1. Introduction
Plants represent one of the primary sources of raw materials for the food, pharmaceutical, and cosmetic industries, with many nutraceuticals and cosmeceuticals deriving their medicinal value from plant-based constituents [
1]. Among various plant-derived products, essential oil (EO), a complex mixture of hydrophobic volatile compounds originating from plant secondary metabolism, is a promising molecular library for drug discovery [
2]. This oil exhibits specific bioactivities, including antimicrobial, anti-inflammatory, and antioxidant properties [
3,
4]. Furthermore, the emergence of green consumerism has increased consumer demand for natural-extracted pharmaceutical agents with minimal environmental footprints. As natural products, EO has been widely utilized in traditional medicine to promote human health and treat diseases, owing to its intriguing biochemical profiles and high added value in environmental sustainability [
5].
In exploring the bioactivity of EO, its antioxidant capacity is often regarded as paramount, given the central role of oxidative stress in various pathological processes [
6]. Oxidative stress arises from either excessive production of free radicals or compromised antioxidant defense systems [
7,
8]. Free radicals, characterized by unpaired electrons, are highly reactive and readily interact with cellular components [
9]. While they participate in critical cellular processes, an overabundance of free radicals can inflict widespread damage on cellular structures [
1]. This damage is implicated in the pathogenesis of numerous chronic and degenerative diseases, including Alzheimer’s disease (AD), diabetes mellitus (DM), and cardiovascular disorders [
10]. Nevertheless, this detrimental process can be modulated through exogenous antioxidants derived from natural sources, such as plant EO and extracts, which offer a promising therapeutic avenue [
11].
AD is an age-related neurodegenerative disorder characterized by three hallmark features: the deposition of amyloid-β (Aβ) plaques, hyperphosphorylation of the cytoskeletal protein tau, and reduced levels of acetylcholine (ACh), leading to neuronal dysfunction [
12]. Among these, Aβ accumulation plays a pivotal role in driving oxidative stress [
13]. Concurrently, AD patients exhibit decreased levels of antioxidants, such as uric acid, vitamin C, and vitamin E, and reduced activity of antioxidant enzymes like superoxide dismutase and catalase [
14]. These findings underscore oxidative stress as a key contributor to AD pathogenesis, suggesting that antioxidant supplementation may offer therapeutic benefits [
15]. Furthermore, acetylcholinesterase (AChE), a serine protease that hydrolyzes ACh in the brain, contributes to AD progression by diminishing ACh levels and disrupting neuronal signaling [
16]. Notably, EO can cross the blood–brain barrier and modulate the central nervous system, potentially alleviating AD symptoms due to its small molecular size and lipophilic properties [
17]. Therefore, EO is a promising candidate for developing novel therapeutics targeting neurodegenerative diseases.
DM is a chronic disease characterized by elevated blood glucose levels due to insulin deficiency or impaired insulin function [
18]. Oxidative stress is widely recognized as a pivotal contributor to the pathogenesis of DM, primarily through mechanisms such as mitochondrial H
2O
2 production and NADPH oxidase activation, which disrupt insulin signaling and promote insulin resistance [
19]. Consequently, antioxidants demonstrate beneficial effects in DM treatment. In addition, the inhibition of α-glucosidase, a carbohydrate-hydrolyzing enzyme located on the intestinal brush border, represents a critical therapeutic strategy. By delaying the digestion of oligosaccharides into absorbable monosaccharides, α-glucosidase inhibitors effectively modulate postprandial glucose absorption, thereby mitigating glycemic fluctuations and slowing disease progression [
20]. Recent studies have identified EO derived from 20 plant species with significant potential for DM management, attributed to their dual antioxidant and α-glucosidase inhibitory activities [
21]. EO has already been utilized in therapeutic formulations and has demonstrated a favorable safety profile with no significant adverse effects reported [
22]. Consequently, EO may offer a promising avenue for discovering and developing novel antidiabetic agents.
