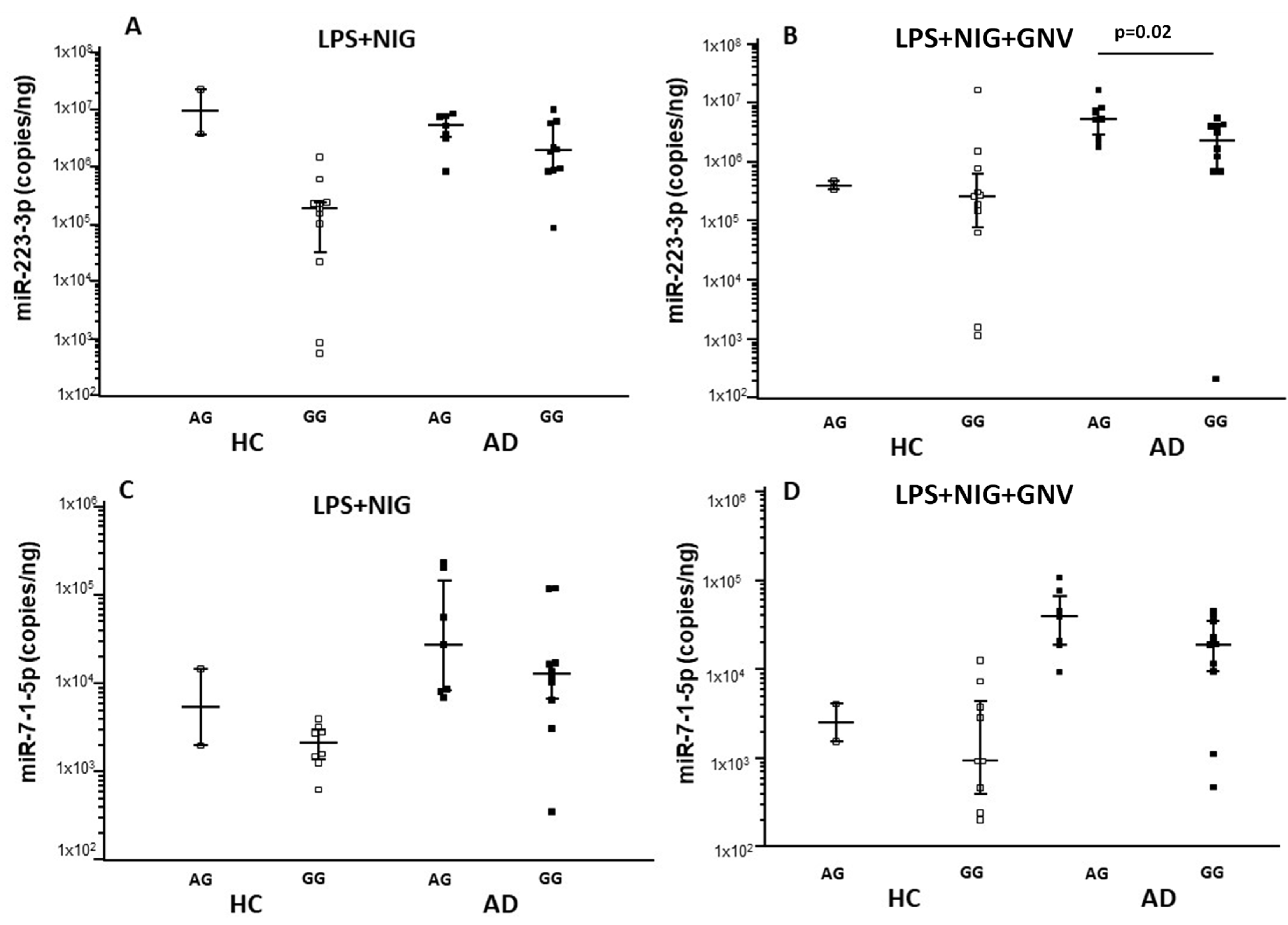
3.6. Association Among NLRP3 Polymorphisms, miRNAs and Gene Expression, and Protein Concentrations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.K.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 19, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 2011, 11, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Le Page, A.; Dupuis, G.; Frost, E.H.; Pawelec, G.; Witkowski, J.M.; Fulop, T. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp. Gerontol. 2018, 107, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Lucin, K.M.; O’Brien, C.E.; Bieri, G.; Czirr, E.; Mosher, K.I.; Abbey, R.J.; Mastroeni, D.F.; Rogers, J.; Spencer, B.; Masliah, E.; et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 2013, 79, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitutes a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Del Rio-Hortega, P. Microglia. In Cytology and Cellular Pathology of the Nervous System; Penfield, W., Ed.; P.B. Hoeber, Inc.: New York, NY, USA, 1932; Volume 2, pp. 483–534. [Google Scholar]
- Sierra, A.; de Castro, F.; Del Río-Hortega, J.; Rafael Iglesias-Rozas, J.; Garrosa, M.; Kettenmann, H. The “Big-Bang” for modern glial biology: Translation and comments on Pío del Río-Hortega 1919 series of papers on microglia. Glia 2016, 64, 1801–1840. [Google Scholar] [CrossRef] [PubMed]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, E5665–E5674. [Google Scholar] [CrossRef] [PubMed]
- Villacampa, N.; Heneka, M.T. Microglia in Alzheimer’s disease: Local heroes! J. Exp. Med. 2020, 217, e20192311. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, A.G.; Wishart, C.L.; Pamphlett, R.; Locatelli, G.; King, N.J. Microglia and monocytes in inflammatory CNS disease: Integrating phenotype and function. Acta Neuropathol. 2022, 143, 179–224. [Google Scholar] [CrossRef] [PubMed]
- Gallizioli, M.; Miro-Mur, F.; Otxoa-de-Amezaga, A.; Cugota, R.; Salas-Perdomo, A.; Justicia, C.; Brait, V.H.; Ruiz-Jaen, F.; Arbaizar-Rovirosa, M.; Pedragosa, J.; et al. Dendritic Cells and Microglia Have Non-redundant Functions in the Inflamed Brain with Protective Effects of Type 1 cDCs. Cell Rep. 2020, 33, 108291. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Samusik, N.; Wieghofer, P.; Ho, P.P.; Crotti, A.; Bjornson, Z.; Prinz, M.; Fantl, W.J.; Nolan, G.P.; Steinman, L. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 2018, 21, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Reed-Geaghan, E.G.; Croxford, A.L.; Becher, B.; Landreth, G.E. Plaque-associated myeloid cells derive from resident microglia in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20191374. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Nie, W.; Yue, Y.; Hu, J. The role of monocytes and macrophages in the progression of Alzheimer’s disease. Front. Immunol. 2025, 16, 1590909. [Google Scholar] [CrossRef] [PubMed]
