
Enhancing Membrane Repair Using Recombinant MG53/TRIM72 (rhMG53) Reduces Neurotoxicity in Alzheimer’s Disease Models


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Recombinant Proteins
2.2. Patient Cerebrospinal Fluid Samples
2.3. Mice
2.4. Infrared Laser Damage Assay
2.5. Neurotoxicity Assays
2.6. Statistical Analyses
3. Results
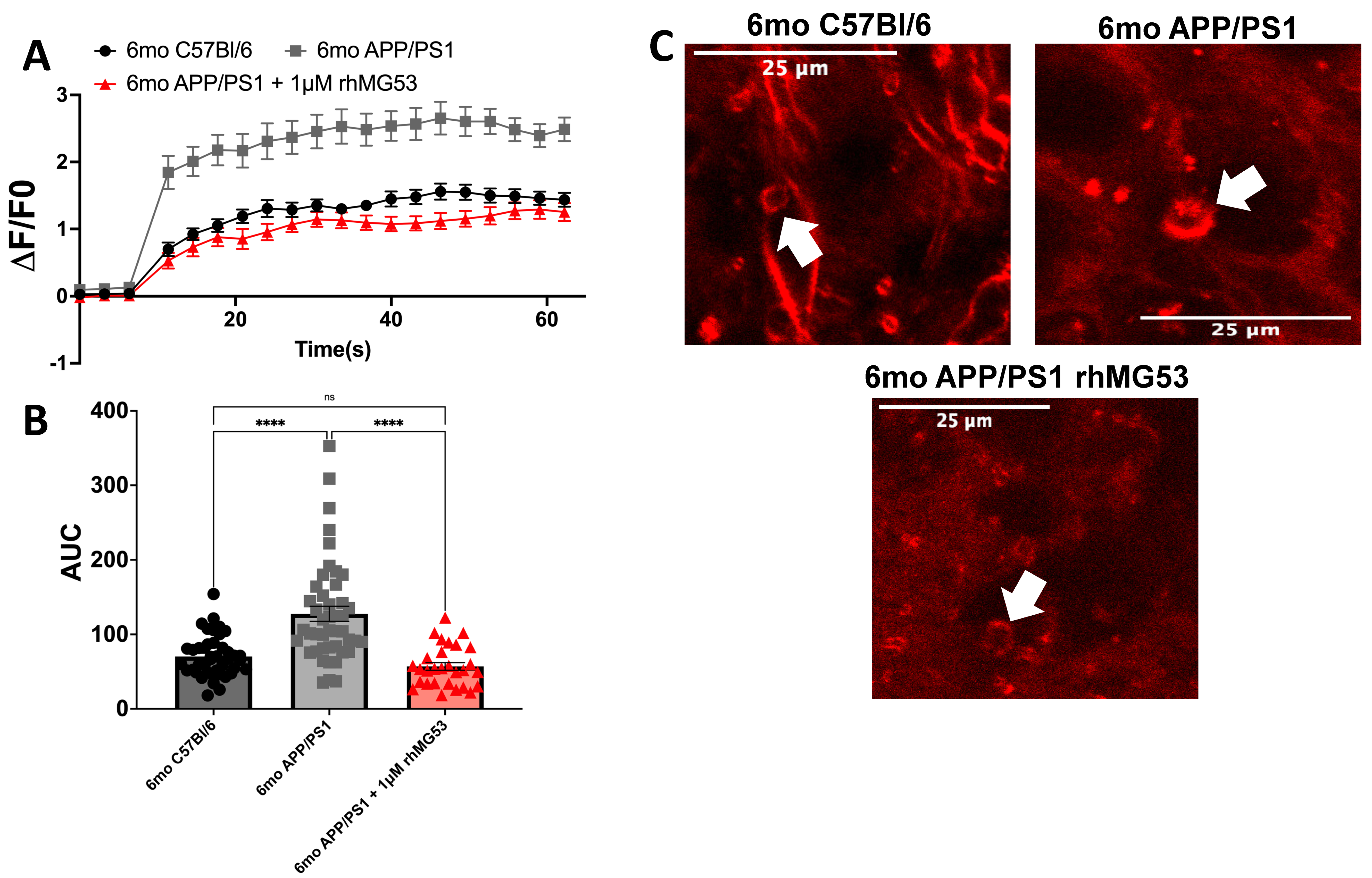
3.1. APP/PS1 Brain Slices Treated with rhMG53 Rescues Repair Capacity
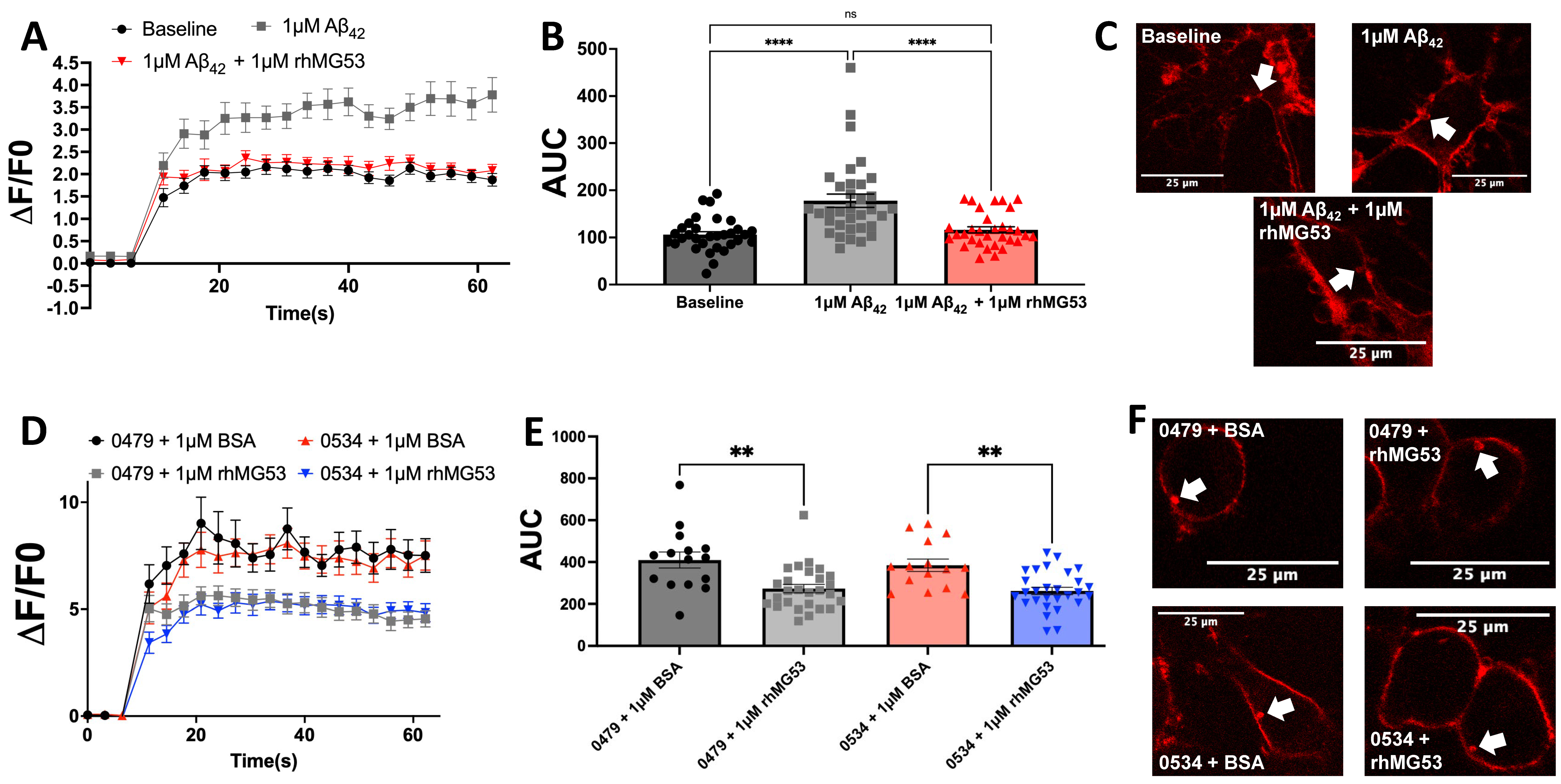
3.2. Exogenous rhMG53 Enhances Membrane Repair in Aβ42 and Patient CSF-Treated Neurons
3.3. rhMG53 Reduces Neurotoxicity in Aβ42-Treated Neuroblastomas
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta |
| AD | Alzheimer’s Disease |
| AUC | Area Under the Curve |
| BSA | Bovine serum albumin |
| CSF | Cerebrospinal fluid |
| N2A | Mouse neuroblastoma |
| rhMG53 | Recombinant human MG53 |
References
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Better, M.A. 2024 Alzheimer’s Disease Facts and Figures. Alzheimers Dement 2023, 19, 1598–1695. [Google Scholar]
- Harvey, R.J.; Skelton-Robinson, M.; Rossor, M.N. The prevalence and causes of dementia in people under the age of 65 years. Neurol. Neurosurg. Psychiatry 2003, 74, 1206–1209. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tomberlin, C.; Roberts, C.M.; Akram, A.; Stein, G.H.; Silverman, M.A.; Link, C.D. In vivo induction of membrane damage by beta-amyloid peptide oligomers. Acta Neuropathol. Commun. 2018, 6, 131. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Matharu, B.; Gibson, G.; Parsons, R.; Huckerby, T.N.; Moore, S.A.; Cooper, L.J.; Millichamp, R.; Allsop, D.; Austen, B. Galantamine inhibits β-amyloid aggregation and cytotoxicity. J. Neurol. Sci. 2009, 280, 49–58. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Lin, H.; Lal, R. Fresh and nonfibrillar amyloid β protein (1–40) induces rapid cellular degeneration in aged human fibroblasts: Evidence for AβP-channel-mediated cellular toxicity. FASEB J. 2000, 14, 1244–1254. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. B-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Bulgart, H.R.; Perez, M.A.L.; Tucker, A.; Giarrano, G.N.; Banford, K.; Miller, O.; Bonser, S.W.G.; Wold, L.E.; Scharre, D.; Weisleder, N. Plasma membrane repair defect in Alzheimer’s disease neurons is driven by the reduced dysferlin expression. FASEB J. 2024, 38, e70099. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205. [Google Scholar] [CrossRef]
