
Senior–Loken Syndrome: Ocular Perspectives on Genetics, Pathogenesis, and Management

Abstract
1. Introduction
2. Clinical Features of SLSN
2.1. Retina
2.2. Kidney
2.3. Others
3. Primary Cilium
3.1. General Structure and Function
3.2. Primary Cilia in the Eye
3.3. Primary Cilium in Retinal Photoreceptors
4. Pathogenesis Mechanisms
4.1. The SLSN Genes
4.1.1. NPHP1
4.1.2. NPHP3
4.1.3. NPHP4
4.1.4. IQCB1/NPHP5
4.1.5. CEP290/NPHP6
4.1.6. SDCCAG8/SLSN7/NPHP10
4.1.7. WDR19
4.1.8. TRAF3IP1
4.2. The SLSN Interactome
4.2.1. NPHP1-4-8 Complex
4.2.2. NPHP5-6 Complex
5. Genetic Basis
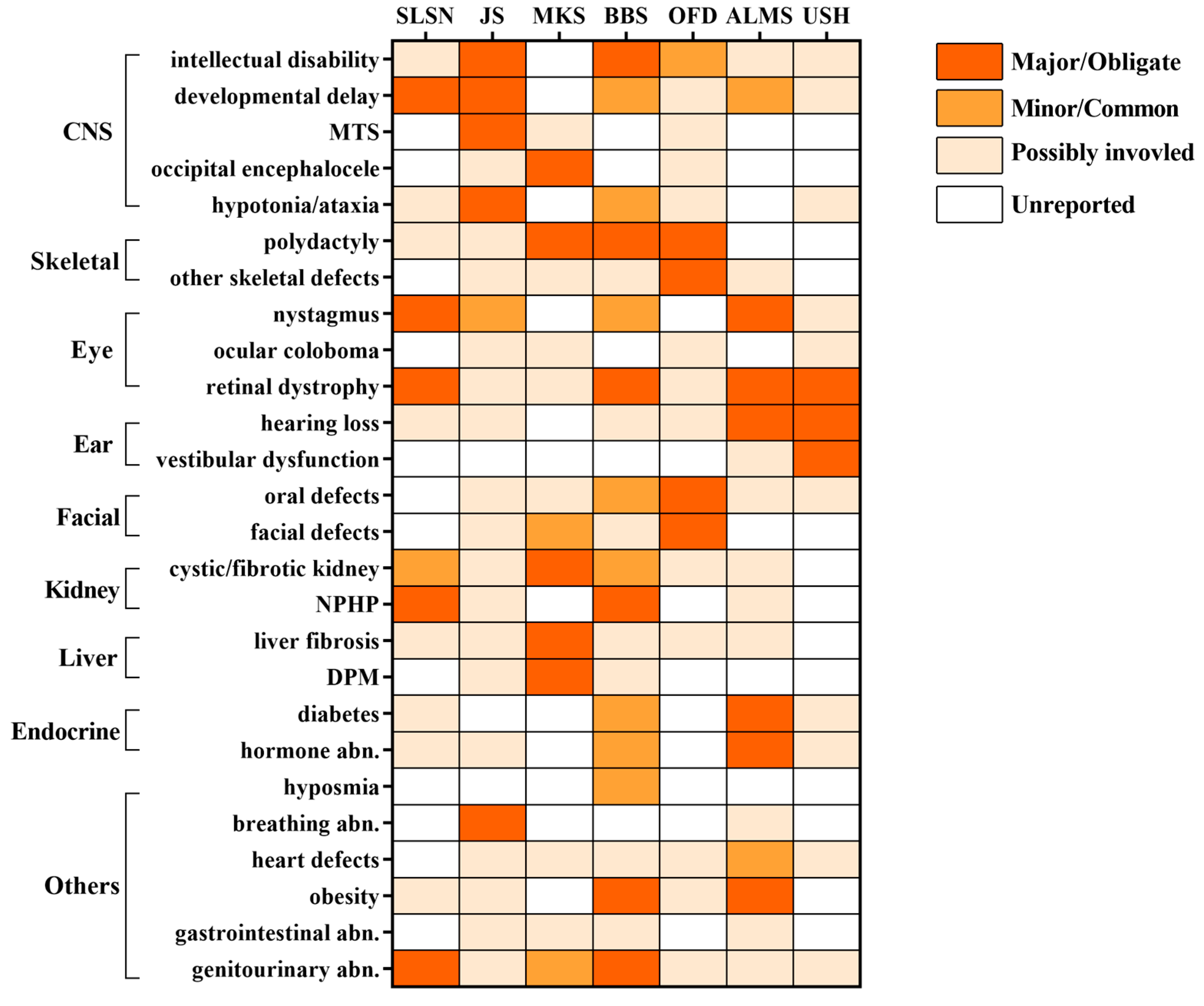
5.1. Shared Phenotypes Among Ciliopathies
5.2. Genotype–Phenotype Correlations
5.2.1. Locus Heterogeneity
5.2.2. Allelic Heterogeneity
5.2.3. Genetic Modifiers and Epistatic Effects
5.2.4. Oligogenic Inheritance
6. Treatments
6.1. Gene Therapy
6.2. Drug Discovery
6.3. Other Discoveries
7. Animal Models
8. Discussion
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| SLSN | Senior–Loken syndrome |
| LCA | Leber’s congenital amaurosis |
| NPHP | Nephronophthisis |
| ESRD | end-stage renal disease |
| TZ | transition zone |
| RPE | retinal pigment epithelium |
| OS | outer segment |
| IS | inner segment |
| OMIM | Online Mendelian Inheritance |
| MSS | Mainzer–Saldino syndrome |
| SRTD | Short-rib thoracic dysplasia |
| ATD | Asphyxiating thoracic dystrophy |
| SRPS | Short-rib polydactyly syndrome |
| IFT | Intraflagellar ciliary transport |
| RPGR | Retinitis GTPase Regulator |
| RPGRIP1 | RPGR-Interacting Protein 1 |
| IQCB1 | IQ calmodulin-binding motif-containing protein 1 |
| JS | Joubert Syndrome |
| MKS | Meckel syndrome |
| BBS | Bardet–Biedl syndrome |
| OFD | Oral–facial–digital syndrome |
| ALMS | Alström Syndrome |
| USH | Usher syndrome |
| CNS | Central Nervous System |
| MTS | Molar Tooth Sign |
| DPM | Ductal plate malformation |
| AAV | Adeno-associated virus |
References
- Hemachandar, R. Senior- Loken Syndrome—A Ciliopathy. J. Clin. Diagn. Res. 2014, 8, MD04–MD05. [Google Scholar] [CrossRef]
- Otto, E.A.; Loeys, B.; Khanna, H.; Hellemans, J.; Sudbrak, R.; Fan, S.; Muerb, U.; O’Toole, J.F.; Helou, J.; Attanasio, M.; et al. Nephrocystin-5, a Ciliary IQ Domain Protein, Is Mutated in Senior-Loken Syndrome and Interacts with RPGR and Calmodulin. Nat. Genet. 2005, 37, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Baehr, W.; Hanke-Gogokhia, C.; Sharif, A.; Reed, M.; Dahl, T.; Frederick, J.M.; Ying, G. Insights into Photoreceptor Ciliogenesis Revealed by Animal Models. Prog. Retin. Eye Res. 2019, 71, 26–56. [Google Scholar] [CrossRef]
- Ran, J.; Zhou, J. Targeting the Photoreceptor Cilium for the Treatment of Retinal Diseases. Acta Pharmacol. Sin. 2020, 41, 1410–1415. [Google Scholar] [CrossRef]
- Huang, C.-H.; Yang, C.-M.; Yang, C.-H.; Hou, Y.-C.; Chen, T.-C. Leber’s Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes 2021, 12, 1261. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Zhou, W. Nephronophthisis-Associated Ciliopathies. J. Am. Soc. Nephrol. 2007, 18, 1855–1871. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber Congenital Amaurosis/Early-Onset Severe Retinal Dystrophy: Clinical Features, Molecular Genetics and Therapeutic Interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Georgiou, M.; Robson, A.G.; Fujinami, K.; de Guimarães, T.A.C.; Fujinami-Yokokawa, Y.; Daich Varela, M.; Pontikos, N.; Kalitzeos, A.; Mahroo, O.A.; Webster, A.R.; et al. Phenotyping and Genotyping Inherited Retinal Diseases: Molecular Genetics, Clinical and Imaging Features, and Therapeutics of Macular Dystrophies, Cone and Cone-Rod Dystrophies, Rod-Cone Dystrophies, Leber Congenital Amaurosis, and Cone Dysfunction Syndromes. Prog. Retin. Eye Res. 2024, 100, 101244. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef]
- Stokman, M.F.; Saunier, S.; Benmerah, A. Renal Ciliopathies: Sorting Out Therapeutic Approaches for Nephronophthisis. Front. Cell Dev. Biol. 2021, 9, 653138. [Google Scholar] [CrossRef] [PubMed]
- Simms, R.J.; Eley, L.; Sayer, J.A. Nephronophthisis. Eur. J. Hum. Genet. 2009, 17, 406–416. [Google Scholar] [CrossRef]
- Salomon, R.; Saunier, S.; Niaudet, P. Nephronophthisis. Pediatr. Nephrol. 2008, 24, 2333. [Google Scholar] [CrossRef] [PubMed]
- Othman, M.; Rashed, A.; Bakr, A. The Senior-Loken Syndrome: Two Cases from the State of Qatar. J. Clin. Diagn. Res. 2012, 6, 1411–1413. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, Y.; Liu, Y.; Ding, J.; Xie, Y.; Li, N. A Novel Pathogenic Variant of CEP164 in an Infant with Senior-Loken Syndrome. Pediatr. Investig. 2023, 7, 140–143. [Google Scholar] [CrossRef]
- Mainzer, F.; Saldino, R.M.; Ozonoff, M.B.; Minagi, H. Familial Nephropathy Associatdd with Retinitis Pigmentosa, Cerebellar Ataxia and Skeletal Abnormalities. Am. J. Med. 1970, 49, 556–562. [Google Scholar] [CrossRef]
- Tay, S.A.; Vincent, A.L. Senior-Løken Syndrome and Intracranial Hypertension. Ophthalmic Genet. 2020, 41, 354–357. [Google Scholar] [CrossRef]
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the Cell’s Antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular Signalling by Primary Cilia in Development, Organ Function and Disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Reddy Palicharla, V.; Mukhopadhyay, S. Molecular and Structural Perspectives on Protein Trafficking to the Primary Cilium Membrane. Biochem. Soc. Trans. 2024, 52, 1473–1487. [Google Scholar] [CrossRef]
- Bai, Y.; Wei, C.; Li, P.; Sun, X.; Cai, G.; Chen, X.; Hong, Q. Primary Cilium in Kidney Development, Function and Disease. Front. Endocrinol. 2022, 13, 952055. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.T.; Chong, W.M.; Wang, W.-J.; Mazo, G.; Tanos, B.; Chen, Z.; Tran, T.M.N.; Chen, Y.-D.; Weng, R.R.; Huang, C.-E.; et al. Super-Resolution Architecture of Mammalian Centriole Distal Appendages Reveals Distinct Blade and Matrix Functional Components. Nat. Commun. 2018, 9, 2023. [Google Scholar] [CrossRef]
- Marshall, W.F. Basal Bodies Platforms for Building Cilia. Curr. Top. Dev. Biol. 2008, 85, 1–22. [Google Scholar] [CrossRef]
- Sorokin, S. Centrioles and the Formation of Rudimentary Cilia by Fibroblasts and Smooth Muscle Cells. J. Cell Biol. 1962, 15, 363–377. [Google Scholar] [CrossRef]
- Sorokin, S.P. Reconstructions of Centriole Formation and Ciliogenesis in Mammalian Lungs. J. Cell Sci. 1968, 3, 207–230. [Google Scholar] [CrossRef]
- Wang, L.; Wen, X.; Wang, Z.; Lin, Z.; Li, C.; Zhou, H.; Yu, H.; Li, Y.; Cheng, Y.; Chen, Y.; et al. Ciliary Transition Zone Proteins Coordinate Ciliary Protein Composition and Ectosome Shedding. Nat. Commun. 2022, 13, 3997. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.R.; Reiter, J.F. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Cold Spring Harb. Perspect. Biol. 2017, 9, a028134. [Google Scholar] [CrossRef]
- May-Simera, H.; Nagel-Wolfrum, K.; Wolfrum, U. Cilia—The Sensory Antennae in the Eye. Prog. Retin. Eye Res. 2017, 60, 144–180. [Google Scholar] [CrossRef]
