
Early-Life Adversity and Epigenetic Aging: Findings from a 17-Year Longitudinal Study

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cohort Description
2.2. Measures
2.2.1. Perinatal Adversity Index
2.2.2. Child and Adolescent Adversity Index
2.3. DNA Methylation Quantification
Cell Type Estimation
2.4. Epigenetic Age Acceleration
Assessment of Epigenetic Age Measure Performance
2.5. Covariates
2.6. Statistical Analyses
3. Results
3.1. Exposure to Adversity Was Low Overall and Differed by Sex
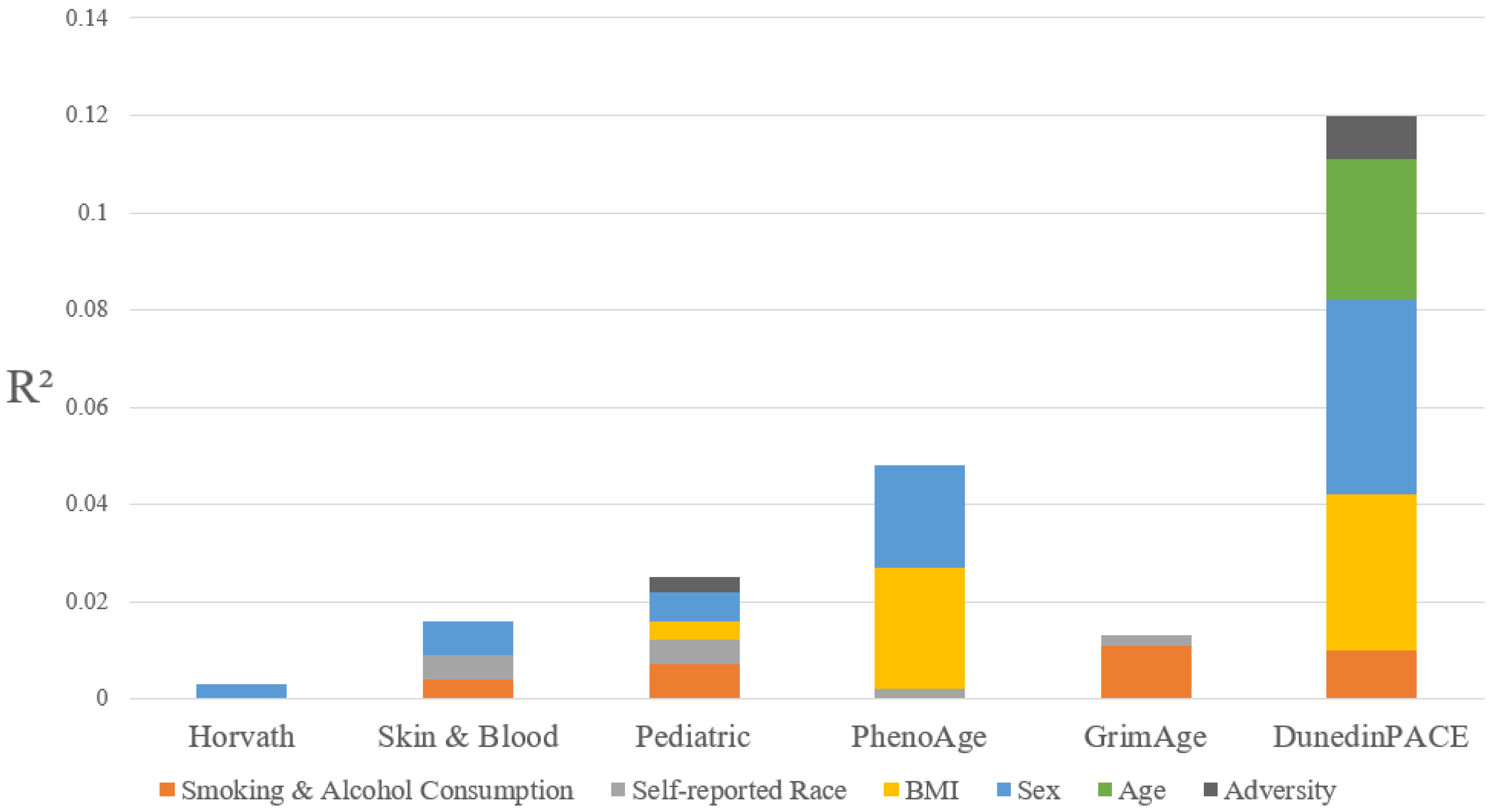
3.2. Measures of Epigenetic Age Among Adolescents
3.3. Only Perinatal Adversity Is Associated with Epigenetic Age Acceleration
3.4. Limited Evidence for the Stress Sensitization Hypothesis
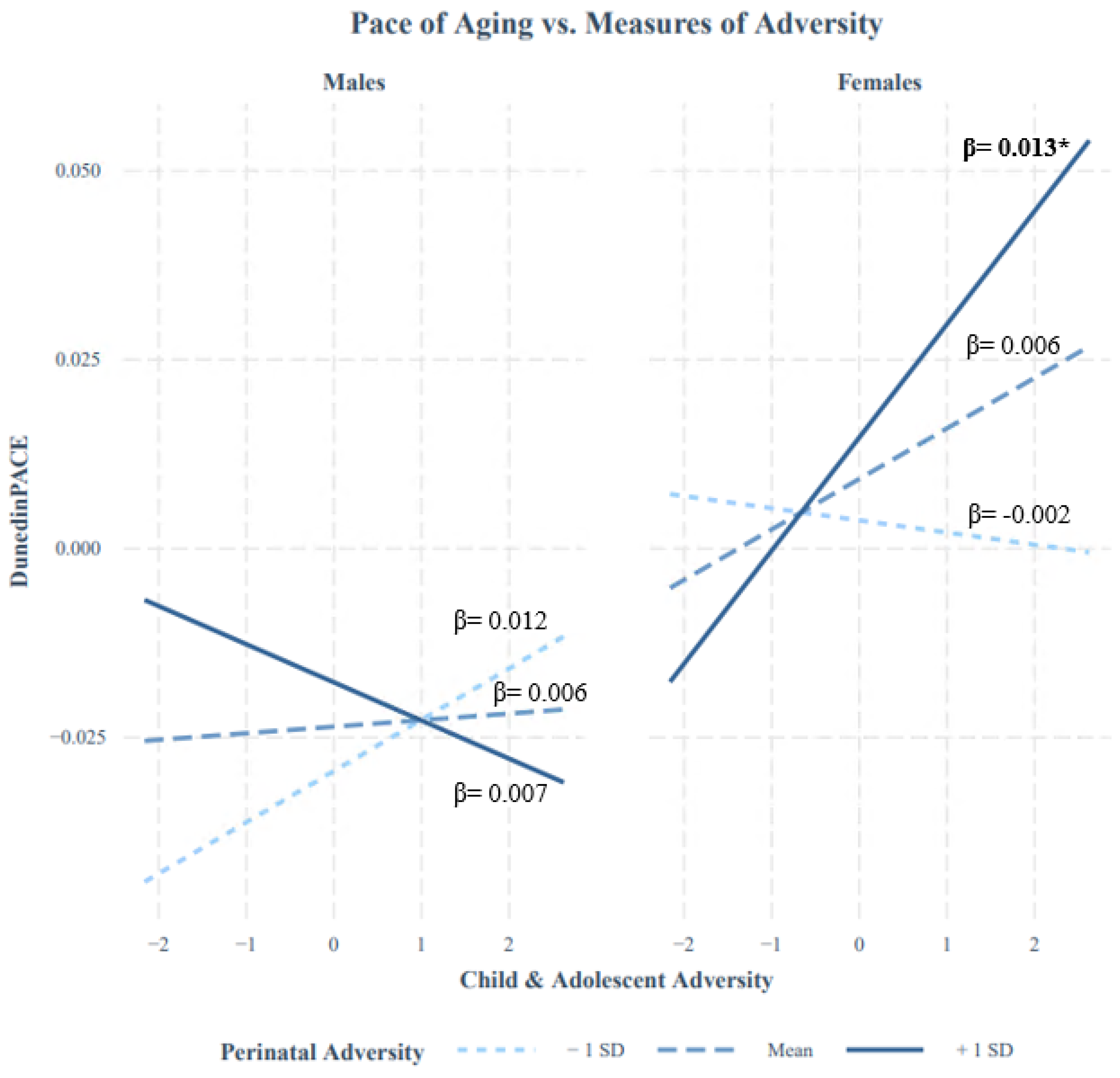
3.5. Epigenetic Age Differs by Sex, but Evidence of Sex Moderation Is Limited
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ELA | Early-Life Adversity |
| DNAm | DNA Methylation |
References
- McLaughlin, K.A. Future Directions in Childhood Adversity and Youth Psychopathology. J. Clin. Child Adolesc. Psychol. 2016, 45, 361–382. [Google Scholar] [CrossRef]
- Afifi, T.O. Considerations for expanding the definition of ACEs. In Adverse Childhood Experiences; Elsevier: Amsterdam, The Netherlands, 2020; pp. 35–44. [Google Scholar]
- Gunnar, M.R. Early adversity, stress, and neurobehavioral development. Dev. Psychopathol. 2020, 32, 1555–1562. [Google Scholar] [CrossRef]
- Boyce, W.T.; Levitt, P.; Martinez, F.D.; McEwen, B.S.; Shonkoff, J.P. Genes, Environments, and Time: The Biology of Adversity and Resilience. Pediatrics 2021, 147, e20201651. [Google Scholar] [CrossRef]
- Arseneault, L. Annual Research Review: The persistent and pervasive impact of being bullied in childhood and adolescence: Implications for policy and practice. J. Child Psychol. Psychiatry 2018, 59, 405–421. [Google Scholar] [CrossRef]
- Forte, A.; Orri, M.; Galera, C.; Pompili, M.; Turecki, G.; Boivin, M.; Tremblay, R.E.; Côté, S.M. Developmental trajectories of childhood symptoms of hyperactivity/inattention and suicidal behavior during adolescence. Eur. Child Adolesc. Psychiatry 2020, 29, 145–151. [Google Scholar] [CrossRef]
- Hertzman, C. Putting the concept of biological embedding in historical perspective. Proc. Natl. Acad. Sci. USA 2012, 109 (Suppl. S2), 17160–17167. [Google Scholar] [CrossRef]
- Engert, V.; Efanov, S.I.; Dedovic, K.; Dagher, A.; Pruessner, J.C. Increased cortisol awakening response and afternoon/evening cortisol output in healthy young adults with low early life parental care. Psychopharmacology 2011, 214, 261–268. [Google Scholar] [CrossRef]
- Trickett, P.K.; Noll, J.G.; Susman, E.J.; Shenk, C.E.; Putnam, F.W. Attenuation of cortisol across development for victims of sexual abuse. Tissue Eng. 2007, 23, 165–175. [Google Scholar] [CrossRef]
- Gunnar, M.R.; Vazquez, D.M. Low cortisol and a flattening of expected daytime rhythm: Potential indices of risk in human development. Dev. Psychopathol. 2001, 13, 515–538. [Google Scholar] [CrossRef]
