
Cholesterol Sulfate: Pathophysiological Implications and Potential Therapeutics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
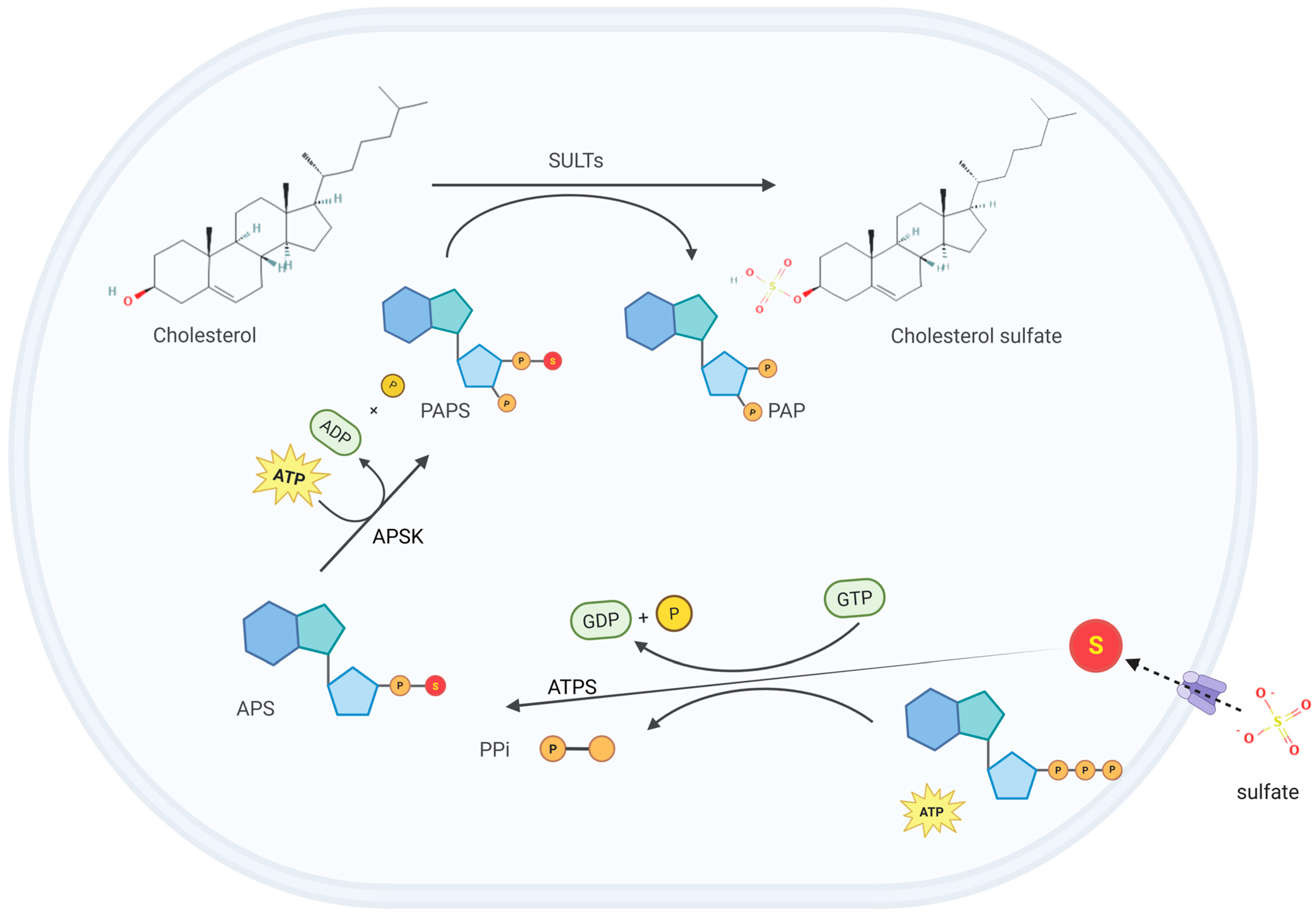
2. CS Biosynthesis and Metabolism
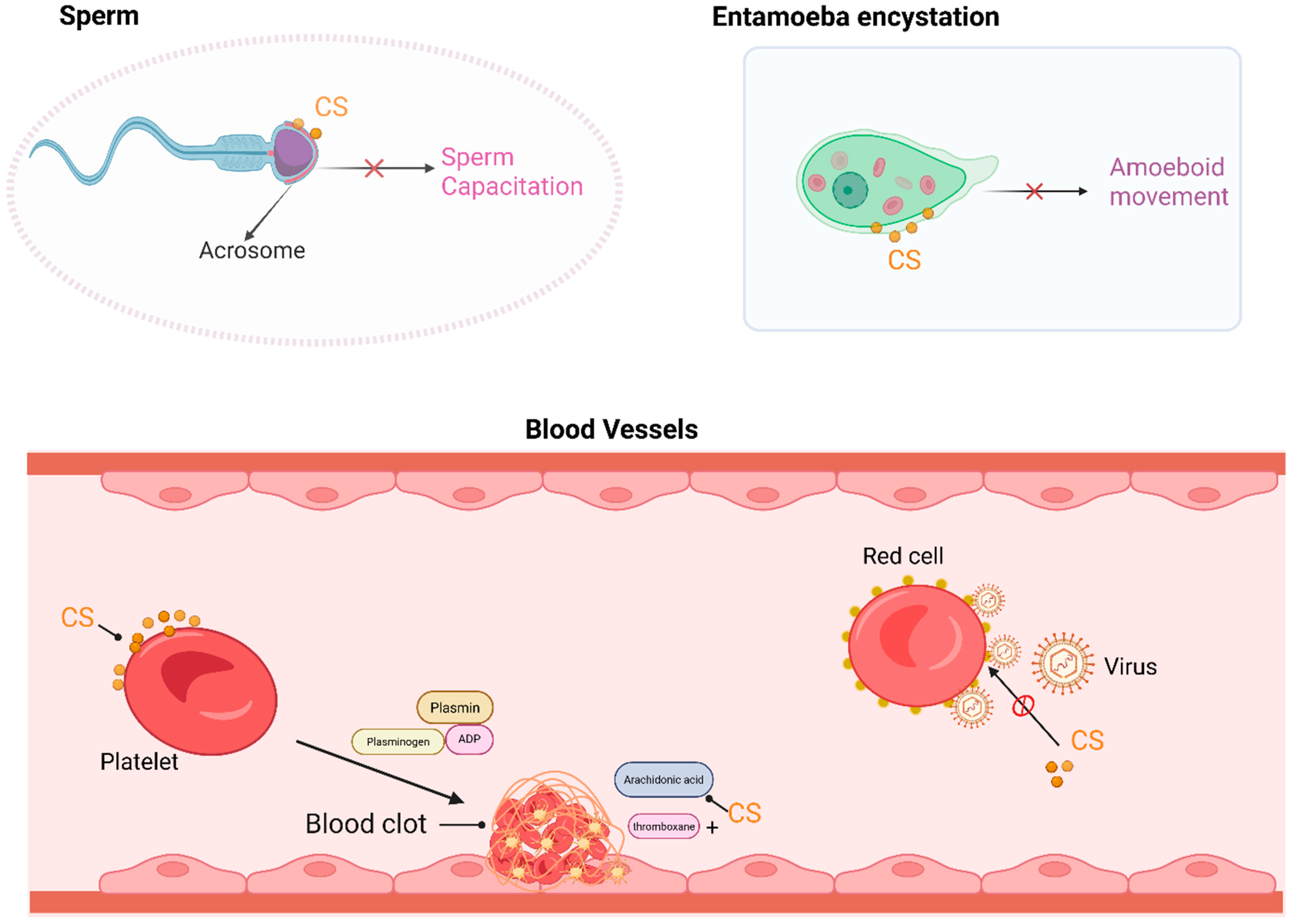
3. Physiological Functions
3.1. CS Regulates Keratinocyte Differentiation and Skin Development
3.2. CS, as a Ligand, Plays Fundamental Roles in Signaling Pathways
3.3. CS Maintains Biological Membrane Homeostasis
3.4. CS Modulates Inflammatory and Immune Responses
3.5. CS Regulates Glucose and Lipid Metabolism
3.6. CS Alters Gut Microbiota-Derived Metabolites
3.7. CS Regulates Neurosteroid Synthesis and Affects Nervous System Function
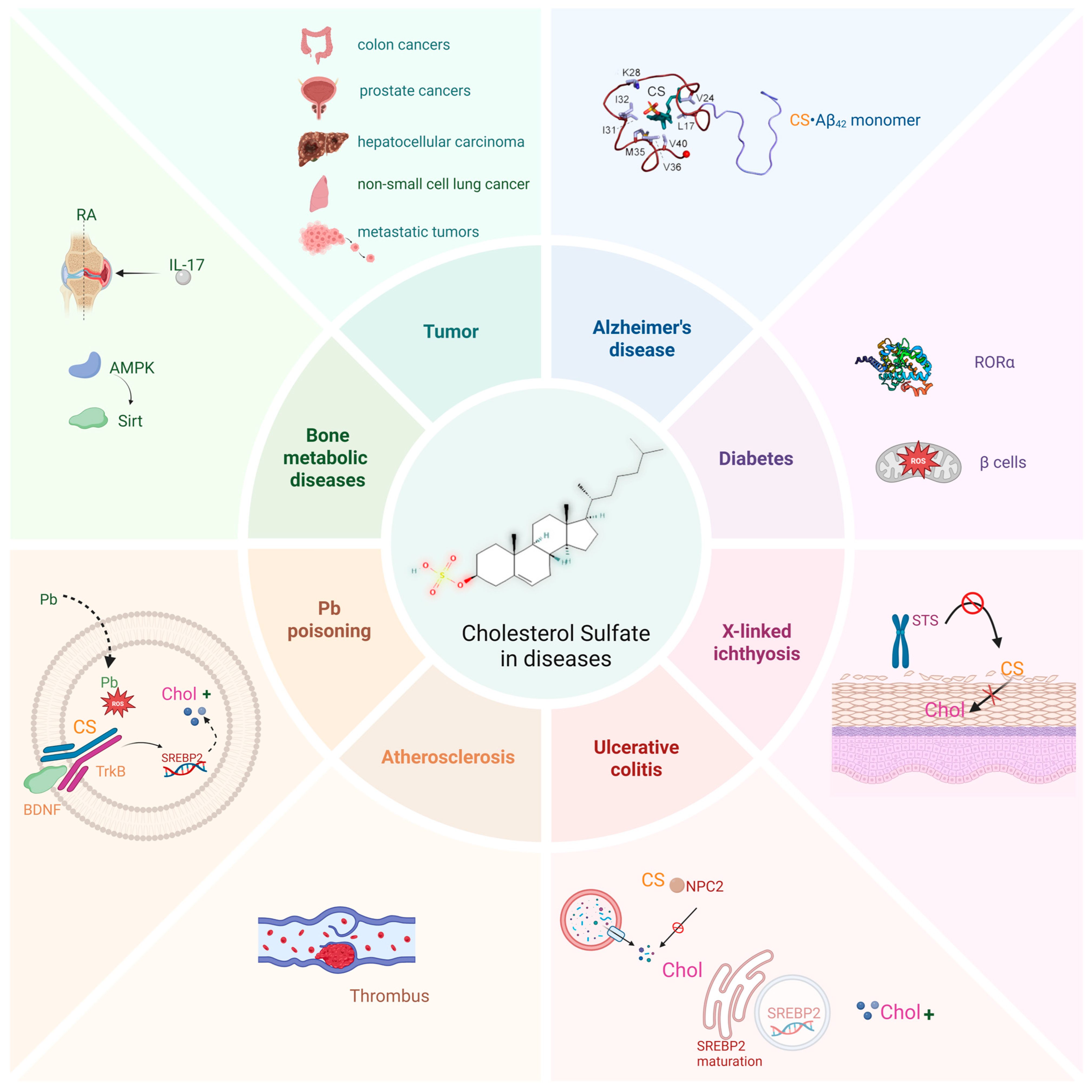
4. CS and Diseases
4.1. X-Linked Ichthyosis
4.2. Diabetes
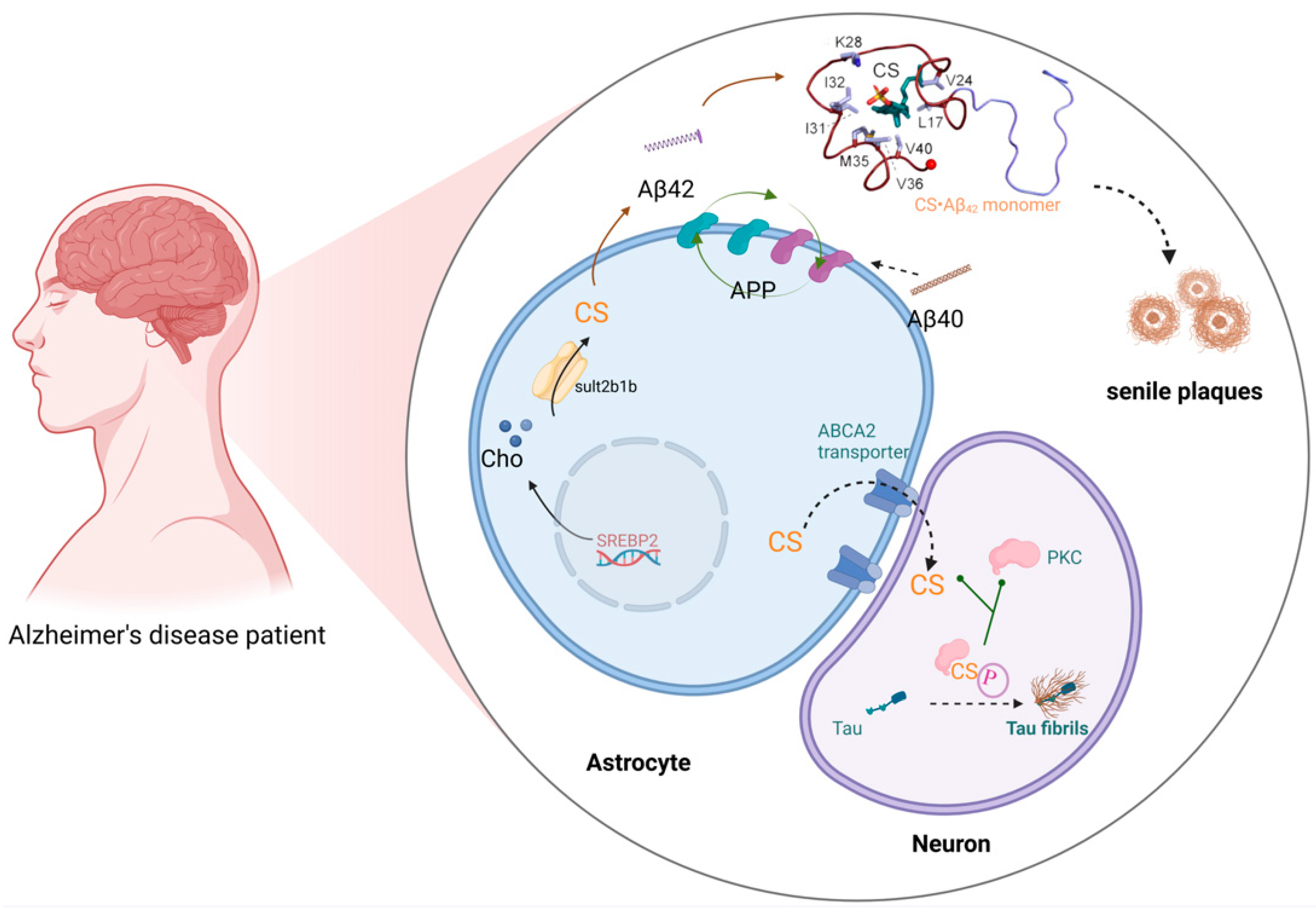
4.3. Alzheimer’s Disease
4.4. Ulcerative Colitis
4.5. Bone Metabolic Diseases
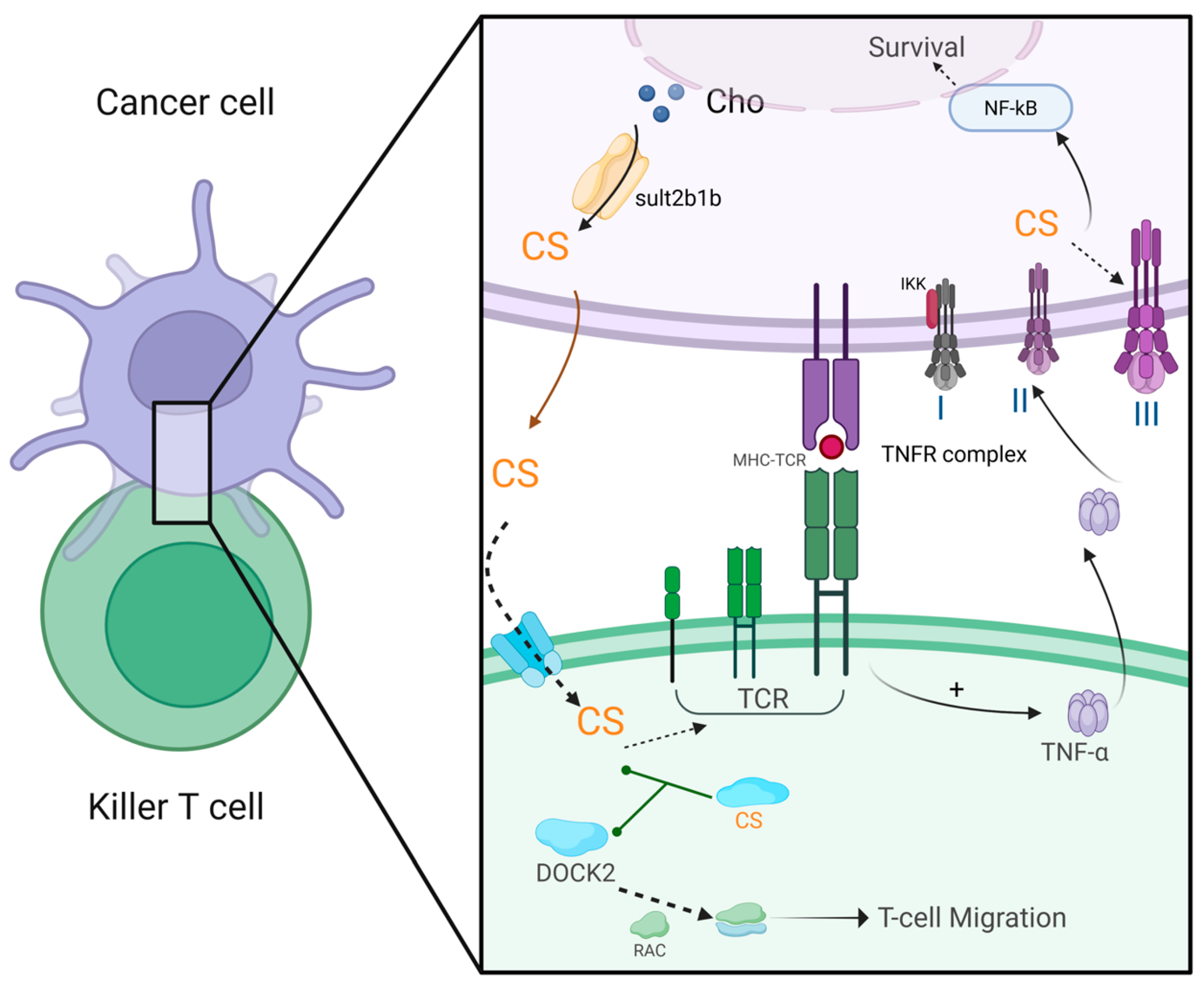
4.6. Cancer
4.7. Atherosclerosis
4.8. Lead Poisoning
5. CS Quantification Strategies
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vitku, J.; Hampl, R. Steroid Conjugates and Their Physiological Role. Physiol. Res. 2023, 72, S317–S322. [Google Scholar] [CrossRef]
- Gunal, S.; Hardman, R.; Kopriva, S.; Mueller, J.W. Sulfation pathways from red to green. J. Biol. Chem. 2019, 294, 12293–12312. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard-Olesen, C.; Ghisari, M.; Kjeldsen, L.S.; Wielsoe, M.; Bonefeld-Jorgensen, E.C. Estrone sulfate and dehydroepiandrosterone sulfate: Transactivation of the estrogen and androgen receptor. Steroids 2016, 105, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Strott, C.A.; Higashi, Y. Cholesterol sulfate in human physiology: What’s it all about? J. Lipid Res. 2003, 44, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.D.; Pontini, L.; Marinozzi, M.; Sanchez-Aranguren, L.C.; Reis, A.; Dias, I.H.K. Cholesterol and oxysterol sulfates: Pathophysiological roles and analytical challenges. Br. J. Pharmacol. 2021, 178, 3327–3341. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, D.; Cui, D.; Liu, Y.; He, S.; Zhang, H.; Xie, Y.; Yu, X.; Yang, S.; Chen, Y.; et al. Cholesterol Sulfate Exerts Protective Effect on Pancreatic beta-Cells by Regulating beta-Cell Mass and Insulin Secretion. Front. Pharmacol. 2022, 13, 840406. [Google Scholar]
- Leyh, T.S. The physical biochemistry and molecular genetics of sulfate activation. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 515–542. [Google Scholar] [CrossRef]
- Mueller, J.W.; Gilligan, L.C.; Idkowiak, J.; Arlt, W.; Foster, P.A. The Regulation of Steroid Action by Sulfation and Desulfation. Endocr. Rev. 2015, 36, 526–563. [Google Scholar] [CrossRef]
- Suiko, M.; Kurogi, K.; Hashiguchi, T.; Sakakibara, Y.; Liu, M.C. Updated perspectives on the cytosolic sulfotransferases (SULTs) and SULT-mediated sulfation. Biosci. Biotechnol. Biochem. 2017, 81, 63–72. [Google Scholar] [CrossRef]
- Nowell, S.; Falany, C.N. Pharmacogenetics of human cytosolic sulfotransferases. Oncogene 2006, 25, 1673–1678. [Google Scholar] [CrossRef]
- Lindsay, J.; Wang, L.L.; Li, Y.; Zhou, S.F. Structure, function and polymorphism of human cytosolic sulfotransferases. Curr. Drug Metab. 2008, 9, 99–105. [Google Scholar] [PubMed]
- Javitt, N.B.; Lee, Y.C.; Shimizu, C.; Fuda, H.; Strott, C.A. Cholesterol and hydroxycholesterol sulfotransferases: Identification, distinction from dehydroepiandrosterone sulfotransferase, and differential tissue expression. Endocrinology 2001, 142, 2978–2984. [Google Scholar] [CrossRef] [PubMed]
- Her, C.; Wood, T.C.; Eichler, E.E.; Mohrenweiser, H.W.; Ramagli, L.S.; Siciliano, M.J.; Weinshilboum, R.M. Human hydroxysteroid sulfotransferase SULT2B1: Two enzymes encoded by a single chromosome 19 gene. Genomics 1998, 53, 284–295. [Google Scholar] [CrossRef]
- Sugawara, T.; Nomura, E.; Hoshi, N. Cholesterol sulphate affects production of steroid hormones by reducing steroidogenic acute regulatory protein level in adrenocortical cells. J. Endocrinol. 2007, 195, 451–458. [Google Scholar] [CrossRef]
- Rizner, T.L. The Important Roles of Steroid Sulfatase and Sulfotransferases in Gynecological Diseases. Front. Pharmacol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Polito, M.P.; Marini, G.; Palamenghi, M.; Enzo, E. Decoding the Human Epidermal Complexity at Single-Cell Resolution. Int. J. Mol. Sci. 2023, 24, 8544. [Google Scholar] [CrossRef] [PubMed]