- Blazek, A.D.; Paleo, B.J.; Weisleder, N. Plasma Membrane Repair: A Central Process for Maintaining Cellular Homeostasis. Physiology 2015, 30, 438. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, B.; Zhu, H.; Li, H.; Han, Y.; Chen, K.; Wang, Z.; Zeng, J.; Liu, Y.; Wang, X.; et al. MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget 2016, 7, 22474–22485. [Google Scholar] [CrossRef]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H.; Ma, J. MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.-K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.K.; Lee, K.J.; Cai, C.; Huang, M.; Cho, C.-H.; Ma, J.; Lee, E.H. Mitsugumin 53 regulates extracellular Ca2+ entry and intracellular Ca2+ release via Orai1 and RyR1 in skeletal muscle. Sci. Rep. 2016, 6, 36909. [Google Scholar] [CrossRef]
- Wu, M.; Li, H.; He, J.; Liang, J.; Liu, Y.; Zhang, W. TRIM72 Alleviates Muscle Inflammation in mdx Mice via Promoting Mitophagy-Mediated NLRP3 Inflammasome Inactivation. Oxid. Med. Cell Longev. 2023, 2023, 8408574. [Google Scholar] [CrossRef]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra85. [Google Scholar] [CrossRef]
- Gushchina, L.V.; Bhattacharya, S.; McElhanon, K.E.; Choi, J.H.; Manring, H.; Beck, E.X.; Alloush, J.; Weisleder, N. Treatment with Recombinant Human MG53 Protein Increases Membrane Integrity in a Mouse Model of Limb Girdle Muscular Dystrophy 2B. Mol. Ther. 2017, 25, 2360–2371. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, H.; Zheng, Y.; Xu, Z.; Li, L.; Tan, T.; Park, K.H.; Hou, J.; Zhang, C.; Li, D.; et al. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J. Mol. Cell Cardiol. 2015, 80, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 2015, 7, 279ra36. [Google Scholar] [CrossRef]
- Paleo, B.J.; Madalena, K.M.; Mital, R.; McElhanon, K.E.; Kwiatkowski, T.A.; Rose, A.L.; Lerch, J.K.; Weisleder, N. Enhancing membrane repair increases regeneration in a sciatic injury model. PLoS ONE 2020, 15, e0231194. [Google Scholar] [CrossRef] [PubMed]