- Pietronigro, E.; Zenaro, E.; Constantin, G. Imaging of Leukocyte Trafficking in Alzheimer’s Disease. Front. Immunol. 2016, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Cheng, J.; Tang, Y. Brain perivascular macrophages: Current understanding and future prospects. Brain 2024, 147, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Kim, K.W.; Xiao, Q.; Ma, X.; Czerniewski, L.R.; Liu, H.; Rawnsley, D.R.; Yan, Y.; Randolph, G.J.; Epelman, S.; et al. Peripheral monocyte-derived cells counter amyloid plaque pathogenesis in a mouse model of Alzheimer’s disease. J. Clin. Invest. 2022, 132, e152565. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Suuronen, T.; Kaarniranta, K.; Kauppinen, A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J. Cell. Mol. Med. 2008, 12, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammosomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; O’Neill, L.A. Disease-associated amyloid and misfolded protein aggregates activate the inflammasome. Trends Mol. Med. 2011, 17, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes topathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid β induces IL-1β processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef] [PubMed]
- Awad, F.; Assrawi, E.; Jumeau, C.; Georgin-Lavialle, S.; Cobret, L.; Duquesnoy, P.; Piterboth, W.; Thomas, L.; Stankovic-Stojanovic, K.; Louvrier, C.; et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS ONE 2017, 12, e0175336. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. microRNA-7 targets nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jin, H.S.; Suh, H.W.; Jo, E.K. Negative regulators and their mechanisms in NLRP3 inflammasome activation and signaling. Immunol. Cell Biol. 2017, 95, 584–592. [Google Scholar] [CrossRef]
- Gogoi, D.; Yu, H.; Casey, M.; Baird, R.; Yusuf, A.; Forde, L.; O’Brien, M.E.; West, J.R.; Flagg, T.; McElvaney, N.G.; et al. Monocyte NLRP3 inflammasome and interleukin-1β activation modulated by alpha-1 antitrypsin therapy in deficient individuals. Thorax 2024, 79, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gong, C. NLRP3 inflammasome in Alzheimer’s disease: Molecular mechanisms and emerging therapies. Front. Immunol. 2025, 16, 1583886. [Google Scholar] [CrossRef] [PubMed]
- Giofrè, S.; Renda, A.; Sesana, S.; Formicola, B.; Vergani, B.; Leone, B.E.; Denti, V.; Paglia, G.; Groppuso, S.; Romeo, V.; et al. Dual Functionalized Liposomes for Selective Delivery of Poorly Soluble Drugs to Inflamed Brain Regions. Pharmaceutics 2022, 14, 2402. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Citterio, L.A.; Agostini, S.; Marventano, I.; La Rosa, F.; Re, F.; Seneci, P.; Saresella, M.; Clerici, M. Glibenclamide-Loaded Nanoparticles Reduce NLRP3 Inflammasome Activation and Modulate miR-223-3p/miR-7-1-5p Expression in THP-1 Cells. Pharmaceuticals 2023, 16, 1590. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Zoia, C.P.; La Rosa, F.; Bazzini, C.; Sala, G.; Grassenis, E.; Marventano, I.; Hernis, A.; Piancone, F.; Conti, E.; et al. Glibenclamide-Loaded Engineered Nanovectors (GNVs) Modulate Autophagy and NLRP3-Inflammasome activation. Pharmaceuticals 2023, 16, 1725. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Service Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-IV-R. 1994. Available online: https://img3.reoveme.com/m/2ab8dabd068b16a5.pdf (accessed on 18 June 2025).