β-Lactam antibiotics are widely regarded as one of the most effective antibacterial agents due to their broad spectrum of activity, favorable pharmacokinetics, and safety profile [
23]. However, the extensive use of β-lactams has led to the emergence and dissemination of resistance, mediated through diverse mechanisms, including target modification, downregulation of porins required for β-lactam entry, overexpression of efflux systems, and enzymatic modification or degradation [
24]. Among these, enzyme-mediated resistance, driven by β-lactamase activity, poses a significant threat. β-Lactamases, produced by both Gram-positive and Gram-negative bacteria, hydrolyze β-lactam antibiotics, rendering one of the most significant threats to antibacterial efficacy [
24,
25]. In our previous work,
Spermacoce alata EO exhibited potent β-lactamase inhibitory activity [
26]. Therefore, developing EO-based adjunctive therapies may represent a promising strategy to counteract β-lactam resistance and enhance the efficacy of existing antibiotics [
27].
Artemisia, one of the largest and most widely distributed genera within the Asteraceae family, comprises over 500 species predominantly found in temperate regions of Europe, Asia, and North America [
28].
Artemisia species are renowned for their diverse bioactivities, including antimicrobial, anticancer, anti-inflammatory, antioxidant, and antipyretic properties, making them highly valued in ethnopharmacology [
29,
30]. The 2015 Nobel Prize in Medicine, awarded for the discovery of artemisinin, a potent antimalarial sesquiterpene lactone isolated from
Artemisia annua, renewed scientific focus on these plants, including
Artemisia schmidtiana Maxim. [
30].
A. schmidtiana, a perennial herbaceous plant native to alpine or rocky environments, is characterized by its slender, silver–white hairy stems and leaves, reaching heights of approximately 10 cm, and produces small white flowers in July and August. It has historically been prized for its medicinal properties and distinctive aromatic qualities, and it is commonly cultivated as an ornamental plant in gardens and pots, favored by horticultural enthusiasts for its unique foliage. Beyond its traditional uses, thiophene acetylenes were isolated from
A. schmidtiana in 1986 [
31]. Thiophenes, a class of secondary metabolites, exhibit a broad spectrum of biological activities, including antimicrobial, antiviral, HIV-1 protease inhibitory, and anticancer effects, highlighting the plant’s potential for further pharmaceutical development [
32].
The previous literature has documented the broad bioactivities and high medicinal value of A. schmidtiana. However, the chemical composition and biological activities of its EO remain unexplored. To address this gap, we investigated the chemical profile, antioxidant activity, and inhibitory effects of A. schmidtiana EO on AChE, α-glucosidase, and β-lactamase. Additionally, molecular docking was employed to elucidate the potential mechanisms underlying its enzyme-inhibitory properties.
2. Materials and Methods
2.1. Plant Material
Plant samples of aerial parts at the vegetative stage were collected from a commercial nursery in Shanghai City, China (31.2304° N, 121.4737° E) in September 2023. The species was identified as A. schmidtiana by Pro. Zhao Hong, through morphological characteristics. The voucher specimen is stored at the Center for Bioscience Analysis and Testing, Shandong University, Weihai, China, with registration number EO2325. The plant samples were refrigerated at −18 °C until EO extraction.
2.2. EO Extraction
Fresh leaves and stems (140 g) of the plant material were finely crushed and placed into a 5 L round-bottom flask, and then 3 L of ultrapure water. Hydrodistillation was carried out for approximately 4 h using a Clevenger-type apparatus to isolate the EO from the plant material. The EO was then separated from the aqueous layer using diethyl ether. Subsequently, the extracted EO was dried using sodium sulfate and concentrated using a Termovap sample concentrator (MD200-1, Shanghai Huyi Technology Co., Ltd., Shanghai, China). The obtained EO was stored at a low temperature (4 °C) for further analysis.