- Blitzer, A.L.; Panagis, L.; Gusella, G.L.; Danias, J.; Mlodzik, M.; Iomini, C. Primary Cilia Dynamics Instruct Tissue Patterning and Repair of Corneal Endothelium. Proc. Natl. Acad. Sci. USA 2011, 108, 2819–2824. [Google Scholar] [CrossRef]
- Chrystal, P.W.; Lambacher, N.J.; Doucette, L.P.; Bellingham, J.; Schiff, E.R.; Noel, N.C.L.; Li, C.; Tsiropoulou, S.; Casey, G.A.; Zhai, Y.; et al. The Inner Junction Protein CFAP20 Functions in Motile and Non-Motile Cilia and Is Critical for Vision. Nat. Commun. 2022, 13, 6595. [Google Scholar] [CrossRef]
- Sun, C.; Zhou, J.; Meng, X. Primary Cilia in Retinal Pigment Epithelium Development and Diseases. J. Cell. Mol. Med. 2021, 25, 9084–9088. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pacwa, A.; Bresciani, G.; Swierczynska, M.; Dorecka, M.; Smedowski, A. Retinal Primary Cilia and Their Dysfunction in Retinal Neurodegenerative Diseases: Beyond Ciliopathies. Mol. Med. 2024, 30, 109. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Kelley, R.A.; Li, T.; Swaroop, A. Primary Cilia Biogenesis and Associated Retinal Ciliopathies. Semin. Cell Dev. Biol. 2021, 110, 70–88. [Google Scholar] [CrossRef] [PubMed]
- Ghossoub, R.; Molla-Herman, A.; Bastin, P.; Benmerah, A. The Ciliary Pocket: A Once-Forgotten Membrane Domain at the Base of Cilia. Biol. Cell 2011, 103, 131–144. [Google Scholar] [CrossRef]
- Chandra, B.; Tung, M.L.; Hsu, Y.; Scheetz, T.; Sheffield, V.C. Retinal Ciliopathies through the Lens of Bardet-Biedl Syndrome: Past, Present and Future. Prog. Retin. Eye Res. 2022, 89, 101035. [Google Scholar] [CrossRef]
- Løken, A.C.; Hanssen, O.; Halvorsen, S.; Jølster, N.J. Hereditary Renal Dysplasia and Blindness. Acta Paediatr. 1961, 50, 177–184. [Google Scholar] [CrossRef]
- Senior, B.; Friedmann, A.I.; Braudo, J.L. Juvenile Familial Nephropathy with Tapetoretinal Degeneration. A New Oculorenal Dystrophy. Am. J. Ophthalmol. 1961, 52, 625–633. [Google Scholar] [CrossRef]
- Home—OMIM. Available online: https://www.omim.org/ (accessed on 26 April 2025).
- UniProt. Available online: https://www.uniprot.org (accessed on 26 April 2025).
- Gene. Available online: https://www.ncbi.nlm.nih.gov/datasets/gene/ (accessed on 26 April 2025).
- Jauregui, A.R.; Nguyen, K.C.Q.; Hall, D.H.; Barr, M.M. The Caenorhabditis Elegans Nephrocystins Act as Global Modifiers of Cilium Structure. J. Cell Biol. 2008, 180, 973–988. [Google Scholar] [CrossRef]
- Caridi, G.; Dagnino, M.; Rossi, A.; Valente, E.M.; Bertini, E.; Fazzi, E.; Emma, F.; Murer, L.; Verrina, E.; Ghiggeri, G.M. Nephronophthisis Type 1 Deletion Syndrome with Neurological Symptoms: Prevalence and Significance of the Association. Kidney Int. 2006, 70, 1342–1347. [Google Scholar] [CrossRef]
- Lindstrand, A.; Davis, E.E.; Carvalho, C.M.B.; Pehlivan, D.; Willer, J.R.; Tsai, I.-C.; Ramanathan, S.; Zuppan, C.; Sabo, A.; Muzny, D.; et al. Recurrent CNVs and SNVs at the NPHP1 Locus Contribute Pathogenic Alleles to Bardet-Biedl Syndrome. Am. J. Hum. Genet. 2014, 94, 745–754. [Google Scholar] [CrossRef]
- Yahalom, C.; Volovelsky, O.; Macarov, M.; Altalbishi, A.; Alsweiti, Y.; Schneider, N.; Hanany, M.; Khan, M.I.; Cremers, F.P.M.; Anteby, I.; et al. SENIOR-LØKEN SYNDROME: A Case Series and Review of the Renoretinal Phenotype and Advances of Molecular Diagnosis. Retina 2021, 41, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Roig, J. NEK8, a NIMA-Family Protein Kinase at the Core of the Ciliary INV Complex. Cell Commun. Signal. 2025, 23, 170. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Fliegauf, M.; Brüchle, N.O.; Frank, V.; Olbrich, H.; Kirschner, J.; Schermer, B.; Schmedding, I.; Kispert, A.; Kränzlin, B.; et al. Loss of Nephrocystin-3 Function Can Cause Embryonic Lethality, Meckel-Gruber-like Syndrome, Situs Inversus, and Renal-Hepatic-Pancreatic Dysplasia. Am. J. Hum. Genet. 2008, 82, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Lin, H.; Li, M.; Wang, H.; Liu, T.; Hu, M.; Chen, H.; Tong, H.; Sun, L. Clinical and Pathological Features and Varied Mutational Spectra of Pathogenic Genes in 55 Chinese Patients with Nephronophthisis. Clin. Chim. Acta 2020, 506, 136–144. [Google Scholar] [CrossRef]
- Omran, H.; Sasmaz, G.; Häffner, K.; Volz, A.; Olbrich, H.; Melkaoui, R.; Otto, E.; Wienker, T.F.; Korinthenberg, R.; Brandis, M.; et al. Identification of a Gene Locus for Senior-Løken Syndrome in the Region of the Nephronophthisis Type 3 Gene. J. Am. Soc. Nephrol. 2002, 13, 75–79. [Google Scholar] [CrossRef]
- Tang, X.; Liu, C.; Liu, X.; Chen, J.; Fan, X.; Liu, J.; Ma, D.; Cao, G.; Chen, Z.; Xu, D.; et al. Phenotype and Genotype Spectra of a Chinese Cohort with Nephronophthisis-Related Ciliopathy. J. Med. Genet. 2022, 59, 147–154. [Google Scholar] [CrossRef]
- Penchev, V.; Boueva, A.; Kamenarova, K.; Roussinov, D.; Tzveova, R.; Ivanova, M.; Dimitrova, V.; Kremensky, I.; Mitev, V.; Kaneva, R.; et al. A Familial Case of Severe Infantile Nephronophthisis Explained by Oligogenic Inheritance. Eur. J. Med. Genet. 2017, 60, 321–325. [Google Scholar] [CrossRef]
- Arts, H.H.; Doherty, D.; van Beersum, S.E.C.; Parisi, M.A.; Letteboer, S.J.F.; Gorden, N.T.; Peters, T.A.; Märker, T.; Voesenek, K.; Kartono, A.; et al. Mutations in the Gene Encoding the Basal Body Protein RPGRIP1L, a Nephrocystin-4 Interactor, Cause Joubert Syndrome. Nat. Genet. 2007, 39, 882–888. [Google Scholar] [CrossRef]
- Kruczek, K.; Qu, Z.; Welby, E.; Shimada, H.; Hiriyanna, S.; English, M.A.; Zein, W.M.; Brooks, B.P.; Swaroop, A. In Vitro Modeling and Rescue of Ciliopathy Associated with IQCB1/NPHP5 Mutations Using Patient-Derived Cells. Stem Cell Rep. 2022, 17, 2172–2186. [Google Scholar] [CrossRef]
- Tory, K.; Lacoste, T.; Burglen, L.; Morinière, V.; Boddaert, N.; Macher, M.-A.; Llanas, B.; Nivet, H.; Bensman, A.; Niaudet, P.; et al. High NPHP1 and NPHP6 Mutation Rate in Patients with Joubert Syndrome and Nephronophthisis: Potential Epistatic Effect of NPHP6 and AHI1 Mutations in Patients with NPHP1 Mutations. J. Am. Soc. Nephrol. 2007, 18, 1566–1575. [Google Scholar] [CrossRef]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; De Baere, E. CEP290, a Gene with Many Faces: Mutation Overview and Presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.-J.F.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Airik, R.; Slaats, G.G.; Guo, Z.; Weiss, A.-C.; Khan, N.; Ghosh, A.; Hurd, T.W.; Bekker-Jensen, S.; Schrøder, J.M.; Elledge, S.J.; et al. Renal-Retinal Ciliopathy Gene Sdccag8 Regulates DNA Damage Response Signaling. J. Am. Soc. Nephrol. 2014, 25, 2573–2583. [Google Scholar] [CrossRef] [PubMed]
- Insolera, R.; Shao, W.; Airik, R.; Hildebrandt, F.; Shi, S.-H. SDCCAG8 Regulates Pericentriolar Material Recruitment and Neuronal Migration in the Developing Cortex. Neuron 2014, 83, 805–822. [Google Scholar] [CrossRef]
- Bredrup, C.; Saunier, S.; Oud, M.M.; Fiskerstrand, T.; Hoischen, A.; Brackman, D.; Leh, S.M.; Midtbø, M.; Filhol, E.; Bole-Feysot, C.; et al. Ciliopathies with Skeletal Anomalies and Renal Insufficiency Due to Mutations in the IFT-A Gene WDR19. Am. J. Hum. Genet. 2011, 89, 634–643. [Google Scholar] [CrossRef]