- Adam, E.K.; Quinn, M.E.; Tavernier, R.; McQuillan, M.T.; Dahlke, K.A.; Gilbert, K.E. Diurnal cortisol slopes and mental and physical health outcomes: A systematic review and meta-analysis. Psychoneuroendocrinology 2017, 83, 25–41. [Google Scholar] [CrossRef]
- Young, E.S.; Farrell, A.K.; Carlson, E.A.; Englund, M.M.; Miller, G.E.; Gunnar, M.R.; Roisman, G.I.; Simpson, J.A. The Dual Impact of Early and Concurrent Life Stress on Adults’ Diurnal Cortisol Patterns: A Prospective Study. Psychol. Sci. 2019, 30, 739–747. [Google Scholar] [CrossRef]
- Tarullo, A.R.; Tuladhar, C.T.; Kao, K.; Drury, E.B.; Meyer, J. Cortisol and socioeconomic status in early childhood: A multidimensional assessment. Dev. Psychopathol. 2020, 32, 1876–1887. [Google Scholar] [CrossRef]
- Ouellet-Morin, I.; Cantave, C.; Lupien, S.; Geoffroy, M.C.; Brendgen, M.; Vitaro, F.; Tremblay, R.E.; Boivin, M.; Côté, S.M. Cumulative exposure to socioeconomic and psychosocial adversity and hair cortisol concentration: A longitudinal study from 5 months to 17 years of age. Psychoneuroendocrinology 2021, 126, 105153. [Google Scholar] [CrossRef]
- Ioannidis, K.; Askelund, A.D.; Kievit, R.A.; van Harmelen, A.L. The complex neurobiology of resilient functioning after childhood maltreatment. BMC Med. 2020, 18, 32. [Google Scholar] [CrossRef]
- Weder, N.; Zhang, H.; Jensen, K.; Yang, B.Z.; Simen, A.; Jackowski, A.; Lipschitz, D.; Douglas-Palumberi, H.; Ge, M.; Perepletchikova, F.; et al. Child Abuse, Depression, and Methylation in Genes Involved With Stress, Neural Plasticity, and Brain Circuitry. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 417–424. [Google Scholar] [CrossRef]
- Parade, S.H.; Huffhines, L.; Daniels, T.E.; Stroud, L.R.; Nugent, N.R.; Tyrka, A.R. A systematic review of childhood maltreatment and DNA methylation: Candidate gene and epigenome-wide approaches. Transl. Psychiatry 2021, 11, 134. [Google Scholar] [CrossRef]
- Provençal, N.; Binder, E.B. The effects of early life stress on the epigenome: From the womb to adulthood and even before. Exp. Neurol. 2015, 268, 10–20. [Google Scholar] [CrossRef]
- Wiechmann, T.; Röh, S.; Sauer, S.; Czamara, D.; Arloth, J.; Ködel, M.; Beintner, M.; Knop, L.; Menke, A.; Binder, E.B.; et al. Identification of dynamic glucocorticoid-induced methylation changes at the FKBP5 locus. Clin. Epigenet. 2019, 11, 83. [Google Scholar] [CrossRef]
- McEwen, B.S. Allostasis and the Epigenetics of Brain and Body Health Over the Life Course. JAMA Psychiatry 2017, 74, 551. [Google Scholar] [CrossRef]
- Martin, D.I.K.; Cropley, J.E.; Suter, C.M. Epigenetics in disease: Leader or follower? Epigenetics 2011, 6, 843–848. [Google Scholar] [CrossRef]
- Oh, D.L.; Jerman, P.; Silvério Marques, S.; Koita, K.; Purewal Boparai, S.K.; Burke Harris, N.; Bucci, M. Systematic review of pediatric health outcomes associated with childhood adversity. BMC Pediatr. 2018, 18, 83. [Google Scholar] [CrossRef]