- Rearick, J.I.; Jetten, A.M. Accumulation of cholesterol 3-sulfate during in vitro squamous differentiation of rabbit tracheal epithelial cells and its regulation by retinoids. J. Biol. Chem. 1986, 261, 13898–13904. [Google Scholar] [CrossRef] [PubMed]
- Sjovall, P.; Gregoire, S.; Wargniez, W.; Skedung, L.; Luengo, G.S. 3D Molecular Imaging of Stratum Corneum by Mass Spectrometry Suggests Distinct Distribution of Cholesteryl Esters Compared to Other Skin Lipids. Int. J. Mol. Sci. 2022, 23, 13799. [Google Scholar] [CrossRef]
- Fandrei, F.; Engberg, O.; Opalka, L.; Jancalkova, P.; Pullmannova, P.; Steinhart, M.; Kovacik, A.; Vavrova, K.; Huster, D. Cholesterol sulfate fluidizes the sterol fraction of the stratum corneum lipid phase and increases its permeability. J. Lipid Res. 2022, 63, 100177. [Google Scholar] [CrossRef]
- Nemes, Z.; Demeny, M.; Marekov, L.N.; Fesus, L.; Steinert, P.M. Cholesterol 3-sulfate interferes with cornified envelope assembly by diverting transglutaminase 1 activity from the formation of cross-links and esters to the hydrolysis of glutamine. J. Biol. Chem. 2000, 275, 2636–2646. [Google Scholar] [CrossRef]
- Takahashi, H.; Asano, K.; Manabe, A.; Kinouchi, M.; Ishida-Yamamoto, A.; Iizuka, H. The alpha and eta isoforms of protein kinase C stimulate transcription of human involucrin gene. J. Investig. Dermatol. 1998, 110, 218–223. [Google Scholar] [CrossRef]
- Kuroki, T.; Ikuta, T.; Kashiwagi, M.; Kawabe, S.; Ohba, M.; Huh, N.; Mizuno, K.; Ohno, S.; Yamada, E.; Chida, K. Cholesterol sulfate, an activator of protein kinase C mediating squamous cell differentiation: A review. Mutat. Res. 2000, 462, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, O.; Nakae, H.; Miida, T.; Higashi, Y.; Fuda, H.; Endo, M.; Kohjitani, A.; Sone, H.; Strott, C.A. Cholesterol sulfate induces expression of the skin barrier protein filaggrin in normal human epidermal keratinocytes through induction of RORalpha. Biochem. Biophys. Res. Commun. 2012, 428, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Rearick, J.I.; Hesterberg, T.W.; Jetten, A.M. Human bronchial epithelial cells synthesize cholesterol sulfate during squamous differentiation in vitro. J. Cell. Physiol. 1987, 133, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.L.; Rutherford, S.L.; Ponec, M.; Hincenbergs, M.; Placzek, D.R.; Elias, P.M. Density-dependent variations in the lipid content and metabolism of cultured human keratinocytes. J. Investig. Dermatol. 1988, 91, 86–91. [Google Scholar] [CrossRef]
- Nematisouldaragh, D.; Kirshenbaum, E.; Uzonna, M.; Kirshenbaum, L.; Rabinovich-Nikitin, I. The Role of Retinoic-Acid-Related Orphan Receptor (RORs) in Cellular Homeostasis. Int. J. Mol. Sci. 2024, 25, 11340. [Google Scholar] [CrossRef]
- Kallen, J.; Schlaeppi, J.M.; Bitsch, F.; Delhon, I.; Fournier, B. Crystal structure of the human RORα ligand binding domain in complex with cholesterol sulfate at 2.2 Å. J. Biol. Chem. 2004, 279, 14033–14038. [Google Scholar] [CrossRef]
- Woscholski, R.; Kodaki, T.; Palmer, R.H.; Waterfield, M.D.; Parker, P.J. Modulation of the substrate specificity of the mammalian phosphatidylinositol 3-kinase by cholesterol sulfate and sulfatide. Biochemistry 1995, 34, 11489–11493. [Google Scholar] [CrossRef]
- Su, W.Y.; Tian, L.Y.; Guo, L.P.; Huang, L.Q.; Gao, W.Y. PI3K signaling-regulated metabolic reprogramming: From mechanism to application. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188952. [Google Scholar] [CrossRef]
- Reis, A.; Dias, I.H.K. Oxysterol sulfates in fluids, cells and tissues: How much do we know about their clinical significance, biological relevance and biophysical implications? Essays Biochem. 2024, 68, 401–410. [Google Scholar]
- Cheetham, J.J.; Epand, R.M.; Andrews, M.; Flanagan, T.D. Cholesterol sulfate inhibits the fusion of Sendai virus to biological and model membranes. J. Biol. Chem. 1990, 265, 12404–12409. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, J.J.; Nir, S.; Johnson, E.; Flanagan, T.D.; Epand, R.M. The effects of membrane physical properties on the fusion of Sendai virus with human erythrocyte ghosts and liposomes. Analysis of kinetics and extent of fusion. J. Biol. Chem. 1994, 269, 5467–5472. [Google Scholar] [CrossRef] [PubMed]
- Blache, D.; Becchi, M.; Davignon, J. Occurrence and biological effects of cholesteryl sulfate on blood platelets. Biochim. Biophys. Acta 1995, 1259, 291–296. [Google Scholar] [CrossRef]
- Merten, M.; Dong, J.F.; Lopez, J.A.; Thiagarajan, P. Cholesterol sulfate: A new adhesive molecule for platelets. Circulation 2001, 103, 2032–2034. [Google Scholar] [CrossRef]