- Tang, X.; Yi, S.; Qin, Z.; Yi, S.; Chen, J.; Yang, Q.; Li, S.; Luo, J. Compound Heterozygous WDR19 Variants Associated with Nephronophthisis, Caroli Disease, Refractory Epilepsy and Congenital Bilateral Central Blindness: Case Report. Heliyon 2024, 10, e23257. [Google Scholar] [CrossRef]
- Coussa, R.G.; Otto, E.A.; Gee, H.Y.; Arthurs, P.; Ren, H.; Lopez, I.; Keser, V.; Fu, Q.; Faingold, R.; Khan, A.; et al. WDR19: An Ancient, Retrograde, Intraflagellar Ciliary Protein Is Mutated in Autosomal Recessive Retinitis Pigmentosa and in Senior-Loken Syndrome. Clin. Genet. 2013, 84, 150–159. [Google Scholar] [CrossRef]
- Liu, L.; Huang, Y.; Fang, F.; Zhou, H.; Liu, X. A Case Report of Intrahepatic Bile Duct Dilatation Caused by WDR19 Gene Mutation and Presented as Caroli Syndrome. Transl. Pediatr. 2024, 13, 682–689. [Google Scholar] [CrossRef]
- Bizet, A.A.; Becker-Heck, A.; Ryan, R.; Weber, K.; Filhol, E.; Krug, P.; Halbritter, J.; Delous, M.; Lasbennes, M.-C.; Linghu, B.; et al. Mutations in TRAF3IP1/IFT54 Reveal a New Role for IFT Proteins in Microtubule Stabilization. Nat. Commun. 2015, 6, 8666. [Google Scholar] [CrossRef]
- Zhang, W.; Taylor, S.P.; Ennis, H.A.; Forlenza, K.N.; Duran, I.; Li, B.; Sanchez, J.A.O.; Nevarez, L.; Nickerson, D.A.; Bamshad, M.; et al. Expanding the Genetic Architecture and Phenotypic Spectrum in the Skeletal Ciliopathies. Hum. Mutat. 2018, 39, 152–166. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Jiang, Y.; Wang, Y.; Ouyang, J.; Yi, Z.; Sun, W.; Jia, X.; Xiao, X.; Wang, P.; et al. Pathogenic Variants in CEP290 or IQCB1 Cause Earlier-Onset Retinopathy in Senior-Loken Syndrome Compared to Those in INVS, NPHP3, or NPHP4. Am. J. Ophthalmol. 2023, 252, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Otto, E.; Rensing, C.; Nothwang, H.G.; Vollmer, M.; Adolphs, J.; Hanusch, H.; Brandis, M. A Novel Gene Encoding an SH3 Domain Protein Is Mutated in Nephronophthisis Type 1. Nat. Genet. 1997, 17, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Lienkamp, S.; Ganner, A.; Walz, G. Inversin, Wnt Signaling and Primary Cilia. Differentiation 2012, 83, S49–S55. [Google Scholar] [CrossRef] [PubMed]
- Delous, M.; Hellman, N.E.; Gaudé, H.-M.; Silbermann, F.; Le Bivic, A.; Salomon, R.; Antignac, C.; Saunier, S. Nephrocystin-1 and Nephrocystin-4 Are Required for Epithelial Morphogenesis and Associate with PALS1/PATJ and Par6. Hum. Mol. Genet. 2009, 18, 4711–4723. [Google Scholar] [CrossRef]
- Sang, L.; Miller, J.J.; Corbit, K.C.; Giles, R.H.; Brauer, M.J.; Otto, E.A.; Baye, L.M.; Wen, X.; Scales, S.J.; Kwong, M.; et al. Mapping the NPHP-JBTS-MKS Protein Network Reveals Ciliopathy Disease Genes and Pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef]
- Jiang, S.-T.; Chiou, Y.-Y.; Wang, E.; Chien, Y.-L.; Ho, H.-H.; Tsai, F.-J.; Lin, C.-Y.; Tsai, S.-P.; Li, H. Essential Role of Nephrocystin in Photoreceptor Intraflagellar Transport in Mouse. Hum. Mol. Genet. 2009, 18, 1566–1577. [Google Scholar] [CrossRef]
- Olbrich, H.; Fliegauf, M.; Hoefele, J.; Kispert, A.; Otto, E.; Volz, A.; Wolf, M.T.; Sasmaz, G.; Trauer, U.; Reinhardt, R.; et al. Mutations in a Novel Gene, NPHP3, Cause Adolescent Nephronophthisis, Tapeto-Retinal Degeneration and Hepatic Fibrosis. Nat. Genet. 2003, 34, 455–459. [Google Scholar] [CrossRef]
- Wright, K.J.; Baye, L.M.; Olivier-Mason, A.; Mukhopadhyay, S.; Sang, L.; Kwong, M.; Wang, W.; Pretorius, P.R.; Sheffield, V.C.; Sengupta, P.; et al. An ARL3-UNC119-RP2 GTPase Cycle Targets Myristoylated NPHP3 to the Primary Cilium. Genes. Dev. 2011, 25, 2347–2360. [Google Scholar] [CrossRef]
- Nakata, K.; Shiba, D.; Kobayashi, D.; Yokoyama, T. Targeting of Nphp3 to the Primary Cilia Is Controlled by an N-Terminal Myristoylation Site and Coiled-Coil Domains. Cytoskeleton 2012, 69, 221–234. [Google Scholar] [CrossRef]
- Schuermann, M.J.; Otto, E.; Becker, A.; Saar, K.; Rüschendorf, F.; Polak, B.C.; Ala-Mello, S.; Hoefele, J.; Wiedensohler, A.; Haller, M.; et al. Mapping of Gene Loci for Nephronophthisis Type 4 and Senior-Løken Syndrome, to Chromosome 1p36. Am. J. Hum. Genet. 2002, 70, 1240–1246. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.A.; Swaroop, A.; Khanna, H. RPGR-Containing Protein Complexes in Syndromic and Non-Syndromic Retinal Degeneration Due to Ciliary Dysfunction. J. Genet. 2009, 88, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Patil, H.; Tserentsoodol, N.; Saha, A.; Hao, Y.; Webb, M.; Ferreira, P.A. Selective Loss of RPGRIP1-Dependent Ciliary Targeting of NPHP4, RPGR and SDCCAG8 Underlies the Degeneration of Photoreceptor Neurons. Cell Death Dis. 2012, 3, e355. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, T.; Hoff, S.; Schell, C.; Helmstädter, M.; Kretz, O.; Kuechlin, S.; Yakulov, T.A.; Engel, C.; Müller, B.; Bensch, R.; et al. The Polarity Protein Inturned Links NPHP4 to Daam1 to Control the Subapical Actin Network in Multiciliated Cells. J. Cell Biol. 2015, 211, 963–973. [Google Scholar] [CrossRef]
- Habbig, S.; Bartram, M.P.; Müller, R.U.; Schwarz, R.; Andriopoulos, N.; Chen, S.; Sägmüller, J.G.; Hoehne, M.; Burst, V.; Liebau, M.C.; et al. NPHP4, a Cilia-Associated Protein, Negatively Regulates the Hippo Pathway. J. Cell Biol. 2011, 193, 633–642. [Google Scholar] [CrossRef]
- Burcklé, C.; Gaudé, H.-M.; Vesque, C.; Silbermann, F.; Salomon, R.; Jeanpierre, C.; Antignac, C.; Saunier, S.; Schneider-Maunoury, S. Control of the Wnt Pathways by Nephrocystin-4 Is Required for Morphogenesis of the Zebrafish Pronephros. Hum. Mol. Genet. 2011, 20, 2611–2627. [Google Scholar] [CrossRef]
- Borgal, L.; Habbig, S.; Hatzold, J.; Liebau, M.C.; Dafinger, C.; Sacarea, I.; Hammerschmidt, M.; Benzing, T.; Schermer, B. The Ciliary Protein Nephrocystin-4 Translocates the Canonical Wnt Regulator Jade-1 to the Nucleus to Negatively Regulate β-Catenin Signaling. J. Biol. Chem. 2012, 287, 25370–25380. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Attanasio, M.; Otto, E. Nephronophthisis: Disease Mechanisms of a Ciliopathy. J. Am. Soc. Nephrol. 2009, 20, 23–35. [Google Scholar] [CrossRef]
- Ronquillo, C.C.; Hanke-Gogokhia, C.; Revelo, M.P.; Frederick, J.M.; Jiang, L.; Baehr, W. Ciliopathy-Associated IQCB1/NPHP5 Protein Is Required for Mouse Photoreceptor Outer Segment Formation. FASEB J. 2016, 30, 3400–3412. [Google Scholar] [CrossRef]
- Barbelanne, M.; Song, J.; Ahmadzai, M.; Tsang, W.Y. Pathogenic NPHP5 Mutations Impair Protein Interaction with Cep290, a Prerequisite for Ciliogenesis. Hum. Mol. Genet. 2013, 22, 2482–2494. [Google Scholar] [CrossRef]
- Hsu, Y.; Seo, S.; Sheffield, V.C. Photoreceptor Cilia, in Contrast to Primary Cilia, Grant Entry to a Partially Assembled BBSome. Hum. Mol. Genet. 2021, 30, 87–102. [Google Scholar] [CrossRef]