- Suarez, A.; Lahti, J.; Lahti-Pulkkinen, M.; Girchenko, P.; Czamara, D.; Arloth, J.; Malmberg, A.L.; Hämäläinen, E.; Kajantie, E.; Laivuori, H.; et al. A polyepigenetic glucocorticoid exposure score at birth and childhood mental and behavioral disorders. Neurobiol. Stress 2020, 13, 100275. [Google Scholar] [CrossRef]
- Bick, J.; Naumova, O.; Hunter, S.; Barbot, B.; Lee, M.; Luthar, S.S.; Raefski, A.; Grigorenko, E.L. Childhood adversity and DNA methylation of genes involved in the hypothalamus–pituitary–adrenal axis and immune system: Whole-genome and candidate-gene associations. Dev. Psychopathol. 2012, 24, 1417–1425. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Jain, P.; Binder, A.M.; Chen, B.; Parada, H.; Gallo, L.C.; Alcaraz, J.; Horvath, S.; Bhatti, P.; Whitsel, E.A.; Jordahl, K.; et al. Analysis of Epigenetic Age Acceleration and Healthy Longevity Among Older US Women. JAMA Netw. Open 2022, 5, e2223285. [Google Scholar] [CrossRef]
- Johnstone, S.E.; Gladyshev, V.N.; Aryee, M.J.; Bernstein, B.E. Epigenetic clocks, aging, and cancer. Science 2022, 378, 1276–1277. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- McEwen, L.M.; O’Donnell, K.J.; McGill, M.G.; Edgar, R.D.; Jones, M.J.; MacIsaac, J.L.; Lin, D.T.S.; Ramadori, K.; Morin, A.; Gladish, N.; et al. The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells. Proc. Natl. Acad. Sci. USA 2020, 117, 23329–23335. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Krieger, N.; Chen, J.T.; Testa, C.; Diez Roux, A.; Tilling, K.; Watkins, S.; Simpkin, A.J.; Suderman, M.; Davey Smith, G.; De Vivo, I.; et al. Use of Correct and Incorrect Methods of Accounting for Age in Studies of Epigenetic Accelerated Aging: Implications and Recommendations for Best Practices. Am. J. Epidemiol. 2023, 192, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Belsky, D.W.; Caspi, A.; Arseneault, L.; Baccarelli, A.; Corcoran, D.L.; Gao, X.; Hannon, E.; Harrington, H.L.; Rasmussen, L.J.; Houts, R.; et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. Elife 2020, 9, e54870. [Google Scholar] [CrossRef] [PubMed]
- Belsky, D.W.; Caspi, A.; Corcoran, D.L.; Sugden, K.; Poulton, R.; Arseneault, L.; Baccarelli, A.; Chamarti, K.; Gao, X.; Hannon, E.; et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. eLife 2022, 11, e73420. [Google Scholar] [CrossRef]
- Wolf, E.J.; Logue, M.W.; Morrison, F.G.; Wilcox, E.S.; Stone, A.; Schichman, S.A.; McGlinchey, R.E.; Milberg, W.P.; Miller, M.W. Posttraumatic psychopathology and the pace of the epigenetic clock: A longitudinal investigation. Psychol. Med. 2019, 49, 791–800. [Google Scholar] [CrossRef]
- Mehta, D.; Bruenig, D.; Pierce, J.; Sathyanarayanan, A.; Stringfellow, R.; Miller, O.; Mullens, A.B.; Shakespeare-Finch, J. Recalibrating the epigenetic clock after exposure to trauma: The role of risk and protective psychosocial factors. J. Psychiatr. Res. 2022, 149, 374–381. [Google Scholar] [CrossRef]
- Han, L.K.M.; Aghajani, M.; Clark, S.L.; Chan, R.F.; Hattab, M.W.; Shabalin, A.A.; Zhao, M.; Kumar, G.; Xie, L.Y.; Jansen, R.; et al. Epigenetic Aging in Major Depressive Disorder. Am. J. Psychiatry 2018, 175, 774–782. [Google Scholar] [CrossRef]