- Lopalco, P.; Vitale, R.; Cho, Y.S.; Totaro, P.; Corcelli, A.; Lobasso, S. Alteration of Cholesterol Sulfate/Seminolipid Ratio in Semen Lipid Profile of Men With Oligoasthenozoospermia. Front. Physiol. 2019, 10, 1344. [Google Scholar] [CrossRef]
- Di Nisio, A.; De Toni, L.; Sabovic, I.; Vignoli, A. Lipidomic Profile of Human Sperm Membrane Identifies a Clustering of Lipids Associated with Semen Quality and Function. Int. J. Mol. Sci. 2023, 25, 297. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.A. Capacitation mechanisms, and the role of capacitation as seen in eutherian mammals. Reprod. Fertil. Dev. 1996, 8, 581–594. [Google Scholar] [CrossRef]
- Osheroff, J.E.; Visconti, P.E.; Valenzuela, J.P.; Travis, A.J.; Alvarez, J.; Kopf, G.S. Regulation of human sperm capacitation by a cholesterol efflux-stimulated signal transduction pathway leading to protein kinase A-mediated up-regulation of protein tyrosine phosphorylation. Mol. Hum. Reprod. 1999, 5, 1017–1026. [Google Scholar] [CrossRef]
- Garolla, A.; Sabovic, I.; Tescari, S.; De Toni, L.; Menegazzo, M.; Cosci, I.; De Filippis, V.; Giarola, M.; Foresta, C. Impaired sperm function in infertile men relies on the membrane sterol pattern. Andrology 2018, 6, 325–334. [Google Scholar] [CrossRef]
- Guillen, N. Pathogenicity and virulence of Entamoeba histolytica, the agent of amoebiasis. Virulence 2023, 14, 2158656. [Google Scholar] [CrossRef]
- Mi-Ichi, F.; Tsugawa, H.; Arita, M.; Yoshida, H. Pleiotropic Roles of Cholesteryl Sulfate during Entamoeba Encystation: Involvement in Cell Rounding and Development of Membrane Impermeability. mSphere 2022, 7, e0029922. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Boyce, J.A.; Barrett, N.A. Cysteinyl Leukotrienes in Allergic Inflammation. Annu. Rev. Pathol. 2025, 20, 115–141. [Google Scholar] [CrossRef]
- Chen, F.; Ghosh, A.; Lin, J.; Zhang, C.; Pan, Y.; Thakur, A.; Singh, K.; Hong, H.; Tang, S. 5-lipoxygenase pathway and its downstream cysteinyl leukotrienes as potential therapeutic targets for Alzheimer’s disease. Brain Behav. Immun. 2020, 88, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Antoniu, S.A. Targeting 5-lipoxygenase-activating protein in asthma and chronic obstructive pulmonary disease. Expert. Opin. Ther. Targets 2014, 18, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrov, D.A.; Zagryagskaya, A.N.; Pushkareva, M.A.; Bachschmid, M.; Peters-Golden, M.; Werz, O.; Steinhilber, D.; Sud’ina, G.F. Cholesterol and its anionic derivatives inhibit 5-lipoxygenase activation in polymorphonuclear leukocytes and MonoMac6 cells. FEBS J. 2006, 273, 548–557. [Google Scholar] [CrossRef]
- Park, J.S.; Moon, S.J.; Lim, M.A.; Byun, J.K.; Hwang, S.H.; Yang, S.; Kim, E.K.; Lee, H.; Kim, S.M.; Lee, J.; et al. Retinoic Acid Receptor-Related Receptor Alpha Ameliorates Autoimmune Arthritis via Inhibiting of Th17 Cells and Osteoclastogenesis. Front. Immunol. 2019, 10, 2270. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, H.; Wang, Y.; Yao, Y.; Yang, C.; Meng, J.; Tan, X.; Nie, Y.; Xue, L.; Xu, B.; et al. Sult2b1 deficiency exacerbates ischemic stroke by promoting pro-inflammatory macrophage polarization in mice. Theranostics 2021, 11, 10074–10090. [Google Scholar] [CrossRef]
- Weng, N.P. Numbers and odds: TCR repertoire size and its age changes impacting on T cell functions. Semin. Immunol. 2023, 69, 101810. [Google Scholar] [CrossRef]
- Pathan-Chhatbar, S.; Drechsler, C.; Richter, K.; Morath, A.; Wu, W.; OuYang, B.; Xu, C.; Schamel, W.W. Direct Regulation of the T Cell Antigen Receptor’s Activity by Cholesterol. Front. Cell Dev. Biol. 2020, 8, 615996. [Google Scholar] [CrossRef]
- Wu, H.; Cao, R.; Wei, S.; Pathan-Chhatbar, S.; Wen, M.; Wu, B.; Schamel, W.W.; Wang, S.; OuYang, B. Cholesterol Binds in a Reversed Orientation to TCRbeta-TM in Which Its OH Group is Localized to the Center of the Lipid Bilayer. J. Mol. Biol. 2021, 433, 167328. [Google Scholar] [CrossRef]
- Cai, E.; Marchuk, K.; Beemiller, P.; Beppler, C.; Rubashkin, M.G.; Weaver, V.M.; Gérard, A.; Liu, T.L.; Chen, B.C.; Betzig, E.; et al. Visualizing dynamic microvillar search and stabilization during ligand detection by T cells. Science 2017, 356, aal3118. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Chung, I.-J.; Kim, H.-R.; Jun, C.-D. The Immunosuppressive Potential of Cholesterol Sulfate Through T Cell Microvilli Disruption. Immune Netw. 2023, 23, e29. [Google Scholar] [CrossRef]
- Wang, F.; Beck-Garcia, K.; Zorzin, C.; Schamel, W.W.; Davis, M.M. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat. Immunol. 2016, 17, 844–850. [Google Scholar] [CrossRef]