- Whitson, B.A.; Mulier, K.; Li, H.; Zhou, X.; Cai, C.; Black, S.M.; Tan, T.; Ma, J.; Beilman, G.J. MG53 as a Novel Therapeutic Protein to Treat Acute Lung Injury. Mil. Med. 2021, 186, 339–345. [Google Scholar] [CrossRef]
- Cong, X.; Nagre, N.; Herrera, J.; Pearson, A.C.; Pepper, I.; Morehouse, R.; Ji, H.-L.; Jiang, D.; Hubmayr, R.D.; Zhao, X. TRIM72 promotes alveolar epithelial cell membrane repair and ameliorates lung fibrosis. Respir. Res. 2020, 21, 132. [Google Scholar] [CrossRef]
- Lee, J.J.A.; Maruyama, R.; Sakurai, H.; Yokota, T. Cell Membrane Repair Assay Using a Two-photon Laser Microscope. Immunol. Infect. 2018, 131, 56999. [Google Scholar]
- Guan, F.; Zhou, X.; Li, P.; Wang, Y.; Liu, M.; Li, F.; Cui, Y.; Huang, T.; Yao, M.; Zhang, Y.; et al. MG53 attenuates lipopolysaccharide-induced neurotoxicity and neuroinflammation via inhibiting TLR4/NF-κB pathway in vitro and in vivo. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 95, 109684. [Google Scholar] [CrossRef]
- Guan, F.; Huang, T.; Wang, X.; Xing, Q.; Gumpper, K.; Li, P.; Song, J.; Tan, T.; Yang, G.L.; Zang, X.; et al. The TRIM protein Mitsugumin 53 enhances survival and therapeutic efficacy of stem cells in murine traumatic brain injury. Stem Cell Res. Ther. 2019, 10, 352. [Google Scholar] [CrossRef]
- Song, R.; Peng, W.; Zhang, Y.; Lv, F.; Wu, H.-K.; Guo, J.; Cao, Y.; Pi, Y.; Zhang, X.; Jin, L.; et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 2013, 494, 375–379. [Google Scholar] [CrossRef]
- Liu, F.; Song, R.; Feng, Y.; Guo, J.; Chen, Y.; Zhang, Y.; Chen, T.; Wang, Y.; Huang, Y.; Li, C.Y.; et al. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor α. Circulation 2015, 131, 795–804. [Google Scholar] [CrossRef]
- Bianchi, C.; Vaccaro, O.; Distaso, M.; Franzini, L.; Raggi, F.; Solini, A. MG53 does not mark cardiovascular risk and all-cause mortality in subjects with type 2 diabetes: A prospective, observational study. Diabetes Res. Clin. Pract. 2023, 204, 110916. [Google Scholar] [CrossRef] [PubMed]
- Philouze, C.; Turban, S.; Cremers, B.; Caliez, A.; Lamarche, G.; Bernard, C.; Provost, N.; Delerive, P. MG53 is not a critical regulator of insulin signaling pathway in skeletal muscle. PLoS ONE 2021, 16, e024517. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Yu, Q.; Luo, M.; Wu, J.; Wang, L. Friend or foe: The paradoxical roles of MG53 in diabetes mellitus. Diabetes 2024, 74, 145–152. