
DNA Damage Response Regulation Alleviates Neuroinflammation in a Mouse Model of α-Synucleinopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Mice
2.3. Mouse Model of α-Synucleinopathy
2.4. Behavioral and Motor Function Assessments
2.5. Cylinder Test
2.6. Raised-Beam Task
2.7. CatWalk Gait Analysis
2.8. Relative Quantitative Real-Time Reverse-Transcriptase PCR (RT-qPCR)
2.9. Immunocytochemistry
2.10. Immunofluorescence Staining in Mouse Brain
2.11. Western Blot Analyses
2.12. Statistical Analysis
3. Results
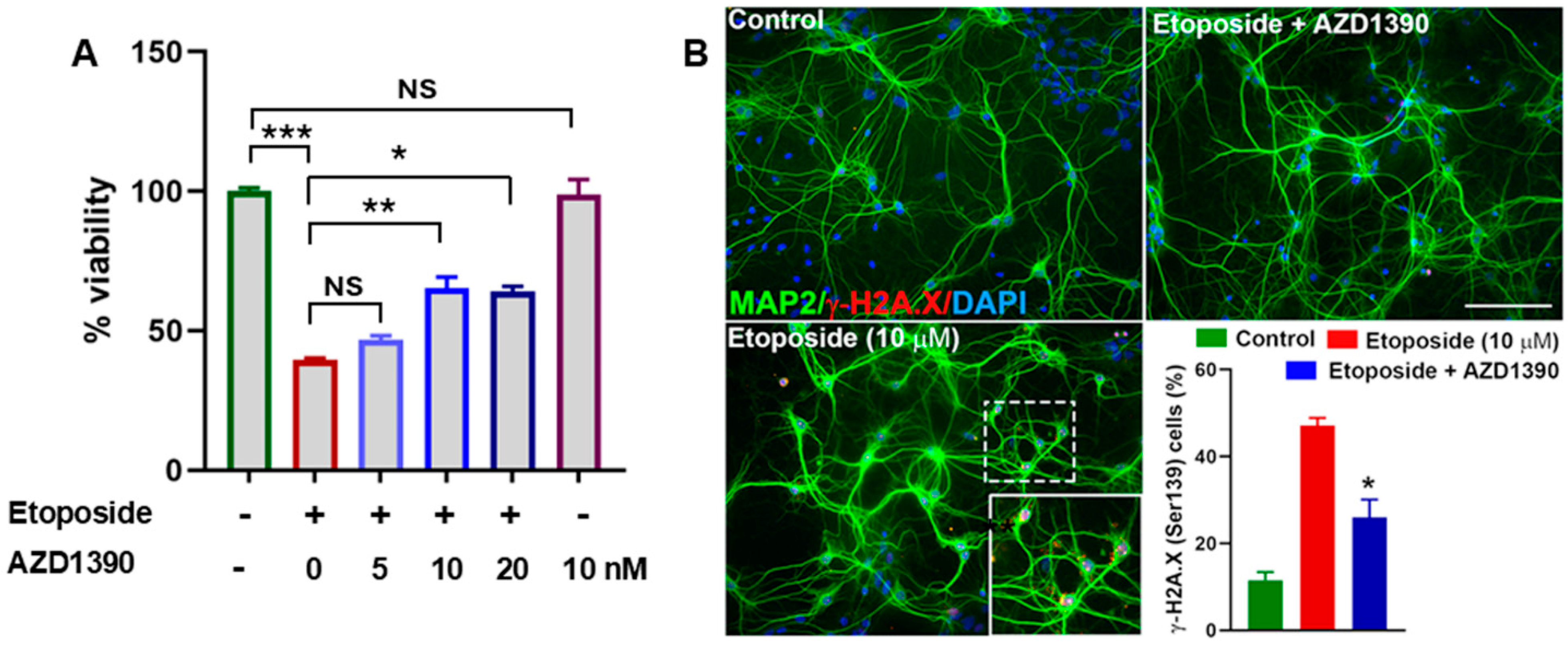
3.1. AZD1390 Mitigates DNA Damage in Etoposide-Exposed Neuronal Cultures and in the Brains of α-Synucleinopathy Mice
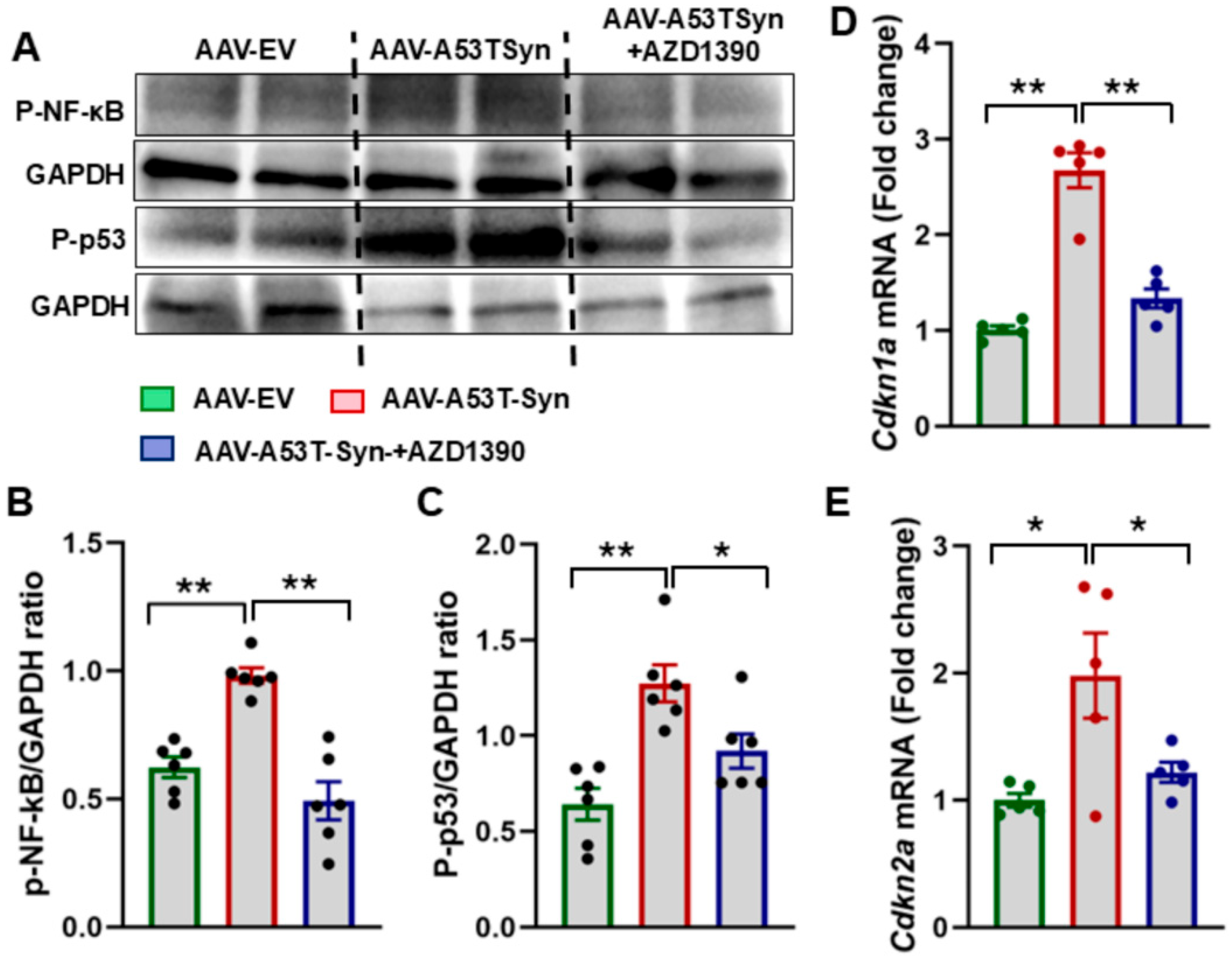
3.2. ATM Inhibition Suppresses Senescence in α-Synuclein-Treated Mice
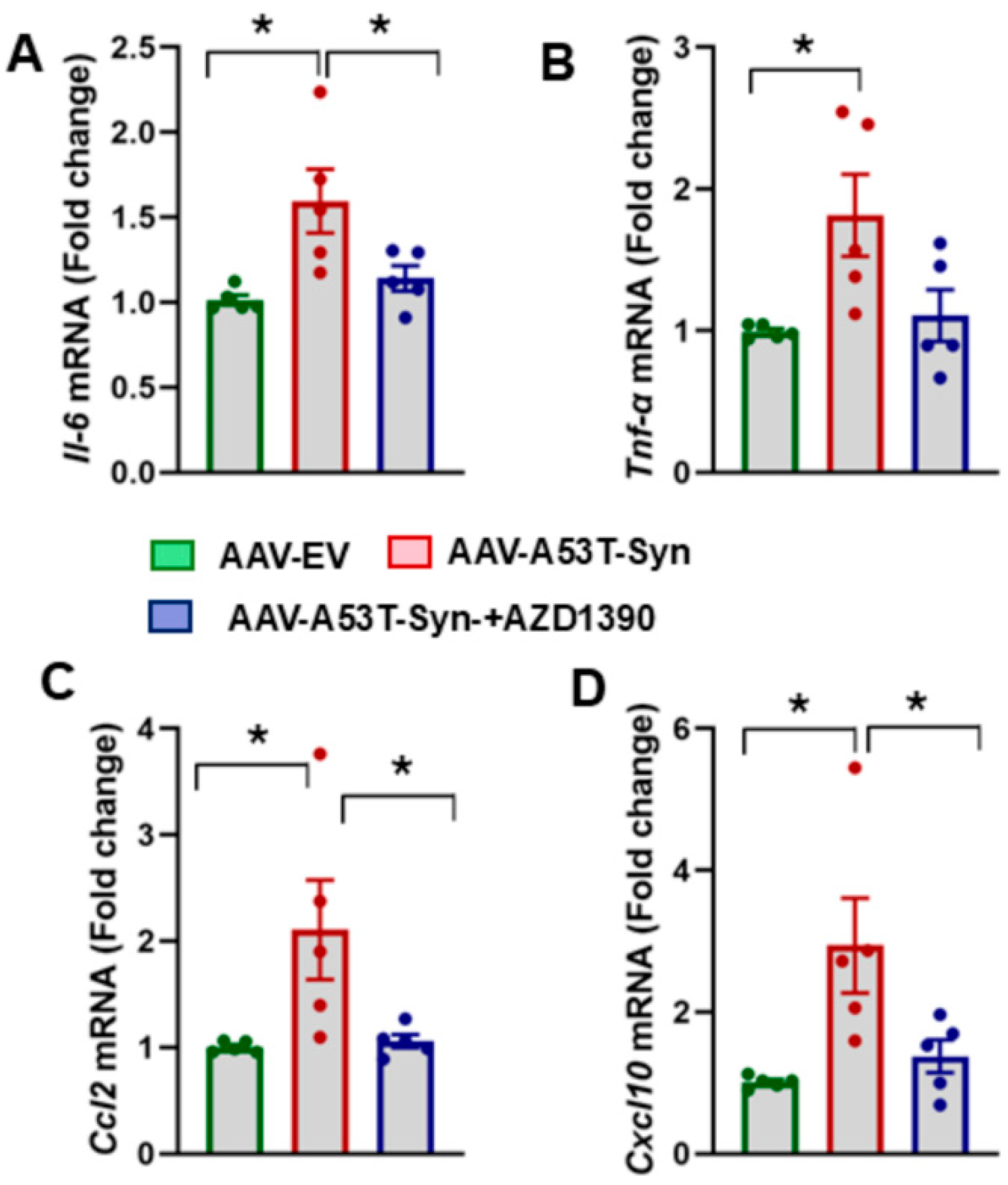
3.3. AZD1390 Reduces Neuroinflammatory Responses in the Brains of α-Synuclein-Treated Mice
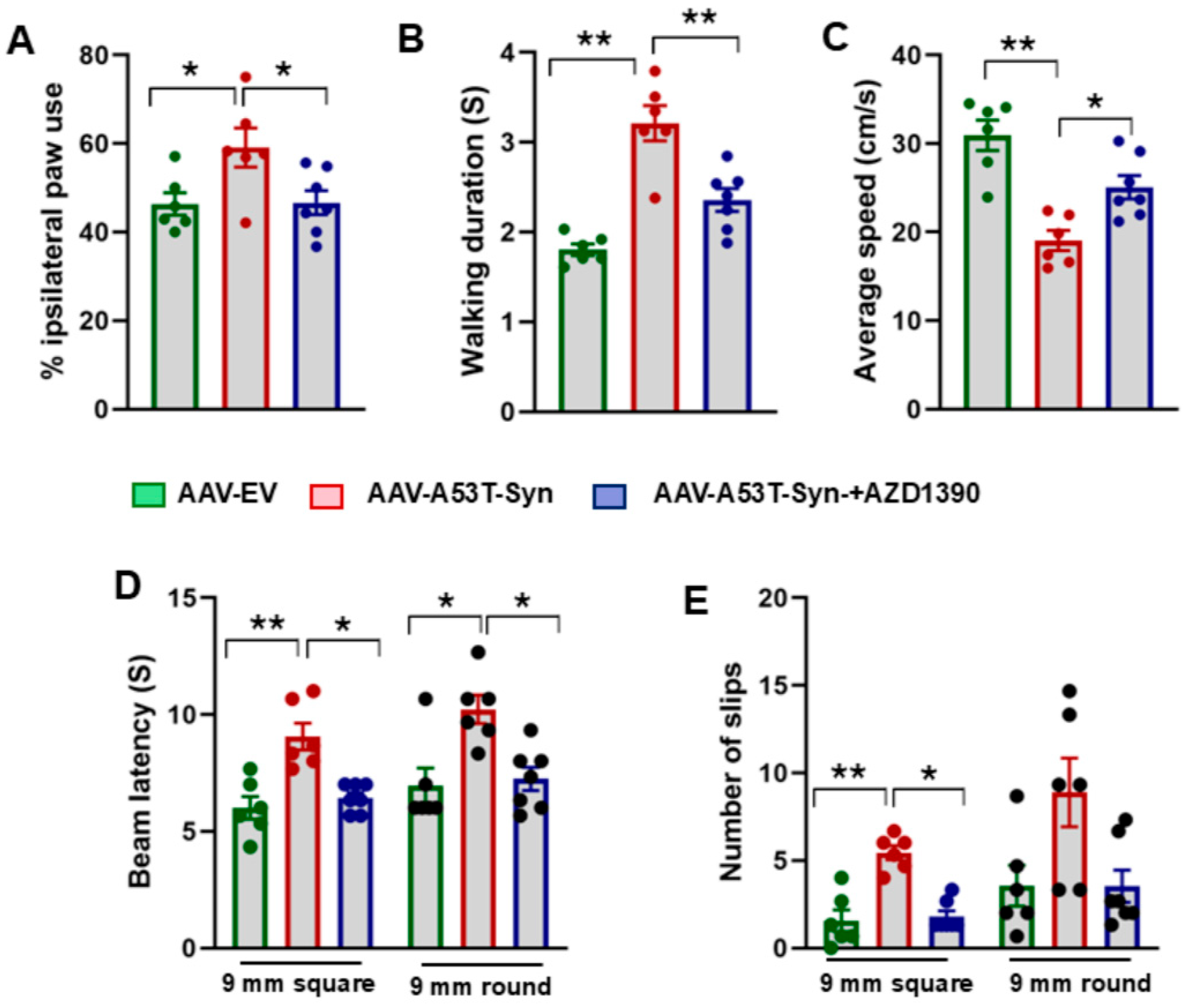
3.4. AZD1390 Improves Behavioral Function in a Mouse Model of α-Synucleinopathy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Su, D.; Cui, Y.; He, C.; Yin, P.; Bai, R.; Zhu, J.; Lam, J.S.T.; Zhang, J.; Yan, R.; Zheng, X.; et al. Projections for prevalence of Parkinson’s disease and its driving factors in 195 countries and territories to 2050: Modelling study of Global Burden of Disease Study 2021. BMJ 2025, 388, e080952. [Google Scholar] [CrossRef] [PubMed]
- Mullaart, E.; Boerrigter, M.E.; Ravid, R.; Swaab, D.F.; Vijg, J. Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol. Aging 1990, 11, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Thadathil, N.; Hori, R.; Xiao, J.; Khan, M.M. DNA double-strand breaks: A potential therapeutic target for neurodegenerative diseases. Chromosome Res. 2019, 27, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Pessina, F.; Gioia, U.; Brandi, O.; Farina, S.; Ceccon, M.; Francia, S.; d’Adda di Fagagna, F. DNA Damage Triggers a New Phase in Neurodegeneration. Trends Genet. 2021, 37, 337–354. [Google Scholar] [CrossRef]