- Kunimura, K.; Uruno, T.; Fukui, Y. DOCK family proteins: Key players in immune surveillance mechanisms. Int. Immunol. 2020, 32, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Uruno, T.; Sugiura, Y.; Tatsuguchi, T.; Yamamura, K.; Ushijima, M.; Hattori, Y.; Kukimoto-Niino, M.; Mishima-Tsumagari, C.; Watanabe, M.; et al. Cholesterol sulfate is a DOCK2 inhibitor that mediates tissue-specific immune evasion in the eye. Sci. Signal. 2018, 11, eaao4874. [Google Scholar] [CrossRef]
- Kostarnoy, A.V.; Gancheva, P.G.; Lepenies, B.; Tukhvatulin, A.I.; Dzharullaeva, A.S.; Polyakov, N.B.; Grumov, D.A.; Egorova, D.A.; Kulibin, A.Y.; Bobrov, M.A.; et al. Receptor Mincle promotes skin allergies and is capable of recognizing cholesterol sulfate. Proc. Natl. Acad. Sci. USA 2017, 114, E2758–E2765. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.H.; Shi, X.J.; Zhu, J.J.; Guan, X.D.; Garbacz, W.G.; Huang, Y.X.; Gao, L.; Yan, J.; Xu, M.S.; Ren, S.R.; et al. Regulation of Cholesterol Sulfotransferase SULT2B1b by Hepatocyte Nuclear Factor 4α Constitutes a Negative Feedback Control of Hepatic Gluconeogenesis. Mol. Cell Biol. 2018, 38, e00654-17. [Google Scholar] [CrossRef]
- Shi, X.J.; Cheng, Q.Q.; Xu, L.Y.; Yan, J.; Jiang, M.X.; He, J.H.; Xu, M.S.; Stefanovic-Racic, M.; Sipula, I.; O’Doherty, R.M.; et al. Cholesterol Sulfate and Cholesterol Sulfotransferase Inhibit Gluconeogenesis by Targeting Hepatocyte Nuclear Factor 4α. Mol. Cell Biol. 2014, 34, 485–497. [Google Scholar] [CrossRef]
- Kim, E.J.; Yoon, Y.S.; Hong, S.; Son, H.Y.; Na, T.Y.; Lee, M.H.; Kang, H.J.; Park, J.; Cho, W.J.; Kim, S.G.; et al. Retinoic acid receptor-related orphan receptor alpha-induced activation of adenosine monophosphate-activated protein kinase results in attenuation of hepatic steatosis. Hepatology 2012, 55, 1379–1388. [Google Scholar] [CrossRef]
- Williams, M.L.; Hughes-Fulford, M.; Elias, P.M. Inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and sterol synthesis by cholesterol sulfate in cultured fibroblasts. Biochim. Biophys. Acta 1985, 845, 349–357. [Google Scholar] [CrossRef]
- Neunzig, J.; Bernhardt, R. Effect of sulfonated steroids on steroidogenic cytochrome P450-dependent steroid hydroxylases. J. Steroid Biochem. Mol. Biol. 2018, 179, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Koay, Y.C.; Wali, J.A.; Luk, A.W.S.; Macia, L.; Cogger, V.C.; Pulpitel, T.J.; Wahl, D.; Solon-Biet, S.M.; Holmes, A.; Simpson, S.J.; et al. Ingestion of resistant starch by mice markedly increases microbiome-derived metabolites. FASEB J. 2019, 33, 8033–8042. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Huang, X.; Liang, X.; Su, M.; Lai, K.P.; Chen, J. Integrated omics analysis reveals the alteration of gut microbe-metabolites in obese adults. Brief. Bioinform. 2021, 22, bbaa165. [Google Scholar] [CrossRef] [PubMed]
- Le, H.H.; Lee, M.T.; Besler, K.R.; Comrie, J.M.C.; Johnson, E.L. Characterization of interactions of dietary cholesterol with the murine and human gut microbiome. Nat. Microbiol. 2022, 7, 1390–1403. [Google Scholar] [CrossRef]
- Yao, L.; D’Agostino, G.D.; Park, J.; Hang, S.; Adhikari, A.A.; Zhang, Y.; Li, W.; Avila-Pacheco, J.; Bae, S.; Clish, C.B.; et al. A biosynthetic pathway for the selective sulfonation of steroidal metabolites by human gut bacteria. Nat. Microbiol. 2022, 7, 1404–1418. [Google Scholar] [CrossRef]
- Liu, Y.; Xiao, H.; Wang, Z.; Pan, Q.; Zhao, X.; Lu, B. Interactions between dietary cholesterol and intestinal flora and their effects on host health. Crit. Rev. Food Sci. Nutr. 2025, 65, 494–506. [Google Scholar] [CrossRef]
- Ivanisevic, J.; Epstein, A.A.; Kurczy, M.E.; Benton, P.H.; Uritboonthai, W.; Fox, H.S.; Boska, M.D.; Gendelman, H.E.; Siuzdak, G. Brain region mapping using global metabolomics. Chem. Biol. 2014, 21, 1575–1584. [Google Scholar] [CrossRef]
- Biagini, G.; Marinelli, C.; Panuccio, G.; Puia, G.; Avoli, M. Glia-Neuron Interactions: Neurosteroids and Epileptogenesis. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; Oxford University Press: Bethesda, MD, USA, 2012. [Google Scholar]
- Biagini, G.; Rustichelli, C.; Curia, G.; Vinet, J.; Lucchi, C.; Pugnaghi, M.; Meletti, S. Neurosteroids and epileptogenesis. J. Neuroendocrinol. 2013, 25, 980–990. [Google Scholar] [CrossRef]
- De Nicola, A.F.; Garay, L.I.; Meyer, M.; Guennoun, R.; Sitruk-Ware, R.; Schumacher, M.; Gonzalez Deniselle, M.C. Neurosteroidogenesis and progesterone anti-inflammatory/neuroprotective effects. J. Neuroendocrinol. 2018, 30, e12502. [Google Scholar] [CrossRef]