- Zhao, C.; Malicki, J. Nephrocystins and MKS Proteins Interact with IFT Particle and Facilitate Transport of Selected Ciliary Cargos. EMBO J. 2011, 30, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The Centrosomal Protein Nephrocystin-6 Is Mutated in Joubert Syndrome and Activates Transcription Factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef]
- Tsang, W.Y.; Bossard, C.; Khanna, H.; Peränen, J.; Swaroop, A.; Malhotra, V.; Dynlacht, B.D. CP110 Suppresses Primary Cilia Formation through Its Interaction with CEP290, a Protein Deficient in Human Ciliary Disease. Dev. Cell 2008, 15, 187–197. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kim, S.; Lin, Y.-C.; Inoue, T.; Dynlacht, B.D. The CP110-Interacting Proteins Talpid3 and Cep290 Play Overlapping and Distinct Roles in Cilia Assembly. J. Cell Biol. 2014, 204, 215–229. [Google Scholar] [CrossRef]
- Frank, V.; den Hollander, A.I.; Brüchle, N.O.; Zonneveld, M.N.; Nürnberg, G.; Becker, C.; Du Bois, G.; Kendziorra, H.; Roosing, S.; Senderek, J.; et al. Mutations of the CEP290 Gene Encoding a Centrosomal Protein Cause Meckel-Gruber Syndrome. Hum. Mutat. 2008, 29, 45–52. [Google Scholar] [CrossRef]
- Barbelanne, M.; Hossain, D.; Chan, D.P.; Peränen, J.; Tsang, W.Y. Nephrocystin Proteins NPHP5 and Cep290 Regulate BBSome Integrity, Ciliary Trafficking and Cargo Delivery. Hum. Mol. Genet. 2015, 24, 2185–2200. [Google Scholar] [CrossRef]
- Otto, E.A.; Hurd, T.W.; Airik, R.; Chaki, M.; Zhou, W.; Stoetzel, C.; Patil, S.B.; Levy, S.; Ghosh, A.K.; Murga-Zamalloa, C.A.; et al. Candidate Exome Capture Identifies Mutation of SDCCAG8 as the Cause of a Retinal-Renal Ciliopathy. Nat. Genet. 2010, 42, 840–850. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Chaya, T.; Tsujii, T.; Furukawa, T. The Carboxyl-Terminal Region of SDCCAG8 Comprises a Functional Module Essential for Cilia Formation as Well as Organ Development and Homeostasis. J. Biol. Chem. 2022, 298, 101686. [Google Scholar] [CrossRef]
- Schaefer, E.; Zaloszyc, A.; Lauer, J.; Durand, M.; Stutzmann, F.; Perdomo-Trujillo, Y.; Redin, C.; Bennouna Greene, V.; Toutain, A.; Perrin, L.; et al. Mutations in SDCCAG8/NPHP10 Cause Bardet-Biedl Syndrome and Are Associated with Penetrant Renal Disease and Absent Polydactyly. Mol. Syndromol. 2011, 1, 273–281. [Google Scholar] [CrossRef]
- Airik, R.; Schueler, M.; Airik, M.; Cho, J.; Ulanowicz, K.A.; Porath, J.D.; Hurd, T.W.; Bekker-Jensen, S.; Schrøder, J.M.; Andersen, J.S.; et al. SDCCAG8 Interacts with RAB Effector Proteins RABEP2 and ERC1 and Is Required for Hedgehog Signaling. PLoS ONE 2016, 11, e0156081. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; White, J.T.; Utleg, A.G.; Wang, S.; Ferguson, C.; True, L.D.; Vessella, R.; Hood, L.; Nelson, P.S. Isolation and Characterization of Human and Mouse WDR19,a Novel WD-Repeat Protein Exhibiting Androgen-Regulated Expression in Prostate Epithelium. Genomics 2003, 82, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Wen, X.; Chih, B.; Nelson, C.D.; Lane, W.S.; Scales, S.J.; Jackson, P.K. TULP3 Bridges the IFT-A Complex and Membrane Phosphoinositides to Promote Trafficking of G Protein-Coupled Receptors into Primary Cilia. Genes. Dev. 2010, 24, 2180–2193. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Goeddel, D.V. MIP-T3, a Novel Protein Linking Tumor Necrosis Factor Receptor-Associated Factor 3 to the Microtubule Network. J. Biol. Chem. 2000, 275, 23852–23860. [Google Scholar] [CrossRef]
- Ghossoub, R.; Hu, Q.; Failler, M.; Rouyez, M.-C.; Spitzbarth, B.; Mostowy, S.; Wolfrum, U.; Saunier, S.; Cossart, P.; Jamesnelson, W.; et al. Septins 2, 7 and 9 and MAP4 Colocalize along the Axoneme in the Primary Cilium and Control Ciliary Length. J. Cell Sci. 2013, 126, 2583–2594. [Google Scholar] [CrossRef]
- Zhang, D.; Aravind, L. Novel Transglutaminase-like Peptidase and C2 Domains Elucidate the Structure, Biogenesis and Evolution of the Ciliary Compartment. Cell Cycle 2012, 11, 3861–3875. [Google Scholar] [CrossRef]
- Remans, K.; Bürger, M.; Vetter, I.R.; Wittinghofer, A. C2 Domains as Protein-Protein Interaction Modules in the Ciliary Transition Zone. Cell Rep. 2014, 8, 1–9. [Google Scholar] [CrossRef]
- Winkelbauer, M.E.; Schafer, J.C.; Haycraft, C.J.; Swoboda, P.; Yoder, B.K. The C. Elegans Homologs of Nephrocystin-1 and Nephrocystin-4 Are Cilia Transition Zone Proteins Involved in Chemosensory Perception. J. Cell Sci. 2005, 118, 5575–5587. [Google Scholar] [CrossRef]
- Shi, X.; Garcia, G.; Van De Weghe, J.C.; McGorty, R.; Pazour, G.J.; Doherty, D.; Huang, B.; Reiter, J.F. Super-Resolution Microscopy Reveals That Disruption of Ciliary Transition-Zone Architecture Causes Joubert Syndrome. Nat. Cell Biol. 2017, 19, 1178–1188. [Google Scholar] [CrossRef]
- Williams, C.L.; Li, C.; Kida, K.; Inglis, P.N.; Mohan, S.; Semenec, L.; Bialas, N.J.; Stupay, R.M.; Chen, N.; Blacque, O.E.; et al. MKS and NPHP Modules Cooperate to Establish Basal Body/Transition Zone Membrane Associations and Ciliary Gate Function during Ciliogenesis. J. Cell Biol. 2011, 192, 1023–1041. [Google Scholar] [CrossRef]
- Adamiok-Ostrowska, A.; Piekiełko-Witkowska, A. Ciliary Genes in Renal Cystic Diseases. Cells 2020, 9, 907. [Google Scholar] [CrossRef]
- Mollet, G.; Silbermann, F.; Delous, M.; Salomon, R.; Antignac, C.; Saunier, S. Characterization of the Nephrocystin/Nephrocystin-4 Complex and Subcellular Localization of Nephrocystin-4 to Primary Cilia and Centrosomes. Hum. Mol. Genet. 2005, 14, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Frederick, J.M.; Hanke-Gogokhia, C.; Ying, G.; Baehr, W. Diffuse or Hitch a Ride: How Photoreceptor Lipidated Proteins Get from Here to There. Biol. Chem. 2020, 401, 573–584. [Google Scholar] [CrossRef]
- Park, K.; Leroux, M.R. Composition, Organization and Mechanisms of the Transition Zone, a Gate for the Cilium. EMBO Rep. 2022, 23, e55420. [Google Scholar] [CrossRef]
- Won, J.; de Evsikova, C.M.; Smith, R.S.; Hicks, W.L.; Edwards, M.M.; Longo-Guess, C.; Li, T.; Naggert, J.K.; Nishina, P.M. NPHP4 Is Necessary for Normal Photoreceptor Ribbon Synapse Maintenance and Outer Segment Formation, and for Sperm Development. Hum. Mol. Genet. 2011, 20, 482–496. [Google Scholar] [CrossRef]
- Khanna, H. Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells 2015, 4, 674–686. [Google Scholar] [CrossRef]
- Vössing, C.; Atigbire, P.; Eilers, J.; Markus, F.; Stieger, K.; Song, F.; Neidhardt, J. The Major Ciliary Isoforms of RPGR Build Different Interaction Complexes with INPP5E and RPGRIP1L. Int. J. Mol. Sci. 2021, 22, 3583. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.A.; Desai, N.J.; Hildebrandt, F.; Khanna, H. Interaction of Ciliary Disease Protein Retinitis Pigmentosa GTPase Regulator with Nephronophthisis-Associated Proteins in Mammalian Retinas. Mol. Vis. 2010, 16, 1373–1381. [Google Scholar]
- Anand, M.; Khanna, H. Ciliary Transition Zone (TZ) Proteins RPGR and CEP290: Role in Photoreceptor Cilia and Degenerative Diseases. Expert. Opin. Ther. Targets 2012, 16, 541–551. [Google Scholar] [CrossRef]