- Delint-Ramirez, I.; Madabhushi, R. DNA damage and its links to neuronal aging and degeneration. Neuron 2025, 113, 7–28. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Sanders, L.H. DNA damage and repair in Parkinson’s disease: Recent advances and new opportunities. J. Neurosci. Res. 2021, 99, 180–189. [Google Scholar] [CrossRef]
- El-Saadi, M.W.; Tian, X.; Grames, M.; Ren, M.; Keys, K.; Li, H.; Knott, E.; Yin, H.; Huang, S.; Lu, X.H. Tracing brain genotoxic stress in Parkinson’s disease with a novel single-cell genetic sensor. Sci. Adv. 2022, 8, eabd1700. [Google Scholar] [CrossRef]
- Thadathil, N.; Delotterie, D.F.; Xiao, J.; Hori, R.; McDonald, M.P.; Khan, M.M. DNA Double-Strand Break Accumulation in Alzheimer’s Disease: Evidence from Experimental Models and Postmortem Human Brains. Mol. Neurobiol. 2021, 58, 118–131. [Google Scholar] [CrossRef]
- Khanna, K.K.; Lavin, M.F.; Jackson, S.P.; Mulhern, T.D. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001, 8, 1052–1065. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776. [Google Scholar] [CrossRef]
- Ahmad, A.; Braden, A.; Khan, S.; Xiao, J.; Khan, M.M. Crosstalk between the DNA damage response and cellular senescence drives aging and age-related diseases. Semin. Immunopathol. 2024, 46, 10. [Google Scholar] [CrossRef]
- Milanese, C.; Cerri, S.; Ulusoy, A.; Gornati, S.V.; Plat, A.; Gabriels, S.; Blandini, F.; Di Monte, D.A.; Hoeijmakers, J.H.; Mastroberardino, P.G. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018, 9, 818. [Google Scholar] [CrossRef]