- Cerveira de Baumont, A.; Hoffmann, M.S.; Bortoluzzi, A.; Fries, G.R.; Lavandoski, P.; Grun, L.K.; Guimarães, L.S.; Guma, F.T.; Salum, G.A.; Barbé-Tuana, F.M.; et al. Telomere length and epigenetic age acceleration in adolescents with anxiety disorders. Sci. Rep. 2021, 11, 7716. [Google Scholar] [CrossRef]
- Jovanovic, T.; Vance, L.A.; Cross, D.; Knight, A.K.; Kilaru, V.; Michopoulos, V.; Klengel, T.; Smith, A.K. Exposure to Violence Accelerates Epigenetic Aging in Children. Sci. Rep. 2017, 7, 8962. [Google Scholar] [CrossRef]
- Lawn, R.B.; Anderson, E.L.; Suderman, M.; Simpkin, A.J.; Gaunt, T.R.; Teschendorff, A.E.; Widschwendter, M.; Hardy, R.; Kuh, D.; Relton, C.L.; et al. Psychosocial adversity and socioeconomic position during childhood and epigenetic age: Analysis of two prospective cohort studies. Hum. Mol. Genet. 2018, 27, 1301–1308. [Google Scholar] [CrossRef]
- Austin, M.K.; Chen, E.; Ross, K.M.; McEwen, L.M.; Maclsaac, J.L.; Kobor, M.S.; Miller, G.E. Early-life socioeconomic disadvantage, not current, predicts accelerated epigenetic aging of monocytes. Psychoneuroendocrinology 2018, 97, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Yusupov, N.; Dieckmann, L.; Erhart, M.; Sauer, S.; Rex-Haffner, M.; Kopf-Beck, J.; Brückl, T.M.; Czamara, D.; Binder, E.B. Transdiagnostic evaluation of epigenetic age acceleration and burden of psychiatric disorders. Neuropsychopharmacology 2023, 48, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Hamlat, E.J.; Prather, A.A.; Horvath, S.; Belsky, J.; Epel, E.S. Early life adversity, pubertal timing, and epigenetic age acceleration in adulthood. Dev. Psychobiol. 2021, 63, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Simons, R.L.; Lei, M.K.; Klopach, E.; Berg, M.; Zhang, Y.; Beach, S.S.R. (Re)Setting Epigenetic Clocks: An Important Avenue Whereby Social Conditions Become Biologically Embedded across the Life Course. J. Health Soc. Behav. 2021, 62, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Raffington, L.; Belsky, D.W.; Kothari, M.; Malanchini, M.; Tucker-Drob, E.M.; Harden, K.P. Socioeconomic Disadvantage and the Pace of Biological Aging in Children. Pediatrics 2021, 147, e2020024406. [Google Scholar] [CrossRef]
- Sumner, J.A.; Colich, N.L.; Uddin, M.; Armstrong, D.; McLaughlin, K.A. Early Experiences of Threat, but Not Deprivation, Are Associated With Accelerated Biological Aging in Children and Adolescents. Biol. Psychiatry 2019, 85, 268–278. [Google Scholar] [CrossRef]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef]
- Marini, S.; Davis, K.A.; Soare, T.W.; Zhu, Y.; Suderman, M.J.; Simpkin, A.J.; Smith, A.D.; Wolf, E.J.; Relton, C.L.; Dunn, E.C. Adversity exposure during sensitive periods predicts accelerated epigenetic aging in children. Psychoneuroendocrinology 2020, 113, 104484. [Google Scholar] [CrossRef]
- Holbrook, B.D. The effects of nicotine on human fetal development. Birth Defects Res. C Embryo Today 2016, 108, 181–192. [Google Scholar] [CrossRef]