- Raghupathy, R.K.; Zhang, X.; Liu, F.; Alhasani, R.H.; Biswas, L.; Akhtar, S.; Pan, L.; Moens, C.B.; Li, W.; Liu, M.; et al. Rpgrip1 Is Required for Rod Outer Segment Development and Ciliary Protein Trafficking in Zebrafish. Sci. Rep. 2017, 7, 16881. [Google Scholar] [CrossRef]
- Chaki, M.; Hoefele, J.; Allen, S.J.; Ramaswami, G.; Janssen, S.; Bergmann, C.; Heckenlively, J.R.; Otto, E.A.; Hildebrandt, F. Genotype-Phenotype Correlation in 440 Patients with NPHP-Related Ciliopathies. Kidney Int. 2011, 80, 1239–1245. [Google Scholar] [CrossRef]
- Liebau, M.C.; Höpker, K.; Müller, R.U.; Schmedding, I.; Zank, S.; Schairer, B.; Fabretti, F.; Höhne, M.; Bartram, M.P.; Dafinger, C.; et al. Nephrocystin-4 Regulates Pyk2-Induced Tyrosine Phosphorylation of Nephrocystin-1 to Control Targeting to Monocilia. J. Biol. Chem. 2011, 286, 14237–14245. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Laclef, C.; Yang, N.; Andreu-Cervera, A.; Lewis, J.; Mao, X.; Li, L.; Snedecor, E.R.; Takemaru, K.-I.; Qin, C.; et al. RPGRIP1L Is Required for Stabilizing Epidermal Keratinocyte Adhesion through Regulating Desmoglein Endocytosis. PLoS Genet. 2019, 15, e1007914. [Google Scholar] [CrossRef] [PubMed]
- Mahuzier, A.; Gaudé, H.-M.; Grampa, V.; Anselme, I.; Silbermann, F.; Leroux-Berger, M.; Delacour, D.; Ezan, J.; Montcouquiol, M.; Saunier, S.; et al. Dishevelled Stabilization by the Ciliopathy Protein Rpgrip1l Is Essential for Planar Cell Polarity. J. Cell Biol. 2012, 198, 927–940. [Google Scholar] [CrossRef]
- Schäfer, T.; Pütz, M.; Lienkamp, S.; Ganner, A.; Bergbreiter, A.; Ramachandran, H.; Gieloff, V.; Gerner, M.; Mattonet, C.; Czarnecki, P.G.; et al. Genetic and Physical Interaction between the NPHP5 and NPHP6 Gene Products. Hum. Mol. Genet. 2008, 17, 3655–3662. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, S.; Jung, Y.; Jung, E.; Kwon, H.J.; Kim, J. Eupatilin Rescues Ciliary Transition Zone Defects to Ameliorate Ciliopathy-Related Phenotypes. J. Clin. Investig. 2018, 128, 3642–3648. [Google Scholar] [CrossRef]
- Tian, X.; Zhao, H.; Zhou, J. Organization, Functions, and Mechanisms of the BBSome in Development, Ciliopathies, and Beyond. eLife 2023, 12, e87623. [Google Scholar] [CrossRef]
- Zuo, X.; Guo, W.; Lipschutz, J.H. The Exocyst Protein Sec10 Is Necessary for Primary Ciliogenesis and Cystogenesis in Vitro. Mol. Biol. Cell 2009, 20, 2522–2529. [Google Scholar] [CrossRef]
- Shimada, H.; Lu, Q.; Insinna-Kettenhofen, C.; Nagashima, K.; English, M.A.; Semler, E.M.; Mahgerefteh, J.; Cideciyan, A.V.; Li, T.; Brooks, B.P.; et al. In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations. Cell Rep. 2017, 20, 384–396. [Google Scholar] [CrossRef]
- Wiegering, A.; Dildrop, R.; Vesque, C.; Khanna, H.; Schneider-Maunoury, S.; Gerhardt, C. Rpgrip1l Controls Ciliary Gating by Ensuring the Proper Amount of Cep290 at the Vertebrate Transition Zone. Mol. Biol. Cell 2021, 32, 675–689. [Google Scholar] [CrossRef]
- Casteels, I.; Spileers, W.; Demaerel, P.; Casaer, P.; De Cock, P.; Dralands, L.; Missotten, L. Leber Congenital Amaurosis--Differential Diagnosis, Ophthalmological and Neuroradiological Report of 18 Patients. Neuropediatrics 1996, 27, 189–193. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Waldherr, R.; Kutt, R.; Brandis, M. The Nephronophthisis Complex: Clinical and Genetic Aspects. Clin. Investig. 1992, 70, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Hayashi, T.; Yoshitake, K.; Murai, N.; Matsui, Z.; Kubo, H.; Satoh, H.; Matsufuji, S.; Takamura, T.; Yokoo, T.; et al. Compound Heterozygous Splice Site Variants in the SCLT1 Gene Highlight an Additional Candidate Locus for Senior-Løken Syndrome. Sci. Rep. 2018, 8, 16733. [Google Scholar] [CrossRef]
- Kuzinska, M.Z.; Lin, S.Y.-Y.; Klämbt, V.; Bufler, P.; Rezvani, M. Ciliopathy Organoid Models: A Comprehensive Review. Am. J. Physiol. Cell Physiol. 2024, 327, C1604–C1625. [Google Scholar] [CrossRef]
- Alzarka, B.; Charnaya, O.; Gunay-Aygun, M. Diseases of the Primary Cilia: A Clinical Characteristics Review. Pediatr. Nephrol. 2025, 40, 611–627. [Google Scholar] [CrossRef]
- Romani, M.; Micalizzi, A.; Valente, E.M. Joubert Syndrome: Congenital Cerebellar Ataxia with the Molar Tooth. Lancet Neurol. 2013, 12, 894–905. [Google Scholar] [CrossRef]
- Bergmann, C. Educational Paper: Ciliopathies. Eur. J. Pediatr. 2012, 171, 1285–1300. [Google Scholar] [CrossRef]
- Garg, S.; Rakesh, K.; Khattri, S.; Mishra, P.; Tikka, S.K. Bipolar Disorder with Intellectual Disability in Joubert Syndrome: A Case Report. Psychiatry Clin. Neurosci. 2019, 73, 761–762. [Google Scholar] [CrossRef]
- Paprocka, J.; Jamroz, E. Joubert Syndrome and Related Disorders. Neurol. Neurochir. Pol. 2012, 46, 379–383. [Google Scholar] [CrossRef]
- Parisi, M.; Glass, I. Joubert Syndrome. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Van De Weghe, J.C.; Gomez, A.; Doherty, D. The Joubert-Meckel-Nephronophthisis Spectrum of Ciliopathies. Annu. Rev. Genom. Hum. Genet. 2022, 23, 301–329. [Google Scholar] [CrossRef]
- Chen, C.-P. Meckel Syndrome: Genetics, Perinatal Findings, and Differential Diagnosis. Taiwan J. Obstet. Gynecol. 2007, 46, 9–14. [Google Scholar] [CrossRef]
- Forsyth, R.; Gunay-Aygun, M. Bardet-Biedl Syndrome Overview. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Forsythe, E.; Beales, P.L. Bardet-Biedl Syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, F.; Franco, B.; Toriello, H.; Neri, G. Oral-Facial-Digital Syndromes: Review and Diagnostic Guidelines. Am. J. Med. Genet. A 2007, 143A, 3314–3323. [Google Scholar] [CrossRef] [PubMed]
- Bruel, A.-L.; Levy, J.; Elenga, N.; Defo, A.; Favre, A.; Lucron, H.; Capri, Y.; Perrin, L.; Passemard, S.; Vial, Y.; et al. INTU-Related Oral-Facial-Digital Syndrome Type VI: A Confirmatory Report. Clin. Genet. 2018, 93, 1205–1209. [Google Scholar] [CrossRef]
- Franco, B.; Bruel, A.-L.; Thauvin-Robinet, C. Oral-Facial-Digital Syndrome Type I. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Alström Syndrome. In Atlas of Inherited Retinal Diseases; Tsang, S.H., Sharma, T., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; pp. 179–180. ISBN 978-3-319-95046-4. [Google Scholar]
- Dassie, F.; Favaretto, F.; Bettini, S.; Parolin, M.; Valenti, M.; Reschke, F.; Danne, T.; Vettor, R.; Milan, G.; Maffei, P. Alström Syndrome: An Ultra-Rare Monogenic Disorder as a Model for Insulin Resistance, Type 2 Diabetes Mellitus and Obesity. Endocrine 2021, 71, 618–625. [Google Scholar] [CrossRef]
- Dassie, F.; Lorusso, R.; Benavides-Varela, S.; Milan, G.; Favaretto, F.; Callus, E.; Cagnin, S.; Reggiani, F.; Minervini, G.; Tosatto, S.; et al. Neurocognitive Assessment and DNA Sequencing Expand the Phenotype and Genotype Spectrum of Alström Syndrome. Am. J. Med. Genet. Part A 2021, 185, 732–742. [Google Scholar] [CrossRef]
- Marshall, J.D.; Bronson, R.T.; Collin, G.B.; Nordstrom, A.D.; Maffei, P.; Paisey, R.B.; Carey, C.; MacDermott, S.; Russell-Eggitt, I.; Shea, S.E.; et al. New Alström Syndrome Phenotypes Based on the Evaluation of 182 Cases. Arch. Intern. Med. 2005, 165, 675–683. [Google Scholar] [CrossRef]