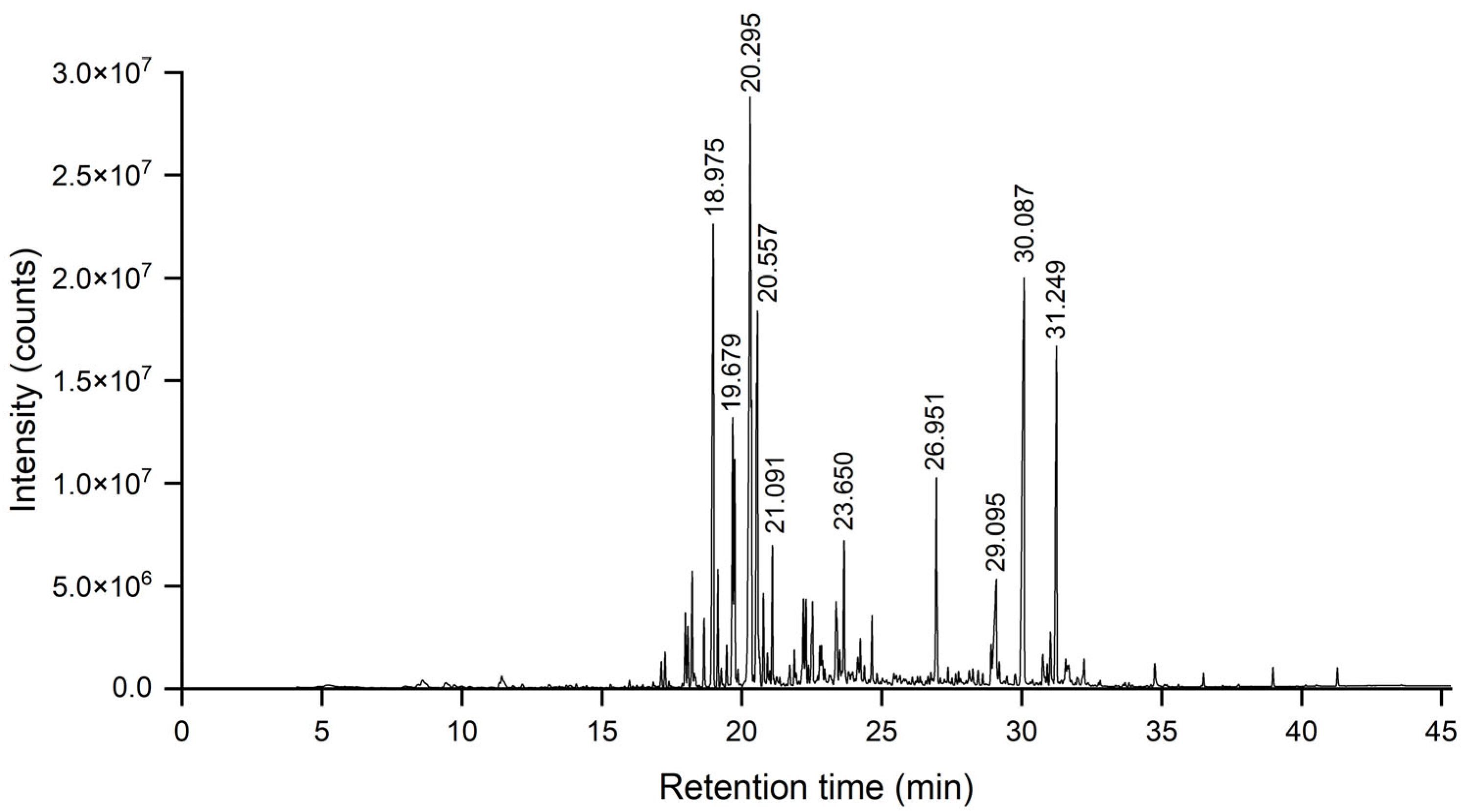
2.3. GC-MS and GC-FID Analyses
The composition and relative content of the EO were analyzed using gas chromatography–mass spectrometry (GC-MS) and gas chromatography with flame ionization detection (GC-FID). GC-MS analysis was conducted on an Agilent 7890–5975C system (Santa Clara, CA, USA) with an HP-5MS fused silica capillary column (30 m × 0.25 mm, 0.25 μm film thickness, Agilent, Santa Clara, CA, USA). The injector and interface temperatures were maintained at 260 °C and 280 °C, respectively. The oven temperature program began at 50 °C for 4 min, followed by a gradient increase to 280 °C at 6 °C/min, and held for 3 min. Ultra-pure helium (99.999%) served as the carrier gas at a 1.0 mL/min flow rate. The mass spectrometer operated in electron ionization (EI) mode at 70 eV, with a scan range of 25–500 amu and a quadrupole temperature of 150 °C. Samples were prepared as a 1% (w/v) solution in dichloromethane, and 0.3 µL was injected in splitless mode. A PerkinElmer Clarus 500 system (Shelton, CT, USA) with an HP-5 column (30 m × 0.25 mm, 0.25 μm film thickness, Agilent, Santa Clara, CA, USA) was used for GC-FID analysis. The injector and detector temperatures were set at 260 °C and 305 °C, respectively. The oven temperature followed the same program as GC-MS, with nitrogen as the carrier gas at 1.1 mL/min.