- Martins, J.; Czamara, D.; Sauer, S.; Rex-Haffner, M.; Dittrich, K.; Dörr, P.; de Punder, K.; Overfeld, J.; Knop, A.; Dammering, F.; et al. Childhood adversity correlates with stable changes in DNA methylation trajectories in children and converges with epigenetic signatures of prenatal stress. Neurobiol. Stress 2021, 15, 100336. [Google Scholar] [CrossRef]
- Post, R.M. Epigenetic basis of sensitization to stress, affective episodes, and stimulants: Implications for illness progression and prevention. Bipolar. Disord. 2016, 18, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Bunea, I.M.; Szentágotai-Tătar, A.; Miu, A.C. Early-life adversity and cortisol response to social stress: A meta-analysis. Transl. Psychiatry 2017, 7, 1274. [Google Scholar] [CrossRef] [PubMed]
- Young, E.S.; Doom, J.R.; Farrell, A.K.; Carlson, E.A.; Englund, M.M.; Miller, G.E.; Gunnar, M.R.; Roisman, G.I.; Simpson, J.A. Life stress and cortisol reactivity: An exploratory analysis of the effects of stress exposure across life on HPA-axis functioning. Dev. Psychopathol. 2021, 33, 301–312. [Google Scholar] [CrossRef]
- McCrory, C.; Fiorito, G.; McLoughlin, S.; Polidoro, S.; Cheallaigh, C.N.; Bourke, N.; Karisola, P.; Alenius, H.; Vineis, P.; Layte, R.; et al. Epigenetic Clocks and Allostatic Load Reveal Potential Sex-Specific Drivers of Biological Aging. J. Gerontol. Ser. A 2019, 75, 495–503. [Google Scholar] [CrossRef]
- Suarez, A.; Lahti, J.; Czamara, D.; Lahti-Pulkkinen, M.; Knight, A.K.; Girchenko, P.; Hämäläinen, E.; Kajantie, E.; Lipsanen, J.; Laivuori, H.; et al. The Epigenetic Clock at Birth: Associations With Maternal Antenatal Depression and Child Psychiatric Problems. J. Am. Acad. Child Adolesc. Psychiatry 2018, 57, 321–328. [Google Scholar] [CrossRef]
- Engelbrecht, H.R.; Merrill, S.M.; Gladish, N.; MacIsaac, J.L.; Lin, D.T.S.; Ecker, S.; Chrysohoou, C.A.; Pes, G.M.; Kobor, M.S.; Rehkopf, D.H. Sex differences in epigenetic age in Mediterranean high longevity regions. Front. Aging 2022, 3, 1007098. [Google Scholar] [CrossRef] [PubMed]
- Orri, M.; Boivin, M.; Chen, C.; Ahun, M.N.; Geoffroy, M.C.; Ouellet-Morin, I.; Tremblay, R.E.; Côté, S.M. Cohort Profile: Quebec Longitudinal Study of Child Development (QLSCD). Soc. Psychiatry Psychiatr. Epidemiol. 2021, 56, 883–894. [Google Scholar] [CrossRef]
- Silveira, P.P.; Pokhvisneva, I.; Parent, C.; Cai, S.; Rema, A.S.S.; Broekman, B.F.P.; Rifkin-Graboi, A.; Pluess, M.; O’Donnell, K.J.; Meaney, M.J. Cumulative prenatal exposure to adversity reveals associations with a broad range of neurodevelopmental outcomes that are moderated by a novel, biologically informed polygenetic score based on the serotonin transporter solute carrier family C6, member 4. Dev. Psychopathol. 2017, 29, 1601–1617. [Google Scholar] [CrossRef]
- Hughes, M.M.; Black, R.E.; Katz, J. 2500-g Low Birth Weight Cutoff: History and Implications for Future Research and Policy. Matern. Child Health J. 2017, 21, 283–289. [Google Scholar] [CrossRef]