- Yoon, Y.S.; You, J.S.; Kim, T.K.; Ahn, W.J.; Kim, M.J.; Son, K.H.; Ricarte, D.; Ortiz, D.; Lee, S.J.; Lee, H.J. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp. Mol. Med. 2022, 54, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.H.; Mattis, V.B.; Wang, N.; Al-Ramahi, I.; van den Berg, N.; Fratantoni, S.A.; Waldvogel, H.; Greiner, E.; Osmand, A.; Elzein, K.; et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 2014, 6, 268ra178. [Google Scholar] [CrossRef]
- Camins, A.; Pizarro, J.G.; Alvira, D.; Gutierrez-Cuesta, J.; de la Torre, A.V.; Folch, J.; Sureda, F.X.; Verdaguer, E.; Junyent, F.; Jordan, J.; et al. Activation of ataxia telangiectasia muted under experimental models and human Parkinson’s disease. Cell Mol. Life Sci. 2010, 67, 3865–3882. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Parker, K.; Zhan, Y.; Miller, M.; Zhu, M.Y. Upregulated DNA Damage-Linked Biomarkers in Parkinson’s Disease Model Mice. ASN Neuro 2023, 15, 17590914231152099. [Google Scholar] [CrossRef]
- Korwek, Z.; Sewastianik, T.; Bielak-Zmijewska, A.; Mosieniak, G.; Alster, O.; Moreno-Villanueva, M.; Burkle, A.; Sikora, E. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair 2012, 11, 864–873. [Google Scholar] [CrossRef]
- Alvira, D.; Yeste-Velasco, M.; Folch, J.; Casadesus, G.; Smith, M.A.; Pallas, M.; Camins, A. Neuroprotective effects of caffeine against complex I inhibition-induced apoptosis are mediated by inhibition of the Atm/p53/E2F-1 path in cerebellar granule neurons. J. Neurosci. Res. 2007, 85, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Wersto, R.P.; Cardozo-Pelaez, F.; Smilenov, L.; Chan, S.L.; Chrest, F.J.; Emokpae, R., Jr.; Gorospe, M.; Mattson, M.P. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 2004, 41, 549–561. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, Z.; Zhang, W.; Gu, M.; Zhou, Z.D.; Li, B.; Li, J.; Tan, E.K.; Zeng, L. LRRK2 interacts with ATM and regulates Mdm2-p53 cell proliferation axis in response to genotoxic stress. Hum. Mol. Genet. 