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Emelin, A.M.; Slesarenko, Y.S.; Limaev, I.S.; Vetrova, I.A.; Belikova, L.D.; Grafskaia, E.N.; Bobrovsky, P.A.; Pokrovsky, M.V.; Kuzubova, E.V.; et al. Dual Adeno-Associated Virus 9 with Codon-Optimized DYSF Gene Promotes In Vivo Muscle Regeneration and May Decrease Inflammatory Response in Limb Girdle Muscular Dystrophy Type R2. Int. J. Mol. Sci. 2023, 24, 13551. [Google Scholar] [CrossRef]
- Lostal, W.; Bartoli, M.; Bourg, N.; Roudaut, C.; Bentaïb, A.; Miyake, K.; Guerchet, N.; Fougerousse, F.; McNeil, P.; Richard, I. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum. Mol. Genet. 2010, 19, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Potter, R.A.; Griffin, D.A.; Sondergaard, P.C.; Johnson, R.W.; Pozsgai, E.R.; Heller, K.N.; Peterson, E.L.; Lehtimäki, K.K.; Windish, H.P.; Mittal, P.J.; et al. Systemic Delivery of Dysferlin Overlap Vectors Provides Long-Term Gene Expression and Functional Improvement for Dysferlinopathy. Hum. Gene Ther. 2018, 29, 749–762. [Google Scholar] [CrossRef]
- Llanga, T.; Nagy, N.; Conatser, L.; Dial, C.; Sutton, R.B.; Hirsch, M.L. Structure-Based Designed Nano-Dysferlin Significantly Improves Dysferlinopathy in BLA/J Mice. Mol. Ther. 2017, 25, 2150–2162. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulgart, H.R.; Lopez Perez, M.A.; Weisleder, N. Enhancing Membrane Repair Using Recombinant MG53/TRIM72 (rhMG53) Reduces Neurotoxicity in Alzheimer’s Disease Models. Biomolecules 2025, 15, 418. https://doi.org/10.3390/biom15030418
Bulgart HR, Lopez Perez MA, Weisleder N. Enhancing Membrane Repair Using Recombinant MG53/TRIM72 (rhMG53) Reduces Neurotoxicity in Alzheimer’s Disease Models. Biomolecules. 2025; 15(3):418. https://doi.org/10.3390/biom15030418
Chicago/Turabian StyleBulgart, Hannah R., Miguel A. Lopez Perez, and Noah Weisleder. 2025. "Enhancing Membrane Repair Using Recombinant MG53/TRIM72 (rhMG53) Reduces Neurotoxicity in Alzheimer’s Disease Models" Biomolecules 15, no. 3: 418. https://doi.org/10.3390/biom15030418
APA StyleBulgart, H. R., Lopez Perez, M. A., & Weisleder, N. (2025). Enhancing Membrane Repair Using Recombinant MG53/TRIM72 (rhMG53) Reduces Neurotoxicity in Alzheimer’s Disease Models. Biomolecules, 15(3), 418. https://doi.org/10.3390/biom15030418