- Kanna, R.M.; Gradil, D.; Boszczyk, B.M. Management of Cervical Myelopathy Due to Ossification of Posterior Longitudinal Ligament in a Patient with Alström Syndrome. Eur. Spine J. 2012, 21, 2418–2424. [Google Scholar] [CrossRef]
- Fuster-García, C.; García-Bohórquez, B.; Rodríguez-Muñoz, A.; Aller, E.; Jaijo, T.; Millán, J.M.; García-García, G. Usher Syndrome: Genetics of a Human Ciliopathy. Int. J. Mol. Sci. 2021, 22, 6723. [Google Scholar] [CrossRef]
- Dammeyer, J. Children with Usher Syndrome: Mental and Behavioral Disorders. Behav. Brain Funct. 2012, 8, 16. [Google Scholar] [CrossRef]
- Wang, H.; Huo, L.; Wang, Y.; Sun, W.; Gu, W. Usher Syndrome Type 2A Complicated with Glycogen Storage Disease Type 3 Due to Paternal Uniparental Isodisomy of Chromosome 1 in a Sporadic Patient. Mol. Genet. Genom. Med. 2021, 9, e1779. [Google Scholar] [CrossRef]
- Mill, P.; Christensen, S.T.; Pedersen, L.B. Primary Cilia as Dynamic and Diverse Signalling Hubs in Development and Disease. Nat. Rev. Genet. 2023, 24, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.-X.; Miao, Y.-T.; Zhang, X.; Huang, M.-Z.; Jahangir, M.; Luo, S.; Lang, B. Primary Cilia-Associated Protein IFT172 in Ciliopathies. Front. Cell Dev. Biol. 2023, 11, 1074880. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Bolz, S.; Junger, K.; Klose, F.; Schubert, T.; Woerz, F.; Boldt, K.; Ueffing, M.; Beyer, T. TTC30A and TTC30B Redundancy Protects IFT Complex B Integrity and Its Pivotal Role in Ciliogenesis. Genes 2022, 13, 1191. [Google Scholar] [CrossRef] [PubMed]
- Cilleros-Rodriguez, D.; Martin-Morales, R.; Barbeito, P.; Deb Roy, A.; Loukil, A.; Sierra-Rodero, B.; Herranz, G.; Pampliega, O.; Redrejo-Rodriguez, M.; Goetz, S.C.; et al. Multiple Ciliary Localization Signals Control INPP5E Ciliary Targeting. eLife 2022, 11, e78383. [Google Scholar] [CrossRef]
- He, R.; Chen, G.; Li, Z.; Li, J. Structure of the N-Terminal Coiled-Coil Domains of the Ciliary Protein Rpgrip1l. iScience 2023, 26, 106249. [Google Scholar] [CrossRef]
- Mitchison, H.M.; Valente, E.M. Motile and Non-Motile Cilia in Human Pathology: From Function to Phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Phelps, I.G.; O’Roak, B.J.; Knutzen, D.M.; Rue, T.C.; Ishak, G.E.; Isabella, C.R.; Gorden, N.; Adkins, J.; et al. Joubert Syndrome: A Model for Untangling Recessive Disorders with Extreme Genetic Heterogeneity. J. Med. Genet. 2015, 52, 514–522. [Google Scholar] [CrossRef]
- Walczak-Sztulpa, J.; Posmyk, R.; Bukowska-Olech, E.M.; Wawrocka, A.; Jamsheer, A.; Oud, M.M.; Schmidts, M.; Arts, H.H.; Latos-Bielenska, A.; Wasilewska, A. Compound Heterozygous IFT140 Variants in Two Polish Families with Sensenbrenner Syndrome and Early Onset End-Stage Renal Disease. Orphanet J. Rare Dis. 2020, 15, 36. [Google Scholar] [CrossRef]
- Ashe, A.; Butterfield, N.C.; Town, L.; Courtney, A.D.; Cooper, A.N.; Ferguson, C.; Barry, R.; Olsson, F.; Liem, K.F.; Parton, R.G.; et al. Mutations in Mouse Ift144 Model the Craniofacial, Limb and Rib Defects in Skeletal Ciliopathies. Hum. Mol. Genet. 2012, 21, 1808–1823. [Google Scholar] [CrossRef]
- Ishida, Y.; Kobayashi, T.; Chiba, S.; Katoh, Y.; Nakayama, K. Molecular Basis of Ciliary Defects Caused by Compound Heterozygous IFT144/WDR19 Mutations Found in Cranioectodermal Dysplasia. Hum. Mol. Genet. 2021, 30, 213–225. [Google Scholar] [CrossRef]
- Keith, B.P.; Robertson, D.L.; Hentges, K.E. Locus Heterogeneity Disease Genes Encode Proteins with High Interconnectivity in the Human Protein Interaction Network. Front. Genet. 2014, 5, 434. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. 2021, 77, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; Pott, J.-W.R.; Collin, R.W.J.; Kroes, H.Y.; Verheij, J.B.G.M.; Blokland, E.A.W.; de Castro Miró, M.; Hoyng, C.B.; Klaver, C.C.W.; Koenekoop, R.K.; et al. A Novel Nonsense Mutation in CEP290 Induces Exon Skipping and Leads to a Relatively Mild Retinal Phenotype. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3646–3652. [Google Scholar] [CrossRef]
- Zhu, T.; Shen, Y.; Sun, Z.; Han, X.; Wei, X.; Li, W.; Lu, C.; Cheng, T.; Zou, X.; Li, H.; et al. Clinical and Molecular Features of a Chinese Cohort With Syndromic and Nonsyndromic Retinal Dystrophies Related to the CEP290 Gene. Am. J. Ophthalmol. 2023, 248, 96–106. [Google Scholar] [CrossRef]
- Baala, L.; Audollent, S.; Martinovic, J.; Ozilou, C.; Babron, M.-C.; Sivanandamoorthy, S.; Saunier, S.; Salomon, R.; Gonzales, M.; Rattenberry, E.; et al. Pleiotropic Effects of CEP290 (NPHP6) Mutations Extend to Meckel Syndrome. Am. J. Hum. Genet. 2007, 81, 170–179. [Google Scholar] [CrossRef]
- Helou, J.; Otto, E.A.; Attanasio, M.; Allen, S.J.; Parisi, M.A.; Glass, I.; Utsch, B.; Hashmi, S.; Fazzi, E.; Omran, H.; et al. Mutation Analysis of NPHP6/CEP290 in Patients with Joubert Syndrome and Senior-Løken Syndrome. J. Med. Genet. 2007, 44, 657–663. [Google Scholar] [CrossRef]
- Williamson, M.; Traboulsi, E.; DeBenedictis, M. Investigation of CEP290 Genotype-Phenotype Correlations in a Patient with Retinitis Pigmentosa, Infertility, End-Stage Renal Disease, and a Novel Mutation. Ophthalmic Genet. 2020, 41, 171–174. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and Molecular Pathways Underpinning Ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Kousi, M.; Katsanis, N. Genetic Modifiers and Oligogenic Inheritance. Cold Spring Harb. Perspect. Med. 2015, 5, a017145. [Google Scholar] [CrossRef]
- Khanna, H.; Davis, E.E.; Murga-Zamalloa, C.A.; Estrada-Cuzcano, A.; Lopez, I.; den Hollander, A.I.; Zonneveld, M.N.; Othman, M.I.; Waseem, N.; Chakarova, C.F.; et al. A Common Allele in RPGRIP1L Is a Modifier of Retinal Degeneration in Ciliopathies. Nat. Genet. 2009, 41, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Louie, C.M.; Caridi, G.; Lopes, V.S.; Brancati, F.; Kispert, A.; Lancaster, M.A.; Schlossman, A.M.; Otto, E.A.; Leitges, M.; Groene, H.-J.; et al. AHI1 Is Required for Outer Segment Development and Is a Modifier for Retinal Degeneration in Nephronophthisis. Nat. Genet. 2010, 42, 175–180. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Solarat, C.; Blanco-Kelly, F.; Sanchez-Navarro, I.; Bea-Mascato, B.; Martin-Salazar, E.; Lorda-Sanchez, I.; Swafiri, S.T.; Avila-Fernandez, A.; Martin-Merida, I.; et al. Allelic Overload and Its Clinical Modifier Effect in Bardet-Biedl Syndrome. NPJ Genom. Med. 2022, 7, 41. [Google Scholar] [CrossRef]
- Ramsbottom, S.A.; Thelwall, P.E.; Wood, K.M.; Clowry, G.J.; Devlin, L.A.; Silbermann, F.; Spiewak, H.L.; Shril, S.; Molinari, E.; Hildebrandt, F.; et al. Mouse Genetics Reveals Barttin as a Genetic Modifier of Joubert Syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 1113–1118. [Google Scholar] [CrossRef]
- Leggatt, G.P.; Seaby, E.G.; Veighey, K.; Gast, C.; Gilbert, R.D.; Ennis, S. A Role for Genetic Modifiers in Tubulointerstitial Kidney Diseases. Genes 2023, 14, 1582. [Google Scholar] [CrossRef]
- Deltas, C. Digenic Inheritance and Genetic Modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef]