Compound identification was achieved by comparing mass spectra and retention indices (RI) with the NIST20 library and published data. RIs were calculated using GC-MS data from a series of n-alkanes (C8–C30) analyzed under identical conditions.
2.4. Antioxidant Activities Evaluation
2.4.1. DPPH Method
The experimental protocol was slightly modified from previously established methods [
33]. 6-Hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) was used as the positive control. Trolox and the EO stock solution concentrations were prepared in ethanol at 0.5 mg/mL and 50 mg/mL, respectively. Aliquots of 50 μL from either the Trolox or gradient-diluted EO solutions were transferred into a 96-well microplate, followed by 200 μL of 0.17 mmol/L 2,2-diphenyl-1-picrylhydrazyl (DPPH) solution. The microplate was shielded from light and incubated at 25 °C for 30 min. Absorbance was subsequently measured at 516 nm using an Epoch microplate spectrophotometer (BioTek Instruments, Minneapolis, MN, USA). The DPPH radical scavenging capacity (RSC%) was determined using Equation (1):
where A
Sample is the absorbance of the tested sample at different concentrations, A
Control is the absorbance of the control (ethanolic DPPH solution), and A
Sample Blank is the absorbance of the ethanol sample without DPPH.
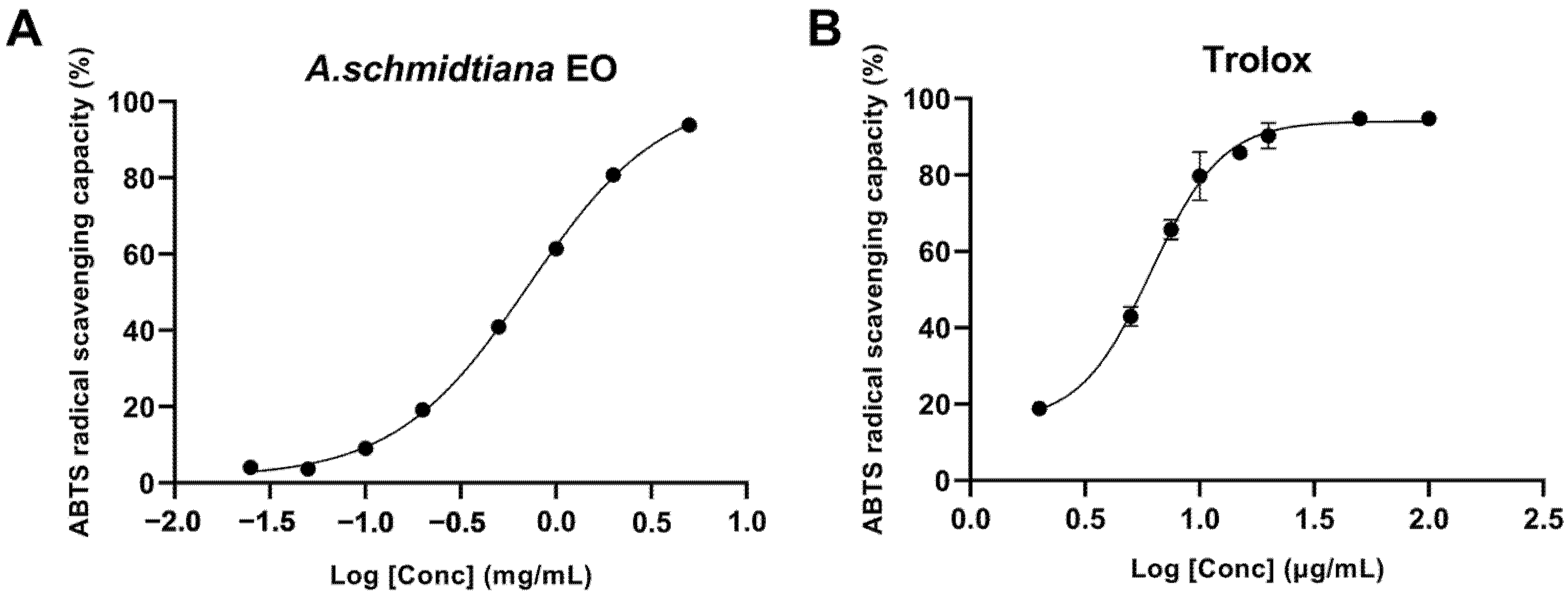
2.4.2. ABTS Method
The experimental protocol was modified slightly based on established methodologies from prior studies [
26]. The ABTS
•+ radical solution was prepared by combining 7.4 mmol/L 2,2-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) with 2.6 mmol/L potassium persulfate. The mixture was incubated in the dark at room temperature for 12 h to ensure complete radical generation. Subsequently, 200 μL of the diluted ABTS
•+ solution was mixed with 50 μL of gradient-diluted ethanolic EO solutions in a 96-well plate. Absorbance was measured at 734 nm six minutes after initiating the reaction. Ethanol served as the blank and dilution solvent, while 0.5 mg/mL Trolox was used as the positive control. The ABTS radical scavenging capacity (RSC%) was calculated to evaluate antioxidant activity using Equation (2):
where A
0 and A are the absorbance of 200 μL diluted ABTS
•+ solution mixed with 50 μL ethanol and 50 μL sample solution, respectively, at 734 nm. IC
50 was then calculated.