2017, 26, 4494–4505. [Google Scholar] [CrossRef]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Tuxworth, R.I. The brain-penetrant ATM inhibitor, AZD1390, promotes axon regeneration and functional recovery in preclinical models of spinal cord injury. Clin. Transl. Med. 2022, 12, e962. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lin, C.C.; Dunn, D.E.; Chen, Y.; Chen, S.Y.; Marchuk, D.A.; Floyd, S.R.; Chi, J.T. Targeting the ferroptosis pathway: A novel compound, AZD1390, protects the brain after ischemic stroke. bioRxiv 2025. [Google Scholar] [CrossRef]
- Lan, Z.; Qu, L.J.; Liang, Y.; Chen, L.Q.; Xu, S.; Ge, J.W.; Xue, Z.W.; Bao, X.Y.; Xia, S.N.; Yang, H.Y.; et al. AZD1390, an ataxia telangiectasia mutated inhibitor, attenuates microglia-mediated neuroinflammation and ischemic brain injury. CNS Neurosci. Ther. 2024, 30, e14696. [Google Scholar] [CrossRef]
- Jucaite, A.; Stenkrona, P.; Cselenyi, Z.; De Vita, S.; Buil-Bruna, N.; Varnas, K.; Savage, A.; Varrone, A.; Johnstrom, P.; Schou, M.; et al. Brain exposure of the ATM inhibitor AZD1390 in humans—A positron emission tomography study. Neuro Oncol. 2021, 23, 687–696. [Google Scholar] [CrossRef]
- Kempuraj, D.; Khan, M.M.; Thangavel, R.; Xiong, Z.; Yang, E.; Zaheer, A. Glia maturation factor induces interleukin-33 release from astrocytes: Implications for neurodegenerative diseases. J. Neuroimmune Pharmacol. 2013, 8, 643–650. [Google Scholar] [CrossRef]
- Ip, C.W.; Klaus, L.C.; Karikari, A.A.; Visanji, N.P.; Brotchie, J.M.; Lang, A.E.; Volkmann, J.; Koprich, J.B. AAV1/2-induced overexpression of A53T-α-synuclein in the substantia nigra results in degeneration of the nigrostriatal system with Lewy-like pathology and motor impairment: A new mouse model for Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 11. [Google Scholar] [CrossRef]