- Prah, J.; Winters, A.; Chaudhari, K.; Hersh, J.; Liu, R.; Yang, S.H. Cholesterol sulfate alters astrocyte metabolism and provides protection against oxidative stress. Brain Res. 2019, 1723, 146378. [Google Scholar] [CrossRef]
- Pierce, S.R.; Germann, A.L.; Steinbach, J.H.; Akk, G. The Sulfated Steroids Pregnenolone Sulfate and Dehydroepiandrosterone Sulfate Inhibit the alpha1beta3gamma2L GABA(A) Receptor by Stabilizing a Novel Nonconducting State. Mol. Pharmacol. 2022, 101, 68–77. [Google Scholar] [CrossRef]
- Clark, B.J.; Klinge, C.M. Structure-function of DHEA binding proteins. Vitam. Horm. 2023, 123, 587–617. [Google Scholar]
- Traish, A.M.; Kang, H.P.; Saad, F.; Guay, A.T. Dehydroepiandrosterone (DHEA)—A precursor steroid or an active hormone in human physiology. J. Sex. Med. 2011, 8, 2960–2982; quiz 2983. [Google Scholar] [CrossRef] [PubMed]
- Vitku, J.; Hill, M.; Kolatorova, L.; Kubala Havrdova, E.; Kancheva, R. Steroid Sulfation in Neurodegenerative Diseases. Front. Mol. Biosci. 2022, 9, 839887. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, A.T.; Stepanov, A.A.; Butkova, T.V.; Malsagova, K.A.; Zakharova, N.V.; Kostyuk, G.P.; Elmuratov, A.U.; Kaysheva, A.L. Consolidation of metabolomic, proteomic, and GWAS data in connective model of schizophrenia. Sci. Rep. 2023, 13, 2139. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liang, C.; Li, P.; Xiao, H. Revisiting X-linked congenital ichthyosis. Int. J. Dermatol. 2024, 64, 51–61. [Google Scholar] [CrossRef]
- McGeoghan, F.; Camera, E.; Maiellaro, M.; Menon, M.; Huang, M.; Dewan, P.; Ziaj, S.; Caley, M.P.; Donaldson, M.; Enright, A.J.; et al. RNA sequencing and lipidomics uncovers novel pathomechanisms in recessive X-linked ichthyosis. Front. Mol. Biosci. 2023, 10, 1176802. [Google Scholar] [CrossRef]
- Lassen, N.; Bateman, J.B.; Estey, T.; Kuszak, J.R.; Nees, D.W.; Piatigorsky, J.; Duester, G.; Day, B.J.; Huang, J.; Hines, L.M.; et al. Multiple and additive functions of ALDH3A1 and ALDH1A1-Cataract phenotype and ocular oxidative damage in Aldh3a1(-/-)/Aldh1a1(-/-) knock-out mice. J. Biol. Chem. 2007, 282, 25668–25676. [Google Scholar] [CrossRef]
- Jacob, S.; Brune, C.W.; Carter, C.S.; Leventhal, B.L.; Lord, C.; Cook, E.H. Association of the oxytocin receptor gene (OXTR) in Caucasian children and adolescents with autism. Neurosci. Lett. 2007, 417, 6–9. [Google Scholar] [CrossRef]
- Wren, G.H.; Davies, W. X-linked ichthyosis: New insights into a multi-system disorder. Skin. Health Dis. 2022, 2, e179. [Google Scholar] [CrossRef]
- Cui, D.; Feng, X.; Lei, S.; Zhang, H.; Hu, W.; Yang, S.; Yu, X.; Su, Z. Pancreatic beta-cell failure, clinical implications, and therapeutic strategies in type 2 diabetes. Chin. Med. J. 2024, 137, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, S.; Zhou, R.; Zhang, X.; Yang, S.; Deng, D.; Zhang, C.; Yu, X.; Chen, Y.; Su, Z. Nuclear Factor-Y in Mouse Pancreatic beta-Cells Plays a Crucial Role in Glucose Homeostasis by Regulating beta-Cell Mass and Insulin Secretion. Diabetes 2021, 70, 1703–1716. [Google Scholar] [CrossRef]
- Perego, C.; Da Dalt, L.; Pirillo, A.; Galli, A.; Catapano, A.L.; Norata, G.D. Cholesterol metabolism, pancreatic beta-cell function and diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, S.; Chen, J.; Su, Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. 2018, 9, 802. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Y.; Zhang, J.; Liu, Y.; Zhang, Y.; Su, Z. Retinoic acid receptor-related orphan receptor alpha stimulates adipose tissue inflammation by modulating endoplasmic reticulum stress. J. Biol. Chem. 2017, 292, 13959–13969. [Google Scholar] [CrossRef]
- Kuang, J.; Hou, X.; Zhang, J.; Chen, Y.; Su, Z. Identification of insulin as a novel retinoic acid receptor-related orphan receptor alpha target gene. FEBS Lett. 2014, 588, 1071–1079. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Ji, Q.Q.; Chen, J.Q.; Li, Y.F.; Tao, E.X.; Zhan, Y.Q. Incidence and prevalence of Alzheimer’s disease in China: A systematic review and meta-analysis. Eur. J. Epidemiol. 2024, 39, 701–714. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef]
- Elbassal, E.A.; Liu, H.; Morris, C.; Wojcikiewicz, E.P.; Du, D. Effects of Charged Cholesterol Derivatives on Abeta40 Amyloid Formation. J. Phys. Chem. B 2016, 120, 59–68. [Google Scholar] [CrossRef]