- Radloff, L.S. The CES-D Scale. Appl. Psychol. Meas. 1977, 1, 385–401. [Google Scholar] [CrossRef]
- Ladd, G.W.; Kochenderfer-Ladd, B. Identifying victims of peer aggression from early to middle childhood: Analysis of cross-informant data for concordance, estimation of relational adjustment, prevalence of victimization, and characteristics of identified victims. Psychol. Assess 2002, 14, 74–96. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.C.; Breeze, C.E.; Beck, S.; Teschendorff, A.E. Identification of differentially methylated cell types in epigenome-wide association studies. Nat. Methods 2018, 15, 1059–1066. [Google Scholar] [CrossRef]
- Lang, M.; Binder, M.; Richter, J.; Schratz, P.; Pfisterer, F.; Coors, S.; Au, Q.; Casalicchio, G.; Kotthoff, L.; Bischl, B.; et al. mlr3: A modern object-oriented machine learning framework in R. J. Open Source Softw. 2019, 4, 1903. [Google Scholar] [CrossRef]
- McCrory, C.; Fiorito, G.; O’Halloran, A.M.; Polidoro, S.; Vineis, P.; Kenny, R.A. Early life adversity and age acceleration at mid-life and older ages indexed using the next-generation GrimAge and Pace of Aging epigenetic clocks. Psychoneuroendocrinology 2022, 137, 105643. [Google Scholar] [CrossRef] [PubMed]
- Schrempft, S.; Stringhini, S. Socioeconomic inequalities in the Pace of Aging. Aging 2023, 15, 1706–1707. [Google Scholar] [CrossRef]
- Perret, L.C.; Geoffroy, M.C.; Barr, E.; Parnet, F.; Provencal, N.; Boivin, M.; O’Donnell, K.J.; Suderman, M.; Power, C.; Turecki, G.; et al. Associations between epigenetic aging and childhood peer victimization, depression, and suicidal ideation in adolescence and adulthood: A study of two population-based samples. Front. Cell Dev. Biol. 2023, 10, 1051556. [Google Scholar] [CrossRef]
- Colich, N.L.; Rosen, M.L.; Williams, E.S.; McLaughlin, K.A. Biological aging in childhood and adolescence following experiences of threat and deprivation: A systematic review and meta-analysis. Psychol. Bull. 2020, 146, 721–764. [Google Scholar] [CrossRef]
- Copeland, W.E.; Shanahan, L.; McGinnis, E.W.; Aberg, K.A.; van den Oord, E.J.C.G. Early adversities accelerate epigenetic aging into adulthood: A 10-year, within-subject analysis. J. Child Psychol. Psychiatry 2022, 63, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Yaroslavsky, I.; Bush, A.H.; France, C.M. Emotion regulation deficits mediate childhood sexual abuse effects on stress sensitization and depression outcomes. Dev. Psychopathol. 2022, 34, 157–170. [Google Scholar] [CrossRef]
- Luo, J.; Willroth, E.C. Values and stress: Examining the relations between values and general and domain-specific stress in two longitudinal studies. J. Pers. Soc. Psychol. 2025, 128, 410–437. [Google Scholar] [CrossRef]
- Stroud, C.B. The Stress Sensitization Model. In The Oxford Handbook of Stress and Mental Health; Harkness, K.L., Hayden, E.P., Eds.; Oxford University Press: Oxford, UK, 2020; pp. 348–370. [Google Scholar]