- Grotemeyer, A.; Fischer, J.F.; Koprich, J.B.; Brotchie, J.M.; Blum, R.; Volkmann, J.; Ip, C.W. Inflammasome inhibition protects dopaminergic neurons from α-synuclein pathology in a model of progressive Parkinson’s disease. J. Neuroinflamm. 2023, 20, 79. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: Waltham, MA, USA, 1982. [Google Scholar]
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 Ganglioside Modifies α-Synuclein Toxicity and is Neuroprotective in a Rat α-Synuclein Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 8362. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Xiao, J.; Patel, D.; LeDoux, M.S. DNA damage and neurodegenerative phenotypes in aged Ciz1 null mice. Neurobiol. Aging 2018, 62, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Zaheer, S.; Thangavel, R.; Patel, M.; Kempuraj, D.; Zaheer, A. Absence of glia maturation factor protects dopaminergic neurons and improves motor behavior in mouse model of parkinsonism. Neurochem. Res. 2015, 40, 980–990. [Google Scholar] [CrossRef]
- Ismael, S.; Nasoohi, S.; Li, L.; Aslam, K.S.; Khan, M.M.; El-Remessy, A.B.; McDonald, M.P.; Liao, F.F.; Ishrat, T. Thioredoxin interacting protein regulates age-associated neuroinflammation. Neurobiol. Dis. 2021, 156, 105399. [Google Scholar] [CrossRef]
- Honig, M.G.; Del Mar, N.A.; Moore, B.M.; Reiner, A. Raloxifene Mitigates Emotional Deficits after Mild Traumatic Brain Injury in Mice. Neurotrauma Rep. 2022, 3, 534–544. [Google Scholar] [CrossRef]
- Javed, H.; Thangavel, R.; Selvakumar, G.P.; Dubova, I.; Schwartz, N.; Ahmed, M.E.; Zaheer, S.; Kempuraj, D.; Iyer, S.; Zaheer, A.; et al. NLRP3 inflammasome and glia maturation factor coordinately regulate neuroinflammation and neuronal loss in MPTP mouse model of Parkinson’s disease. Int. Immunopharmacol. 2020, 83, 106441. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef]