- Cook, I.; Leyh, T.S. Sulfotransferase 2B1b, Sterol Sulfonation, and Disease. Pharmacol. Rev. 2023, 75, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Cook, I.; Leyh, T.S. Sterol-activated amyloid beta fibril formation. J. Biol. Chem. 2023, 299, 105445. [Google Scholar] [CrossRef]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol. Psychiatry 2023, 28, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Maulik, M.; Westaway, D.; Jhamandas, J.H.; Kar, S. Role of cholesterol in APP metabolism and its significance in Alzheimer’s disease pathogenesis. Mol. Neurobiol. 2013, 47, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shou, Y.; Pan, J.; Du, Y.; Liu, C.; Wang, H. The relationship between cholesterol level and Alzheimer’s disease-associated APP proteolysis/Abeta metabolism. Nutr. Neurosci. 2019, 22, 453–463. [Google Scholar] [CrossRef]
- Denning, M.F.; Kazanietz, M.G.; Blumberg, P.M.; Yuspa, S.H. Cholesterol sulfate activates multiple protein kinase C isoenzymes and induces granular cell differentiation in cultured murine keratinocytes. Cell Growth Differ. 1995, 6, 1619–1626. [Google Scholar]
- Tanaka, T.; Tsujio, I.; Nishikawa, T.; Shinosaki, K.; Kudo, T.; Takeda, M. Significance of tau phosphorylation and protein kinase regulation in the pathogenesis of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2000, 14 (Suppl. S1), S18–S24. [Google Scholar] [CrossRef]
- Froger, N. New therapeutic avenues for neurosteroids in psychiatric diseases. Biol. Aujourdhui 2019, 213, 131–140. [Google Scholar] [CrossRef]
- Monnet, F.P.; Maurice, T. The sigma1 protein as a target for the non-genomic effects of neuro(active)steroids: Molecular, physiological, and behavioral aspects. J. Pharmacol. Sci. 2006, 100, 93–118. [Google Scholar] [CrossRef]
- de Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Zhang, M.; Li, X.; Zhang, Q.; Yang, J.; Liu, G. Roles of macrophages on ulcerative colitis and colitis-associated colorectal cancer. Front. Immunol. 2023, 14, 1103617. [Google Scholar] [CrossRef] [PubMed]
- Harbord, M.; Eliakim, R.; Bettenworth, D.; Karmiris, K.; Katsanos, K.; Kopylov, U.; Kucharzik, T.; Molnar, T.; Raine, T.; Sebastian, S.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 2: Current Management. J. Crohn’s Colitis 2017, 11, 769–784. [Google Scholar] [CrossRef]
- Xu, D.; Ma, R.; Ju, Y.; Song, X.; Niu, B.; Hong, W.; Wang, R.; Yang, Q.; Zhao, Z.; Zhang, Y.; et al. Cholesterol sulfate alleviates ulcerative colitis by promoting cholesterol biosynthesis in colonic epithelial cells. Nat. Commun. 2022, 13, 4428. [Google Scholar] [CrossRef]
- Infante, R.E.; Radhakrishnan, A.; Abi-Mosleh, L.; Kinch, L.N.; Wang, M.L.; Grishin, N.V.; Goldstein, J.L.; Brown, M.S. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 2008, 283, 1064–1075. [Google Scholar] [CrossRef]
- Xu, S.; Benoff, B.; Liou, H.L.; Lobel, P.; Stock, A.M. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J. Biol. Chem. 2007, 282, 23525–23531. [Google Scholar] [CrossRef] [PubMed]
- Morino, K.; Kunimura, K.; Sugiura, Y.; Izumi, Y.; Matsubara, K.; Akiyoshi, S.; Maeda, R.; Hirotani, K.; Sakata, D.; Mizuno, S.; et al. Cholesterol sulfate limits neutrophil recruitment and gut inflammation during mucosal injury. Front. Immunol. 2023, 14, 1131146. [Google Scholar] [CrossRef]
- Minoia, A.; Dalle Carbonare, L.; Schwamborn, J.C.; Bolognin, S.; Valenti, M.T. Bone Tissue and the Nervous System: What Do They Have in Common? Cells 2022, 12, 51. [Google Scholar] [CrossRef]
- Hodgkinson, T.; Tsimbouri, P.M.; Llopis-Hernandez, V. The use of nanovibration to discover specific and potent bioactive metabolites that stimulate osteogenic differentiation in mesenchymal stem cells. Sci. Adv. 2021, 7, eabb7921. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, J.; Lee, G.R.; Kwon, M.; Lee, H.I.; Kim, N.; Kim, H.J.; Lee, M.O. Cholesterol sulfate inhibits osteoclast differentiation and survival by regulating the AMPK-Sirt1-NF-κB pathway. J. Cell. Physiol. 2023, 238, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- van de Kamp, J.M.; Bokenkamp, A.; Smith, D.E.C.; Wamelink, M.M.C.; Jansen, E.E.W.; Struys, E.A.; Waisfisz, Q.; Verkleij, M.; Hartmann, M.F.; Wang, R.; et al. Biallelic variants in the SLC13A1 sulfate transporter gene cause hyposulfatemia with a mild spondylo-epi-metaphyseal dysplasia. Clin. Genet. 2023, 103, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.W.; Park, J.Y. Host modulation therapy for improving the osseointegration of dental implants under bone healing-suppressed conditions: A preclinical rodent-model experiment. J. Periodontal Implant. Sci. 2024, 54, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Kim, H.R.; Kim, B.M.; Cho, M.L.; Lee, S.H. Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am. J. Pathol. 2015, 185, 3011–3024. [Google Scholar] [CrossRef]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef]