- Ligthart, G.J.; Corberand, J.X.; Fournier, C.; Galanaud, P.; Hijmans, W.; Kennes, B.; Müller-Hermelink, H.K.; Steinmann, G.G. Admission criteria for immuno immunogerontological studies in man: The SENIEUR protocol. Mech. Ageing Dev. 1984, 28, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Lemire, M.; Fortin, G.; Louis, E.; Silverberg, M.S.; Collette, C.; Baba, N.; Libioulle, C.; Belaiche, J.; Bitton, A.; et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 2009, 41, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, P.; Suprenant, A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1 release through a dye uptake-independent pathway. J. Biol. Chem. 2007, 282, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Taiarol, L.; Bigogno, C.; Sesana, S.; Kavicz, M.; Viale, F.; Pozzi, E.; Monz, L.; Carozzi, V.A.; Meregalli, C.; Valtorta, S.; et al. Givinostat-Liposomes: Anti-Tumor Effect on 2D and 3D Glioblastoma Models and Pharmacokinetics. Cancers 2022, 14, 2978. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.C. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980, 104, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Sesana, S.; Re, F.; Bulbarelli, A.; Salerno, D.; Cazzaniga, E.; Masserini, M. Membrane features and activity of GPI-anchored enzymes: Alkaline phosphatase reconstituted in model membranes. Biochemistry 2008, 47, 5433–5440. [Google Scholar] [CrossRef] [PubMed]
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Santangelo, M.A.; Caputo, D.; Mendozzi, L.; Rovaris, M.; Clerici, M. Monosodium Urate Crystals Activate the Inflammasome in Primary Progressive Multiple Sclerosis. Front. Immunol. 2018, 9, 983. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.F.; Wu, J.P. Responses of microglia and neural progenitors to mechanical brain injury. Neuroreport 1999, 10, 2287–2292. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, L.; Daley, D.; Black, K.L.; Koronyo-Hamaoui, M. Clearance of cerebral A_ in Alzheimer’s disease: Reassessing he role of microglia and monocytes. Cell. Mol. Life Sci. 2017, 74, 2167–2201. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Cooper, M.A.; Graf, T.; Hornung, V. Human monocytes engage an alternative inflammasome pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Marventano, I.; Calabrese, E.; Piancone, F.; Rainone, V.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. A complex proinflammatory role for peripheral monocytes in Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, F.; Zoia, C.P.; Bazzini, C.; Bolognini, A.; Saresella, M.; Conti, E.; Ferrarese, C.; Piancone, F.; Marventano, I.; Galimberti, D.; et al. Modulation of MAPK- and PI3/AKT-Dependent Autophagy Signaling by Stavudine (D4T) in PBMC of Alzheimer’s Disease Patients. Cells 2022, 11, 2180. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, F.; Mancuso, R.; Agostini, S.; Piancone, F.; Marventano, I.; Saresella, M.; Hernis, A.; Fenoglio, C.; Galimberti, D.; Scarpini, E.; et al. Pharmacological and Epigenetic Regulators of NLRP3 Inflammasome Activation in Alzheimer’s Disease. Pharmaceuticals 2021, 14, 1187. [Google Scholar] [CrossRef] [PubMed]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.N.; Johnson, R.E.; O’Banion, M.K. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest. 2007, 117, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Kivisakk, P.; Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed]