- McCarthy, M.M.; Nugent, B.M. At the frontier of epigenetics of brain sex differences. Front. Behav. Neurosci. 2015, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Lim, S.; Gudsnuk, K.; Champagne, F.A. Sex-specific and strain-dependent effects of early life adversity on behavioral and epigenetic outcomes. Front. Psychiatry 2013, 4, 78. [Google Scholar] [CrossRef] [PubMed]
- Aylwin, C.F.; Toro, C.A.; Shirtcliff, E.; Lomniczi, A. Emerging Genetic and Epigenetic Mechanisms Underlying Pubertal Maturation in Adolescence. J. Res. Adolesc. 2019, 29, 54–79. [Google Scholar] [CrossRef]
- Almstrup, K.; Lindhardt Johansen, M.; Busch, A.S.; Hagen, C.P.; Nielsen, J.E.; Petersen, J.H.; Juul, A. Pubertal development in healthy children is mirrored by DNA methylation patterns in peripheral blood. Sci. Rep. 2016, 6, 28657. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Total (n = 696) | Female (n = 385) | Male (n = 311) | Mean Differences | |
|---|---|---|---|---|
| n (%) or Mean (SD) | n (%) or Mean (SD) | n (%) or Mean (SD) | Chi-Square Test (p-Value) or t-test (p-Value) | |
| Specific Indicators | ||||
| Low Birth Weight (<2500 g) | 26 (3.7%) | 14 (3.6%) | 12 (3.9%) | 6.8 × 10−31 (1.00) |
| Pre-Term Birth (<37 weeks) | 92 (13.2%) | 44 (11.4%) | 48 (15.4%) | 2.07 (0.15) |
| Low SES (<CAD 30,000) | 156 (22.4%) | 89 (23.1%) | 67 (21.5%) | 0.16 (0.69) |
| Maternal Smoking (Yes) | 147 (21.1%) | 82 (21.3%) | 65 (20.9%) | 0.001 (0.97) |
| Hospital Admissions (First 5 Months) | 80 (11.5%) | 39 (10.1%) | 41 (13.2%) | 1.29 (0.26) |
| Maternal Depression (CESD > 16) | 31 (4.5%) | 20 (5.2%) | 11 (3.5%) | 0.76 (0.39) |
| Poor Family Functioning | 61 (8.8%) | 35 (9.1%) | 32 (8.4%) | 0.04 (0.84) |
| Total Score | ||||
| Perinatal Adversity (Range 0–2) | 0.74 (0.79) | 0.73 (0.79) | 0.77 (0.78) | 0.73 (0.46) |
| Total (n = 696) | Female (n = 385) | Male (n = 311) | Mean Differences | |
|---|---|---|---|---|
| n (%) or Mean (SD) | n (%) or Mean (SD) | n (%) or Mean (SD) | Chi-Square Test (p-Value) or t-test (p-Value) | |
| Specific Indicators | ||||
| Young Motherhood Yes | 133 (19.1%) | 69 (17.9%) | 64 (20.6%) | 0.62 (0.43) |
| Single-Parent Household Yes | 90 (12.9%) | 46 (11.9%) | 44 (14.1%) | 0.56 (0.46) |
| Socioeconomic Status (Low vs.) Medium High | 283 (40.7%) 174 (25.0%) | 141 (36.6%) 106 (27.5%) | 142 (45.7%) 68 (21.9%) | 6.23 (0.044) |
| Coercive Parenting (Low vs.) Medium High | 327 (47.0%) 84 (12.1%) | 173 (44.9%) 36 (9.35%) | 154 (49.5%) 48 (15.4%) | 10.82 (0.004) |
| Maternal Depression (Low vs.) Medium High | 285 (40.9%) 76 (10.9%) | 152 (39.5%) 42 (10.9%) | 133 (42.7%) 34 (10.9%) | 0.85 (0.66) |
| Maternal Alcohol Consumption (Low vs.) Yes | 143 (20.5%) | 76 (19.7%) | 67 (21.5%) | 0.24 (0.62) |
| Peer Victimization (Low vs.) Medium High | 390 (56.0%) 84 (12.1%) | 199 (51.7%) 36 (9.35%) | 191 (61.4%) 48 (15.4%) | 21.66 (<0.001) |
| Dangerous Neighborhood (Low vs.) Medium High | 561 (80.6%) 42 (6.03%) | 306 (79.5%) 27 (7.01%) | 255 (82.0%) 15 (4.82%) | 1.52 (0.47) |
| Total Score | ||||
| Child and Adolescent Adversity (Range 0–11) | 4.97 (2.30) | 4.76 (2.31) | 5.23 (2.26) | 2.72 (0.007) |