- Rose, E.P.; Osterberg, V.R.; Gorbunova, V.; Unni, V.K. Alpha-synuclein modulates the repair of genomic DNA double-strand breaks in a DNA-PK(cs)-regulated manner. Neurobiol. Dis. 2024, 201, 106675. [Google Scholar] [CrossRef]
- Surguchov, A. α-Synuclein and Mechanisms of Epigenetic Regulation. Brain Sci. 2023, 13, 150. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Liao, C.-Y.; Li, H.; Ke, Y.-C.; Lin, C.-H.; Teng, S.-C. ATM-mediated co-chaperone DNAJB11 phosphorylation facilitates α-synuclein folding upon DNA double-stranded breaks. NAR Mol. Med. 2024, 1, ugae007. [Google Scholar] [CrossRef]
- Shen, Q.Q.; Jv, X.H.; Ma, X.Z.; Li, C.; Liu, L.; Jia, W.T.; Qu, L.; Chen, L.L.; Xie, J.X. Cell senescence induced by toxic interaction between α-synuclein and iron precedes nigral dopaminergic neuron loss in a mouse model of Parkinson’s disease. Acta Pharmacol. Sin. 2024, 45, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Thompson, A.M.; Alhajlah, S.; Tuxworth, R.I.; Ahmed, Z. Inhibition of Chk2 promotes neuroprotection, axon regeneration, and functional recovery after CNS injury. Sci. Adv. 2022, 8, eabq2611. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Lyu, Z.; Ge, L.; Li, Y.; Liu, Y.; Yuan, Y.; Zhao, R.; Huang, L.; Zhao, J.; Huang, H.; et al. Ataxia Telangiectasia Mutated Protects Against Lipopolysaccaride-Induced Blood-Brain Barrier Disruption by Regulating Atk/Drp1-Mediated Mitochondrial Homeostasis. Shock 2023, 60, 100–109. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Lu, A.; Han, Y.; Colangelo, D.; Bukata, C.; Scibetta, A.; Yousefzadeh, M.J.; Li, X.; Gurkar, A.U.; et al. ATM is a key driver of NF-kappaB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging 2020, 12, 4688–4710. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Kuk, M.U.; Kim, J.W.; Lee, Y.S.; Cho, K.A.; Park, J.T.; Park, S.C. Alleviation of Senescence via ATM Inhibition in Accelerated Aging Models. Mol. Cells 2019, 42, 210–217. [Google Scholar]