- Beales, P.L.; Badano, J.L.; Ross, A.J.; Ansley, S.J.; Hoskins, B.E.; Kirsten, B.; Mein, C.A.; Froguel, P.; Scambler, P.J.; Lewis, R.A.; et al. Genetic Interaction of BBS1 Mutations with Alleles at Other BBS Loci Can Result in Non-Mendelian Bardet-Biedl Syndrome. Am. J. Hum. Genet. 2003, 72, 1187–1199. [Google Scholar] [CrossRef]
- Katsanis, N.; Ansley, S.J.; Badano, J.L.; Eichers, E.R.; Lewis, R.A.; Hoskins, B.E.; Scambler, P.J.; Davidson, W.S.; Beales, P.L.; Lupski, J.R. Triallelic Inheritance in Bardet-Biedl Syndrome, a Mendelian Recessive Disorder. Science 2001, 293, 2256–2259. [Google Scholar] [CrossRef]
- Hoefele, J.; Wolf, M.T.F.; O’Toole, J.F.; Otto, E.A.; Schultheiss, U.; Dêschenes, G.; Attanasio, M.; Utsch, B.; Antignac, C.; Hildebrandt, F. Evidence of Oligogenic Inheritance in Nephronophthisis. J. Am. Soc. Nephrol. 2007, 18, 2789–2795. [Google Scholar] [CrossRef]
- Manara, E.; Paolacci, S.; D’Esposito, F.; Abeshi, A.; Ziccardi, L.; Falsini, B.; Colombo, L.; Iarossi, G.; Pilotta, A.; Boccone, L.; et al. Mutation Profile of BBS Genes in Patients with Bardet-Biedl Syndrome: An Italian Study. Ital. J. Pediatr. 2019, 45, 72. [Google Scholar] [CrossRef]
- Pollara, L.; Sottile, V.; Valente, E.M. Patient-Derived Cellular Models of Primary Ciliopathies. J. Med. Genet. 2022, 59, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.S.; Sattar, S.; Massoudi, R.A.; Gleeson, J.G. Co-Occurrence of Distinct Ciliopathy Diseases in Single Families Suggests Genetic Modifiers. Am. J. Med. Genet. A 2011, 155, 3042–3049. [Google Scholar] [CrossRef]
- Shaheen, R.; Szymanska, K.; Basu, B.; Patel, N.; Ewida, N.; Faqeih, E.; Al Hashem, A.; Derar, N.; Alsharif, H.; Aldahmesh, M.A.; et al. Characterizing the Morbid Genome of Ciliopathies. Genome Biol. 2016, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Zaghloul, N.A.; Katsanis, N. Functional Modules, Mutational Load and Human Genetic Disease. Trends Genet. 2010, 26, 168–176. [Google Scholar] [CrossRef]
- Fahim, A. Retinitis Pigmentosa: Recent Advances and Future Directions in Diagnosis and Management. Curr. Opin. Pediatr. 2018, 30, 725–733. [Google Scholar] [CrossRef]
- Robert, M.-A.; Chahal, P.S.; Audy, A.; Kamen, A.; Gilbert, R.; Gaillet, B. Manufacturing of Recombinant Adeno-Associated Viruses Using Mammalian Expression Platforms. Biotechnol. J. 2017, 12, 1600193. [Google Scholar] [CrossRef]
- Aguirre, G.D.; Cideciyan, A.V.; Dufour, V.L.; Ripolles-García, A.; Sudharsan, R.; Swider, M.; Nikonov, R.; Iwabe, S.; Boye, S.L.; Hauswirth, W.W.; et al. Gene Therapy Reforms Photoreceptor Structure and Restores Vision in NPHP5-Associated Leber Congenital Amaurosis. Mol. Ther. 2021, 29, 2456–2468. [Google Scholar] [CrossRef]
- Mookherjee, S.; Chen, H.Y.; Isgrig, K.; Yu, W.; Hiriyanna, S.; Levron, R.; Li, T.; Colosi, P.; Chien, W.; Swaroop, A.; et al. A CEP290 C-Terminal Domain Complements the Mutant CEP290 of Rd16 Mice In Trans and Rescues Retinal Degeneration. Cell Rep. 2018, 25, 611–623.e6. [Google Scholar] [CrossRef]
- Pierce, E.A.; Aleman, T.S.; Jayasundera, K.T.; Ashimatey, B.S.; Kim, K.; Rashid, A.; Jaskolka, M.C.; Myers, R.L.; Lam, B.L.; Bailey, S.T.; et al. Gene Editing for CEP290-Associated Retinal Degeneration. N Engl J Med 2024, 390, 1972–1984. [Google Scholar] [CrossRef]
- Zhao, Q.; Wei, L.; Chen, Y. From Bench to Bedside: Developing CRISPR/Cas-Based Therapy for Ocular Diseases. Pharmacol. Res. 2025, 213, 107638. [Google Scholar] [CrossRef]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a Gene-Editing Approach to Restore Vision Loss in Leber Congenital Amaurosis Type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.-X.; Barry, E.; Yu, D.; Lukason, M.; Cheng, S.H.; Scaria, A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol Ther 2017, 25, 331–341. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense Oligonucleotides and Other Genetic Therapies Made Simple. Pract. Neurol. 2018, 18, 126–131. [Google Scholar] [CrossRef]
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The Challenge and Prospect of mRNA Therapeutics Landscape. Biotechnol. Adv. 2020, 40, 107534. [Google Scholar] [CrossRef]
- Garanto, A.; Chung, D.C.; Duijkers, L.; Corral-Serrano, J.C.; Messchaert, M.; Xiao, R.; Bennett, J.; Vandenberghe, L.H.; Collin, R.W.J. In Vitro and in Vivo Rescue of Aberrant Splicing in CEP290-Associated LCA by Antisense Oligonucleotide Delivery. Hum. Mol. Genet. 2016, 25, 2552–2563. [Google Scholar] [CrossRef]
- Parfitt, D.A.; Lane, A.; Ramsden, C.; Jovanovic, K.; Coffey, P.J.; Hardcastle, A.J.; Cheetham, M.E. Using Induced Pluripotent Stem Cells to Understand Retinal Ciliopathy Disease Mechanisms and Develop Therapies. Biochem. Soc. Trans. 2016, 44, 1245–1251. [Google Scholar] [CrossRef]
- Mathur, P.; Yang, J. Usher Syndrome: Hearing Loss, Retinal Degeneration and Associated Abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef]
- Johansson, I.; Monsen, V.T.; Pettersen, K.; Mildenberger, J.; Misund, K.; Kaarniranta, K.; Schønberg, S.; Bjørkøy, G. The Marine N-3 PUFA DHA Evokes Cytoprotection against Oxidative Stress and Protein Misfolding by Inducing Autophagy and NFE2L2 in Human Retinal Pigment Epithelial Cells. Autophagy 2015, 11, 1636–1651. [Google Scholar] [CrossRef]
- Choi, E.H.; Daruwalla, A.; Suh, S.; Leinonen, H.; Palczewski, K. Retinoids in the Visual Cycle: Role of the Retinal G Protein-Coupled Receptor. J. Lipid Res. 2021, 62, 100040. [Google Scholar] [CrossRef]
- Guadagni, V.; Novelli, E.; Piano, I.; Gargini, C.; Strettoi, E. Pharmacological Approaches to Retinitis Pigmentosa: A Laboratory Perspective. Prog. Retin. Eye Res. 2015, 48, 62–81. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, D.M.; Barnard, A.R.; Singh, M.S.; Martin, C.; Lee, E.J.; Davies, W.I.L.; MacLaren, R.E. CNTF Gene Therapy Confers Lifelong Neuroprotection in a Mouse Model of Human Retinitis Pigmentosa. Mol. Ther. 2015, 23, 1308–1319. [Google Scholar] [CrossRef]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A.; Woo, S.J.; Kwon, Y.J. Molecular Genetics and Emerging Therapies for Retinitis Pigmentosa: Basic Research and Clinical Perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Athanasiou, D.; Afanasyeva, T.A.V.; Chai, N.; Ziaka, K.; Jovanovic, K.; Guarascio, R.; Boldt, K.; Corral-Serrano, J.C.; Kanuga, N.; Roepman, R.; et al. Small Molecule Treatment Alleviates Photoreceptor Cilia Defects in LCA5-Deficient Human Retinal Organoids. Acta Neuropathol. Commun. 2025, 13, 26. [Google Scholar] [CrossRef]
- Smith, C.E.L.; Streets, A.J.; Lake, A.V.R.; Natarajan, S.; Best, S.K.; Szymanska, K.; Karwatka, M.; Stevenson, T.; Trowbridge, R.; Grant, G.; et al. Drug and siRNA Screens Identify ROCK2 as a Therapeutic Target for Ciliopathies. Commun. Med. 2025, 5, 129. [Google Scholar] [CrossRef]
- Howden, S.E.; Thomson, J.A.; Little, M.H. Simultaneous Reprogramming and Gene Editing of Human Fibroblasts. Nat. Protoc. 2018, 13, 875–898. [Google Scholar] [CrossRef]