- Tatsuguchi, T.; Uruno, T.; Sugiura, Y.; Oisaki, K.; Takaya, D.; Sakata, D.; Izumi, Y.; Togo, T.; Hattori, Y.; Kunimura, K.; et al. Pharmacological intervention of cholesterol sulfate-mediated T cell exclusion promotes antitumor immunity. Biochem. Biophys. Res. Commun. 2022, 609, 183–188. [Google Scholar] [CrossRef]
- Tatsuguchi, T.; Uruno, T.; Sugiura, Y.; Sakata, D.; Izumi, Y.; Sakurai, T.; Hattori, Y.; Oki, E.; Kubota, N.; Nishimoto, K.; et al. Cancer-derived cholesterol sulfate is a key mediator to prevent tumor infiltration by effector T cells. Int. Immunol. 2022, 34, 277–289. [Google Scholar] [CrossRef]
- Wang, S.; Wang, R.; Xu, N.; Wei, X.Y.; Yang, Y.J.; Lian, Z.X.; Cen, B.N.; Shen, C.C.; Li, W.Y.; Wang, J.G.; et al. SULT2B1-CS-DOCK2 axis regulates effector T-cell exhaustion in HCC microenvironment. Hepatology 2023, 78, 1064–1078. [Google Scholar] [CrossRef]
- Hu, L.; Yang, G.Z.; Zhang, Y.; Feng, D.; Zhai, Y.X.; Gong, H.; Qi, C.Y.; Fu, H.; Ye, M.M.; Cai, Q.P.; et al. Overexpression of SULT2B1b is an independent prognostic indicator and promotes cell growth and invasion in colorectal carcinoma. Lab. Investig. 2015, 95, 1005–1018. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Matsuda, T.; Shimada, M.; Sato, A.; Akase, T.; Yoshinari, K.; Nagata, K.; Yamazoe, Y. Tumor Necrosis Factor-Alpha-Nuclear Factor-Kappa B-Signaling Enhances St2b2 Expression during 12-Tetradecanoylphorbol-13-acetate-Induced Epidermal Hyperplasia. Biol. Pharm. Bull. 2011, 34, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Micheau, O.; Doucey, M.A.; Tschopp, J.; Bron, C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFα-mediated NF-κB activation. Immunity 2003, 18, 655–664. [Google Scholar] [PubMed]
- Yao, W.; Guan, Y. Ginsenosides in cancer: A focus on the regulation of cell metabolism. Biomed. Pharmacother. 2022, 156, 113756. [Google Scholar] [CrossRef]
- Johnson, C.H.; Santidrian, A.F.; LeBoeuf, S.E.; Kurczy, M.E.; Rattray, N.J.W.; Rattray, Z.; Warth, B.; Ritland, M.; Hoang, L.T.; Loriot, C.; et al. Metabolomics guided pathway analysis reveals link between cancer metastasis, cholesterol sulfate, and phospholipids. Cancer Metab. 2017, 5, 9. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyazaki, K.; Higashi, S. Cholesterol sulfate alters substrate preference of matrix metalloproteinase-7 and promotes degradations of pericellular laminin-332 and fibronectin. J. Biol. Chem. 2010, 285, 28862–28873. [Google Scholar]
- Seneff, S.; Davidson, R.M.; Lauritzen, A.; Samsel, A.; Wainwright, G. A novel hypothesis for atherosclerosis as a cholesterol sulfate deficiency syndrome. Theor. Biol. Med. Model. 2015, 12, 9. [Google Scholar]
- Rohani, M.; Pollack, G.H. Flow through horizontal tubes submerged in water in the absence of a pressure gradient: Mechanistic considerations. Langmuir 2013, 29, 6556–6561. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar]
- Samsel, A.; Seneff, S. Glyphosate, pathways to modern diseases II: Celiac sprue and gluten intolerance. Interdiscip. Toxicol. 2013, 6, 159–184. [Google Scholar] [CrossRef]
- Lonn, E.; Yusuf, S.; Arnold, M.J.; Sheridan, P.; Pogue, J.; Micks, M.; McQueen, M.J.; Probstfield, J.; Fodor, G.; Held, C.; et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. N. Engl. J. Med. 2006, 354, 1567–1577. [Google Scholar]
- Mayans, L. Lead Poisoning in Children. Am. Fam. Physician 2019, 100, 24–30. [Google Scholar]
- Richetti, S.K.; Rosemberg, D.B.; Ventura-Lima, J.; Monserrat, J.M.; Bogo, M.R.; Bonan, C.D. Acetylcholinesterase activity and antioxidant capacity of zebrafish brain is altered by heavy metal exposure. Neurotoxicology 2011, 32, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Ferris, H.A.; Perry, R.J.; Moreira, G.V.; Shulman, G.I.; Horton, J.D.; Kahn, C.R. Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc. Natl. Acad. Sci. USA 2017, 114, 1189–1194. [Google Scholar] [CrossRef]
- Wang, S.S.; Lu, A.X.; Li, W.H.; Zhang, H.; Hu, C.P.; Liu, J.X.; Pan, H.; Wu, M.Q.; Xu, X.; Yan, C.H.; et al. Effects of food-borne cholesterol supplementation on lead-induced neurodevelopmental impairments of rats based on BDNF signaling pathway and cholesterol metabolism. Ecotox Environ. Safe 2023, 259, 115026. [Google Scholar] [CrossRef] [PubMed]