2.4.3. Ferric-Reducing Antioxidant Power (FRAP) Method
The assay was performed following established protocols with minor adaptations. Three stock solutions—(i) acetate buffer (pH 3.6), (ii) 10 mM 2,4,6-tripyridyl-s-triazine (TPTZ), and (iii) 20 mM Fe3+ solution—were mixed in a 10:1:1 ratio and diluted 50-fold with ethanol to prepare the FRAP working solution. A standard curve was constructed using Trolox. EO samples were serially diluted to 5000, 2500, 1000, 500, 250, 100, 50, and 25 μg/mL concentrations. The EO samples were combined with the FRAP working solution and incubated at 37 °C in the dark. Absorbance was measured at 593 nm, and the antioxidant capacity was expressed as Trolox equivalent antioxidant concentration (TEAC) by referencing the standard curve. All experiments were conducted in triplicate, and results were reported as mean values.
2.5. Anti-AChE Activity Test
This assay was conducted using the previously described method with minor modifications [
34]. In total, 145 μL of 0.1 mM phosphate buffered saline (PBS) (pH 8.0), 20 μL of ethanolic EO solution, and 15 μL of AChE solution (0.28 U/mL) were combined in a microplate, with galantamine as the positive control. The mixture was incubated at 4.0 °C for 20 min. Subsequently, 10 μL of 15 mM acetylthiocholine iodide (ATCI) and 10 μL of 2 mM 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) were added, followed by homogenization for 60 s. Absorbance was measured at 412 nm at 60 s intervals for 6 min using a microplate reader (Epoch, BioTek Instruments, Minneapolis, MN, USA). The acetylcholinesterase inhibition rate was calculated based on the absorbance data. The AChE inhibition rate is calculated according to Formula (3):
where K
E is the initial reaction rate of the enzyme without the inhibition, while K
S is the initial reaction rate of the inhibited enzyme.
2.6. Anti-α-Glucosidase Capacity Test
The test was conducted according to previous research with minor modifications [
7]. In total, 20 μL of ethanolic EO solution, 80 μL of 100 mM PBS (pH 6.8), and 40 μL of α-glucosidase solution (0.25 U/mL) were combined in a microplate, with acarbose serving as the control. The mixture was incubated at 30 °C for 10 min. Subsequently, 20 μL of 3.0 mg/mL 4-nitrophenyl-β-D-glucopyranoside (pNPG) was added, followed by homogenization for 60 s and further incubation at 30 °C for 4 min. Absorbance was measured at 410 nm at 60 s intervals for 6 min. The α-glucosidase inhibition rate is expressed using Equation (3). The IC
50 value was calculated using nonlinear regression.
2.7. Test for β-Lactamase Inhibitory Effect
The β-lactamase inhibitory effect was assessed using a previously described method with some modifications [
35]. In total, 20 μL of ethanolic EO solution, 30 μL of 50 mM PBS (pH 7.0), and 100 μL of β-lactamase solution (1000 U/mL) were combined in a microplate, with clavulanate potassium serving as the positive control. The mixture was incubated at 30 °C for 10 min. Subsequently, 50 μL of 0.1 mg/mL nitrocefin was added, followed by an additional 10 min incubation at 30 °C. Absorbance was measured at 489 nm. The β-lactamase inhibition rate was calculated using the following Formula (4):
where A
S is the absorbance of the EO-containing sample, A
sb is the absorbance of the sample blank reaction, A
E is the absorbance of the reaction in which the enzyme was not inhibited, and A
B is the absorbance of the blank response. IC
50 was assessed using nonlinear regression.