- Shaftel, S.S.; Carlson, T.J.; Olschowka, J.A.; Kyrkanides, S.; Matousek, S.B.; O’Banion, M.K. Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J. Neurosci. 2007, 27, 9301–9309. [Google Scholar] [CrossRef] [PubMed]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sorensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 inflammasome expression and activation in human atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AD Patients | HC | |
|---|---|---|
| N | 17 | 13 |
| Gender (M:F) | 6:11 | 4:9 |
| Age (years) | 79.3 ± 2.5 | 76.4 ± 3.0 |
| MMSE Level of education (years) | 20 ± 3 9.1 ± 4.6 | 30 10.0 ± 5.6 |
| Amyloid-β in CSF (pg/mL) | 390 ± 132 | - |
| Total-τ in CSF (pg/mL) | 607 ± 270 | - |
| Phospho-τ in CSF (pg/mL) | 104 ± 40 | - |
| ApoE4-carriers (%) | 17.6 | 11.5 |
| AD Patients | HC | ||||
|---|---|---|---|---|---|
| ApoE | Genotypes | N = 17 | % | N = 13 | % |
| ε2/ε3 | 0 | 0.0 | 2 | 15.4 | |
| ε2/ε4 | 1 | 5.9 | 0 | 0.0 | |
| ε3/ε3 | 11 | 64.7 | 8 | 61.5 | |
| ε3/ε4 | 5 | 29.4 | 3 | 23.1 | |
| pc = 0.6 2 df | |||||
| ε4+ | 6 | 35.3 | 3 | 23.1 | |
| ε4− | 11 | 64.7 | 10 | 76.9 | |
| pf = 0.5 1 | |||||
| NLRP3 | rs35829419 | ||||
| C C | 17 | 100 | 12 | 92.3 | |
| C A | 0 | 0.0 | 1 | 7.7 | |
| A A | 0 | 0.0 | 0 | 0.0 | |
| pf = 0.4 | |||||
| rs10733113 | |||||
| G G | 10 | 58.8 | 11 | 84.6 | |
| G A | 7 | 41.2 | 2 | 15.4 | |
| A A | 0 | 0.0 | 0 | 0.0 | |
| pf = 0.2 | |||||
| rs4925663 | |||||
| C C | 9 | 52.9 | 4 | 30.8 | |
| C T | 6 | 35.3 | 6 | 46.1 | |
| T T | 2 | 11.8 | 3 | 23.1 | |
| pc = 0.4 2 df | |||||
| Diameter (nm) | PDI | ζ-Potential (mV) | EE (%) | |
|---|---|---|---|---|
| GNV | 145.1 ± 2.6 | 0.19 | −29.5 ± 2.3 | |
| 152.5 ± 2.5 | 0.20 | −31.2 ± 3.4 | 75 ± 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Rosa, F.; Agostini, S.; Bolognesi, E.; Marventano, I.; Mancuso, R.; Guerini, F.R.; Hernis, A.; Citterio, L.A.; Piancone, F.; Trimarchi, P.D.; et al. Engineered Glibenclamide-Loaded Nanovectors Hamper Inflammasome Activation in an Ex Vivo Alzheimer’s Disease Model—A Novel Potential Therapy for Neuroinflammation: A Pilot Study. Biomolecules 2025, 15, 1074. https://doi.org/10.3390/biom15081074
La Rosa F, Agostini S, Bolognesi E, Marventano I, Mancuso R, Guerini FR, Hernis A, Citterio LA, Piancone F, Trimarchi PD, et al. Engineered Glibenclamide-Loaded Nanovectors Hamper Inflammasome Activation in an Ex Vivo Alzheimer’s Disease Model—A Novel Potential Therapy for Neuroinflammation: A Pilot Study. Biomolecules. 2025; 15(8):1074. https://doi.org/10.3390/biom15081074
Chicago/Turabian StyleLa Rosa, Francesca, Simone Agostini, Elisabetta Bolognesi, Ivana Marventano, Roberta Mancuso, Franca Rosa Guerini, Ambra Hernis, Lorenzo Agostino Citterio, Federica Piancone, Pietro Davide Trimarchi, and et al. 2025. "Engineered Glibenclamide-Loaded Nanovectors Hamper Inflammasome Activation in an Ex Vivo Alzheimer’s Disease Model—A Novel Potential Therapy for Neuroinflammation: A Pilot Study" Biomolecules 15, no. 8: 1074. https://doi.org/10.3390/biom15081074
APA StyleLa Rosa, F., Agostini, S., Bolognesi, E., Marventano, I., Mancuso, R., Guerini, F. R., Hernis, A., Citterio, L. A., Piancone, F., Trimarchi, P. D., Navarro, J., Rossetto, F., Amenta, A., Seneci, P., Sesana, S., Re, F., Clerici, M., & Saresella, M. (2025). Engineered Glibenclamide-Loaded Nanovectors Hamper Inflammasome Activation in an Ex Vivo Alzheimer’s Disease Model—A Novel Potential Therapy for Neuroinflammation: A Pilot Study. Biomolecules, 15(8), 1074. https://doi.org/10.3390/biom15081074