- Gholkar, A.A.; Gimeno, T.V.; Edgemon, J.E.; Sim, M.S.; Torres, J.Z. MI-181 Modulates Cilia Length and Restores Cilia Length in Cells with Defective Shortened Cilia. ACS Chem. Biol. 2024, 19, 1733–1742. [Google Scholar] [CrossRef]
- Stern, J.H.; Tian, Y.; Funderburgh, J.; Pellegrini, G.; Zhang, K.; Goldberg, J.L.; Ali, R.R.; Young, M.; Xie, Y.; Temple, S. Regenerating Eye Tissues to Preserve and Restore Vision. Cell Stem Cell 2018, 22, 834–849. [Google Scholar] [CrossRef]
- Garita-Hernandez, M.; Lampič, M.; Chaffiol, A.; Guibbal, L.; Routet, F.; Santos-Ferreira, T.; Gasparini, S.; Borsch, O.; Gagliardi, G.; Reichman, S.; et al. Restoration of Visual Function by Transplantation of Optogenetically Engineered Photoreceptors. Nat. Commun. 2019, 10, 4524. [Google Scholar] [CrossRef]
- Pansare, V.; Hejazi, S.; Faenza, W.; Prud’homme, R.K. Review of Long-Wavelength Optical and NIR Imaging Materials: Contrast Agents, Fluorophores and Multifunctional Nano Carriers. Chem. Mater. 2012, 24, 812–827. [Google Scholar] [CrossRef]
- Jones, M.K.; Lu, B.; Girman, S.; Wang, S. Cell-Based Therapeutic Strategies for Replacement and Preservation in Retinal Degenerative Diseases. Prog. Retin. Eye Res. 2017, 58, 1–27. [Google Scholar] [CrossRef] [PubMed]
- da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 Clinical Study of an Embryonic Stem Cell-Derived Retinal Pigment Epithelium Patch in Age-Related Macular Degeneration. Nat. Biotechnol. 2018, 36, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Weiland, J.D.; Roska, B.; Humayun, M.S. Retinal Stimulation Strategies to Restore Vision: Fundamentals and Systems. Prog. Retin. Eye Res. 2016, 53, 21–47. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.O.; Cogan, S.F.; Rizzo, J.F. Retinal Prostheses: Current Challenges and Future Outlook. J. Biomater. Sci. Polym. Ed. 2007, 18, 1031–1055. [Google Scholar] [CrossRef]
- Cardenas-Rodriguez, M.; Austin-Tse, C.; Bergboer, J.G.M.; Molinari, E.; Sugano, Y.; Bachmann-Gagescu, R.; Sayer, J.A.; Drummond, I.A. Genetic Compensation for Cilia Defects in Cep290 Mutants by Upregulation of Cilia-Associated Small GTPases. J. Cell Sci. 2021, 134, jcs258568. [Google Scholar] [CrossRef]
- Zhang, K.; Da Silva, F.; Seidl, C.; Wilsch-Bräuninger, M.; Herbst, J.; Huttner, W.B.; Niehrs, C. Primary Cilia Are WNT-Transducing Organelles Whose Biogenesis Is Controlled by a WNT-PP1 Axis. Dev. Cell 2023, 58, 139–154.e8. [Google Scholar] [CrossRef]
- Ren, Z.-L.; Zhang, H.-B.; Li, L.; Yang, Z.-L.; Jiang, L. Characterization of Two Novel Knock-in Mouse Models of Syndromic Retinal Ciliopathy Carrying Hypomorphic Sdccag8 Mutations. Zool. Res. 2022, 43, 442–456. [Google Scholar] [CrossRef]
- Drivas, T.G.; Holzbaur, E.L.F.; Bennett, J. Disruption of CEP290 Microtubule/Membrane-Binding Domains Causes Retinal Degeneration. J. Clin. Investig. 2013, 123, 4525–4539. [Google Scholar] [CrossRef]
- Jiang, S.-T.; Chiou, Y.-Y.; Wang, E.; Lin, H.-K.; Lee, S.-P.; Lu, H.-Y.; Wang, C.-K.L.; Tang, M.-J.; Li, H. Targeted Disruption of Nphp1 Causes Male Infertility Due to Defects in the Later Steps of Sperm Morphogenesis in Mice. Hum. Mol. Genet. 2008, 17, 3368–3379. [Google Scholar] [CrossRef]
- O’Toole, J.F.; Otto, E.A.; Frishberg, Y.; Hildebrandt, F. Retinitis Pigmentosa and Renal Failure in a Patient with Mutations in INVS. Nephrol. Dial. Transplant. 2006, 21, 1989–1991. [Google Scholar] [CrossRef]
- Vogel, P.; Gelfman, C.M.; Issa, T.; Payne, B.J.; Hansen, G.M.; Read, R.W.; Jones, C.; Pitcher, M.R.; Ding, Z.-M.; DaCosta, C.M.; et al. Nephronophthisis and Retinal Degeneration in Tmem218-/- Mice: A Novel Mouse Model for Senior-Løken Syndrome? Vet. Pathol. 2015, 52, 580–595. [Google Scholar] [CrossRef] [PubMed]
- Hanke-Gogokhia, C.; Chiodo, V.A.; Hauswirth, W.W.; Frederick, J.M.; Baehr, W. Rescue of Cone Function in Cone-Only Nphp5 Knockout Mouse Model with Leber Congenital Amaurosis Phenotype. Mol. Vis. 2018, 24, 834–846. [Google Scholar] [PubMed]
- Oh, A.; Pearce, J.W.; Gandolfi, B.; Creighton, E.K.; Suedmeyer, W.K.; Selig, M.; Bosiack, A.P.; Castaner, L.J.; Whiting, R.E.H.; Belknap, E.B.; et al. Early-Onset Progressive Retinal Atrophy Associated with an IQCB1 Variant in African Black-Footed Cats (Felis Nigripes). Sci. Rep. 2017, 7, 43918. [Google Scholar] [CrossRef] [PubMed]
- Narfström, K. Hereditary Progressive Retinal Atrophy in the Abyssinian Cat. J. Hered. 1983, 74, 273–276. [Google Scholar] [CrossRef]
- May-Simera, H.L.; Wan, Q.; Jha, B.S.; Hartford, J.; Khristov, V.; Dejene, R.; Chang, J.; Patnaik, S.; Lu, Q.; Banerjee, P.; et al. Primary Cilium-Mediated Retinal Pigment Epithelium Maturation Is Disrupted in Ciliopathy Patient Cells. Cell Rep. 2018, 22, 189–205. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Gene | MIM Number | Chromosome Location | Protein | Protein Function | Localization in Cilium | Phenotype |
|---|---|---|---|---|---|---|
| NPHP1 | 266900, 607100 | 2q13 | Nephrocystin-1 | Cell-cell and cell-matrix signaling | Transition zone [41] | NPHP, SLSN, JS, BBS [42,43,44] |
| NPHP3 | 606995, 606995 | 3q22 | Nephrocystin-3 | Wnt signaling | Inversin compartments [45] | NPHP, SLSN, MKS, BBS, JS, MSS, COACH, SRTD [46,47,48,49] |
| NPHP4 | 606996, 607215 | 1p36.31 | Nephrocystin-4 | Wnt signaling | Basal body and transition zone [41] | NPHP, SLSN [50,51] |
| IQCB1 | 609254, 609237 | 3q13.33 | IQ calmodulin-binding motif-containing protein-1 | Ciliogenesis, regulation of BBSome complex integrity | Basal body [27] | LCA, NPHP, SLSN, JS [2,52,53] |
| CEP290 | 610189, 610142 | 12q21.32 | Centrosomal protein of 290 kDa | Ciliogenesis, regulation of BBSome complex integrity | Transition zone [27] | LCA, NPHP, SLSN, JS, MKS, BBS [54,55] |
| SDCCAG8 | 613615, 613524 | 1q43-44 | Serologically defined colon cancer antigen 8 | Ciliogenesis, Hedgehog signaling | Basal body [56] | NPHP, SLSN, BBS [17,57] |
| WDR19 | 616307, 608151 | 4p14 | WD repeat-containing protein 19 | Ciliary transport, assembly | IFT-A complex [58] | NPHP, SLSN, ATD, Sensenbrenner syndrome, Caroli syndrome [58,59,60,61] |
| TRAF3IP1 | 616629, 607380 | 2q37.3 | TRAF-3 interacting protein 1 | Cilia import | IFT-B complex [62] | NPHP, SLSN, SRPS, ATD [63,64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, D.; Zeng, Y.; Luo, W.; Leng, C.; Li, C. Senior–Loken Syndrome: Ocular Perspectives on Genetics, Pathogenesis, and Management. Biomolecules 2025, 15, 667. https://doi.org/10.3390/biom15050667
Zhou D, Zeng Y, Luo W, Leng C, Li C. Senior–Loken Syndrome: Ocular Perspectives on Genetics, Pathogenesis, and Management. Biomolecules. 2025; 15(5):667. https://doi.org/10.3390/biom15050667
Chicago/Turabian StyleZhou, Di, Yi Zeng, Weihan Luo, Chenyang Leng, and Chen Li. 2025. "Senior–Loken Syndrome: Ocular Perspectives on Genetics, Pathogenesis, and Management" Biomolecules 15, no. 5: 667. https://doi.org/10.3390/biom15050667
APA StyleZhou, D., Zeng, Y., Luo, W., Leng, C., & Li, C. (2025). Senior–Loken Syndrome: Ocular Perspectives on Genetics, Pathogenesis, and Management. Biomolecules, 15(5), 667. https://doi.org/10.3390/biom15050667