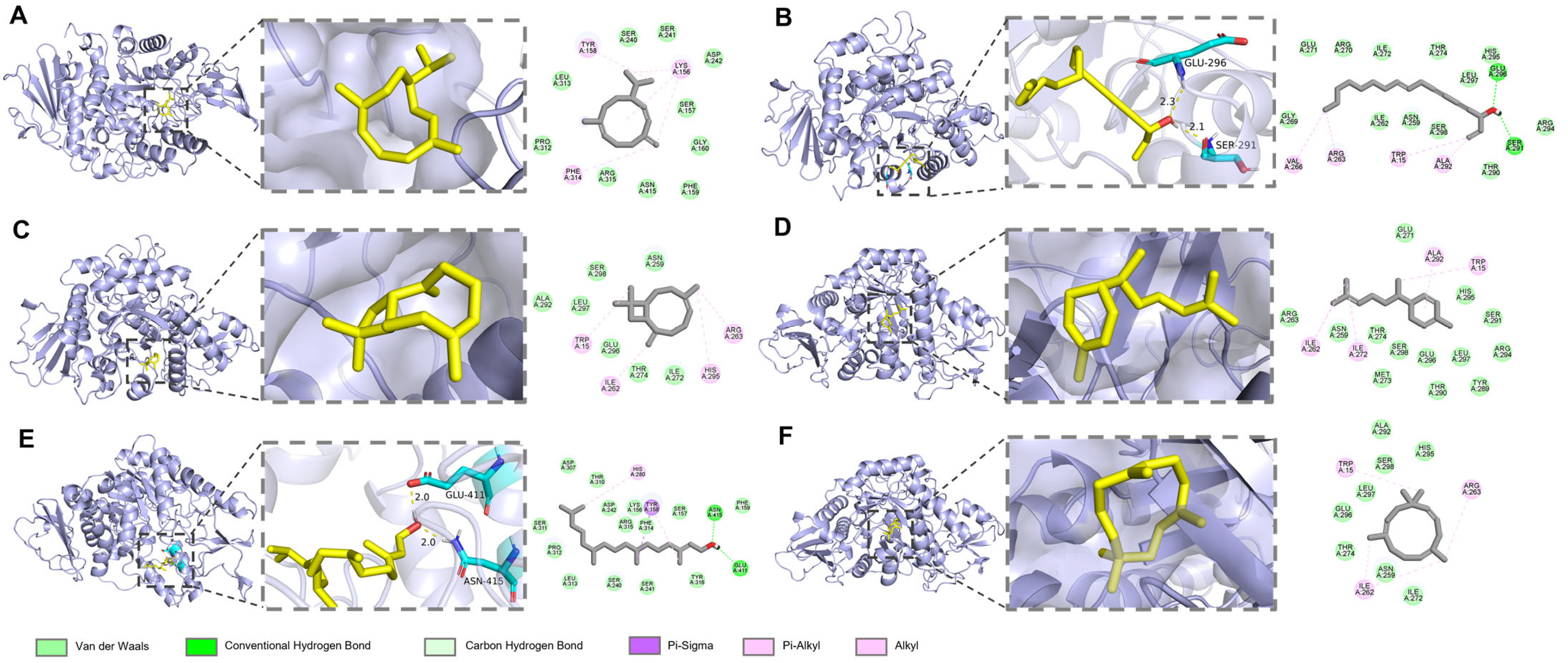
2.8. Molecular Docking
In the molecular docking experiments, complexes of AChE (
Tetronarce californica, PDB code: 1EA5), α-glucosidase (
Saccharomyces cerevisiae, PDB code: 3AJ7), and β-lactamase (
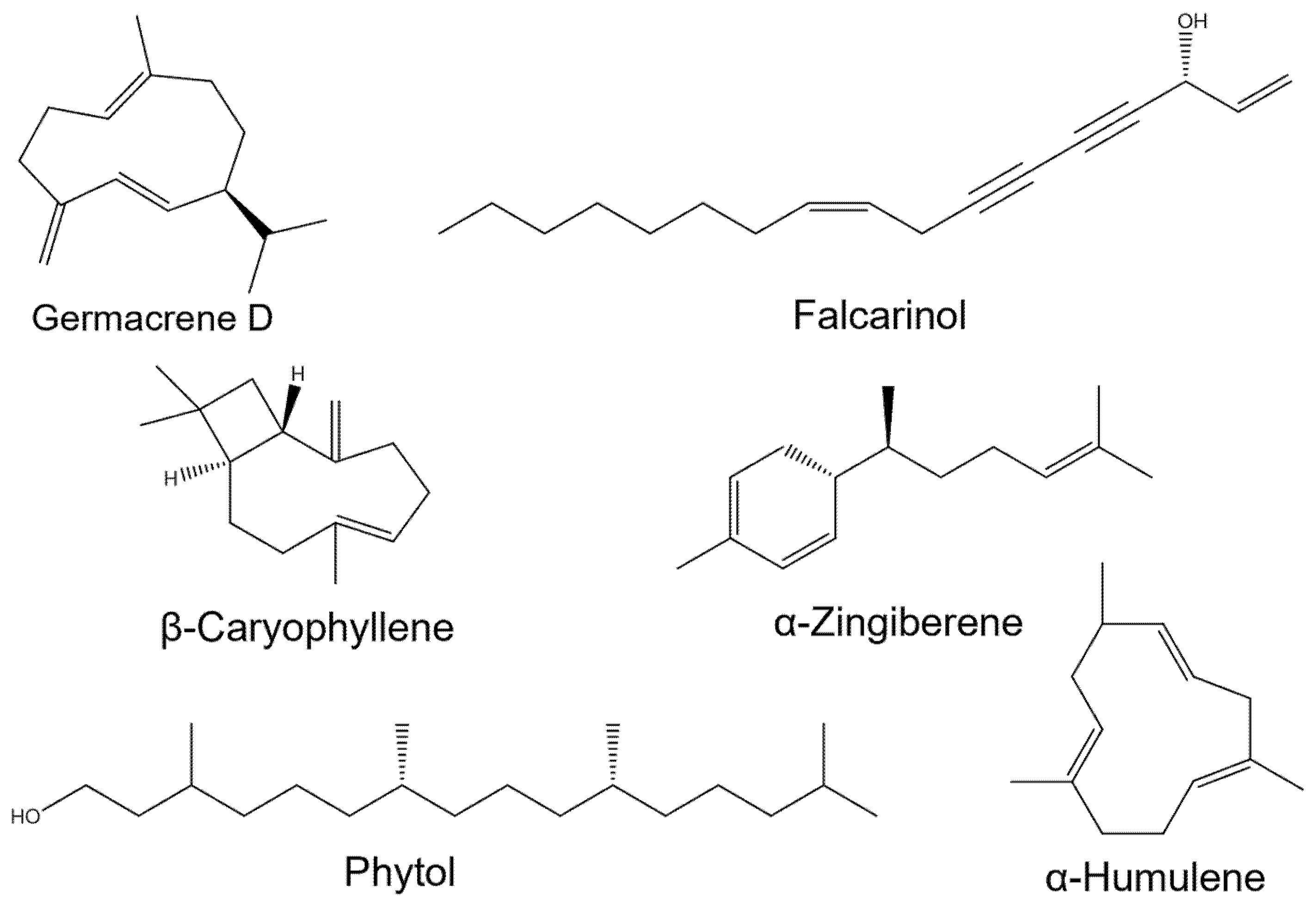
Enterobacter cloacae, PDB code: 7TI1) were collected from the PDB database. The receptor structures were prepared by removing bound water molecules and ligands using PyMol v2.2.0, and hydrogen atoms were subsequently added to ensure proper protonation states. The 3D structures of the major components of
A. schmidtiana EO, used as ligands for interaction with the enzyme receptors, were retrieved from the CAS SciFinder Discovery Platform (
https://scifinder-n.cas.org/, accessed on 10 March 2025) with energy minimized using Chem3D.
Autodock v4.2.6 was employed to perform semi-flexible molecular docking. Each ligand was subjected to at least 50 runs. Results were analyzed using the Lamarckian genetic algorithm. Binding energies and interactions between the ligands and proteins were evaluated during the docking process. Discovery Studio visualizer and PyMol v2.2.0 were used to visualize the docking results.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}