| Model | Horvath | Pediatric Clock | Skin and Blood Clock | PhenoAge | GrimAge | DunedinPACE |
|---|---|---|---|---|---|---|
| Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | |
| Perinatal Adversity | −0.00025 (0.99) | 0.024 (0.55) | 0.020 (0.62) | 0.023 (0.57) | 0.0033 (0.94) | 0.079 (0.045) |
| Child and Adolescent Adversity | −0.035 (0.41) | 0.056 (0.17) | −0.0016 (0.97) | 0.0037 (0.93) | 0.029 (0.50) | 0.064 (0.12) |
| Model | Horvath | Pediatric Clock | Skin and Blood Clock | PhenoAge | GrimAge | DunedinPACE | |
|---|---|---|---|---|---|---|---|
| Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | ||
| Perinatal Adversity + Child and Adolescent Adversity | |||||||
| Perinatal Adversity | 0.011 (0.79) | 0.0070 (0.87) | 0.022 (0.88) | 0.024 (0.58) | −0.0050 (0.91) | 0.065 (0.12) | |
| Child and Adolescent Adversity | −0.039 (0.38) | 0.055 (0.21) | −0.0067 (0.61) | −0.0022 (0.96) | 0.027 (0.55) | 0.045 (0.29) | |
| Perinatal Adversity*Child and Adolescent Adversity | |||||||
| Perinatal Adversity*Child and Adolescent Adversity | −0.029 (0.48) | 0.0077 (0.85) | −0.020 (0.62) | −0.054 (0.18) | 0.010 (0.80) | 0.027 (0.49) | |
| Model | Horvath | Pediatric Clock | Skin and Blood Clock | PhenoAge | GrimAge | DunedinPACE |
|---|---|---|---|---|---|---|
| Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | Estimate (pval) | |
| Perinatal Adversity*Sex | −0.049 (0.23) | −0.002 (0.96) | −0.021 (0.60) | 0.056 (0.15) | - | 0.018 (0.64) |
| Child and Adolescent Adversity*Sex | 0.0080 (0.85) | −0.0097 (0.81) | 0.0077 (0.85) | 0.065 (0.11) | - | 0.033 (0.40) |
| Perinatal Adversity*Child and Adolescent Adversity*Sex | 0.05 (0.22) | −0.049 (0.24) | −0.015 (0.71) | 0.051 (0.22) | - | 0.073 (0.072) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barr, E.; Comtois-Cabana, M.; Coope, A.; Coté, S.M.; Kobor, M.S.; Konwar, C.; Lupien, S.; Geoffroy, M.-C.; Boivin, M.; Provençal, N.; et al. Early-Life Adversity and Epigenetic Aging: Findings from a 17-Year Longitudinal Study. Biomolecules 2025, 15, 887. https://doi.org/10.3390/biom15060887
Barr E, Comtois-Cabana M, Coope A, Coté SM, Kobor MS, Konwar C, Lupien S, Geoffroy M-C, Boivin M, Provençal N, et al. Early-Life Adversity and Epigenetic Aging: Findings from a 17-Year Longitudinal Study. Biomolecules. 2025; 15(6):887. https://doi.org/10.3390/biom15060887
Chicago/Turabian StyleBarr, Emily, Maude Comtois-Cabana, Andressa Coope, Sylvana M. Coté, Michael S. Kobor, Chaini Konwar, Sonia Lupien, Marie-Claude Geoffroy, Michel Boivin, Nadine Provençal, and et al. 2025. "Early-Life Adversity and Epigenetic Aging: Findings from a 17-Year Longitudinal Study" Biomolecules 15, no. 6: 887. https://doi.org/10.3390/biom15060887
APA StyleBarr, E., Comtois-Cabana, M., Coope, A., Coté, S. M., Kobor, M. S., Konwar, C., Lupien, S., Geoffroy, M.-C., Boivin, M., Provençal, N., Catherine, N. L. A., Dennis, J. K., & Ouellet-Morin, I. (2025). Early-Life Adversity and Epigenetic Aging: Findings from a 17-Year Longitudinal Study. Biomolecules, 15(6), 887. https://doi.org/10.3390/biom15060887