- Carito, V.; Ceccanti, M.; Ferraguti, G.; Coccurello, R.; Ciafre, S.; Tirassa, P.; Fiore, M. NGF and BDNF Alterations by Prenatal Alcohol Exposure. Curr. Neuropharmacol. 2019, 17, 308–317. [Google Scholar] [CrossRef]
- Malavika, L.; Mitra, P.; Goyal, T.; Sharma, S.; Purohit, P.; Sharma, P. Association of blood lead levels with neurobehavior and BDNF expression in school going children. J. Trace Elem. Med. Biol. 2021, 66, 126749. [Google Scholar]
- Wang, S.S.; Xu, X.; Lu, A.X.; Li, W.H.; Liu, J.X.; Liu, C.; Yan, C.H. Amelioration of cholesterol sulfate for lead-induced CTX cell apoptosis based on BDNF signaling pathway mediated cholesterol metabolism. Ecotox Environ. Safe 2022, 248, 114307. [Google Scholar] [CrossRef]
- Follis, R.M.; Tep, C.; Genaro-Mattos, T.C.; Kim, M.L.; Ryu, J.C.; Morrison, V.E.; Chan, J.R.; Porter, N.; Carter, B.D.; Yoon, S.O. Metabolic Control of Sensory Neuron Survival by the p75 Neurotrophin Receptor in Schwann Cells. J. Neurosci. 2021, 41, 8710–8724. [Google Scholar] [CrossRef]
- Do, H.T.; Bruelle, C.; Pham, D.D.; Jauhiainen, M.; Eriksson, O.; Korhonen, L.T.; Lindholm, D. Nerve growth factor (NGF) and pro-NGF increase low-density lipoprotein (LDL) receptors in neuronal cells partly by different mechanisms: Role of LDL in neurite outgrowth. J. Neurochem. 2016, 136, 306–315. [Google Scholar] [CrossRef]
- Lee, D.Y.; Kind, T.; Yoon, Y.R.; Fiehn, O.; Liu, K.H. Comparative evaluation of extraction methods for simultaneous mass-spectrometric analysis of complex lipids and primary metabolites from human blood plasma. Anal. Bioanal. Chem. 2014, 406, 7275–7286. [Google Scholar] [CrossRef]
- Sarafian, M.H.; Gaudin, M.; Lewis, M.R.; Martin, F.P.; Holmes, E.; Nicholson, J.K.; Dumas, M.E. Objective Set of Criteria for Optimization of Sample Preparation Procedures for Ultra-High Throughput Untargeted Blood Plasma Lipid Profiling by Ultra Performance Liquid Chromatography-Mass Spectrometry. Anal. Chem. 2014, 86, 5766–5774. [Google Scholar] [CrossRef] [PubMed]
- Reinicke, M.; Schröter, J.; Müller-Klieser, D.; Helmschrodt, C.; Ceglarek, U. Free oxysterols and bile acids including conjugates—Simultaneous quantification in human plasma and cerebrospinal fluid by liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2018, 1037, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guijo, A.; Oji, V.; Hartmann, M.F.; Schuppe, H.C.; Traupe, H.; Wudy, S.A. High levels of oxysterol sulfates in serum of patients with steroid sulfatase deficiency. J. Lipid Res. 2015, 56, 403–412. [Google Scholar] [CrossRef]
- Dias, I.H.K.; Ferreira, R.; Gruber, F.; Vitorino, R.; Rivas-Urbina, A.; Sanchez-Quesada, J.L.; Silva, J.V.; Fardilha, M.; de Freitas, V.; Reis, A. Sulfate-based lipids: Analysis of healthy human fluids and cell extracts. Chem. Phys. Lipids 2019, 221, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.Y.; Shen, S.W.; Ma, Y.J.; Kim, J.K.; Rodriguez-Agudo, D.; Heuman, D.M.; Hylemon, P.B.; Pandak, W.M.; Ren, S.L. 25-Hydroxycholesterol-3-sulfate attenuates inflammatory response via PPARγ signaling in human THP-1 macrophages. Am. J. Physiol-Endoc M. 2012, 302, E788–E799. [Google Scholar] [CrossRef]
- Schuler, G. Steroid sulfates in domestic mammals and laboratory rodents. Domest. Anim. Endocrinol. 2021, 76, 106622. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyazaki, K.; Higashi, S. Pericellular proteolysis by matrix metalloproteinase-7 is differentially modulated by cholesterol sulfate, sulfatide, and cardiolipin. FEBS J. 2014, 281, 3346–3356. [Google Scholar] [CrossRef]
- Geese, W.J.; Raftogianis, R.B. Biochemical characterization and tissue distribution of human SULT2B1. Biochem. Biophys. Res. Commun. 2001, 288, 280–289. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Lei, S.; Shen, Y.; Liu, T.; Li, J.; Wang, J.; Su, Z. Cholesterol Sulfate: Pathophysiological Implications and Potential Therapeutics. Biomolecules 2025, 15, 646. https://doi.org/10.3390/biom15050646
Yu X, Lei S, Shen Y, Liu T, Li J, Wang J, Su Z. Cholesterol Sulfate: Pathophysiological Implications and Potential Therapeutics. Biomolecules. 2025; 15(5):646. https://doi.org/10.3390/biom15050646
Chicago/Turabian StyleYu, Xiaoqian, Siman Lei, Ying Shen, Tao Liu, Jun Li, Jia Wang, and Zhiguang Su. 2025. "Cholesterol Sulfate: Pathophysiological Implications and Potential Therapeutics" Biomolecules 15, no. 5: 646. https://doi.org/10.3390/biom15050646
APA StyleYu, X., Lei, S., Shen, Y., Liu, T., Li, J., Wang, J., & Su, Z. (2025). Cholesterol Sulfate: Pathophysiological Implications and Potential Therapeutics. Biomolecules, 15(5), 646. https://doi.org/10.3390/biom15050646