- Ashraf, S.; Deshpande, N.; Cheung, Q.; Asabere, J.B.; Wong, R.J.; Gauthier, A.G.; Parekh, M.; Adhikari, Y.; Melangath, G.; Jurkunas, U.V. Modulation of ATM enhances DNA repair in G2/M phase of cell cycle and averts senescence in Fuchs endothelial corneal dystrophy. Commun. Biol. 2024, 7, 1482. [Google Scholar] [CrossRef]
- Weiss, F.; Labrador-Garrido, A.; Dzamko, N.; Halliday, G. Immune responses in the Parkinson’s disease brain. Neurobiol. Dis. 2022, 168, 105700. [Google Scholar] [CrossRef]
- Miyamoto, S. Nuclear initiated NF-kappaB signaling: NEMO and ATM take center stage. Cell Res. 2011, 21, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Hall, S.; Surova, Y.; Nielsen, H.M.; Janelidze, S.; Brundin, L.; Hansson, O. Cerebrospinal fluid inflammatory markers in Parkinson’s disease—Associations with depression, fatigue, and cognitive impairment. Brain Behav. Immun. 2013, 33, 183–189. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Singh, H.; Xiao, J.; Khan, M.M. DNA Damage Response Regulation Alleviates Neuroinflammation in a Mouse Model of α-Synucleinopathy. Biomolecules 2025, 15, 907. https://doi.org/10.3390/biom15070907
Khan S, Singh H, Xiao J, Khan MM. DNA Damage Response Regulation Alleviates Neuroinflammation in a Mouse Model of α-Synucleinopathy. Biomolecules. 2025; 15(7):907. https://doi.org/10.3390/biom15070907
Chicago/Turabian StyleKhan, Sazzad, Himanshi Singh, Jianfeng Xiao, and Mohammad Moshahid Khan. 2025. "DNA Damage Response Regulation Alleviates Neuroinflammation in a Mouse Model of α-Synucleinopathy" Biomolecules 15, no. 7: 907. https://doi.org/10.3390/biom15070907
APA StyleKhan, S., Singh, H., Xiao, J., & Khan, M. M. (2025). DNA Damage Response Regulation Alleviates Neuroinflammation in a Mouse Model of α-Synucleinopathy. Biomolecules, 15(7), 907. https://doi.org/10.3390/biom15070907