
TSC-mTORC1 Pathway in Postnatal V-SVZ Neurodevelopment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
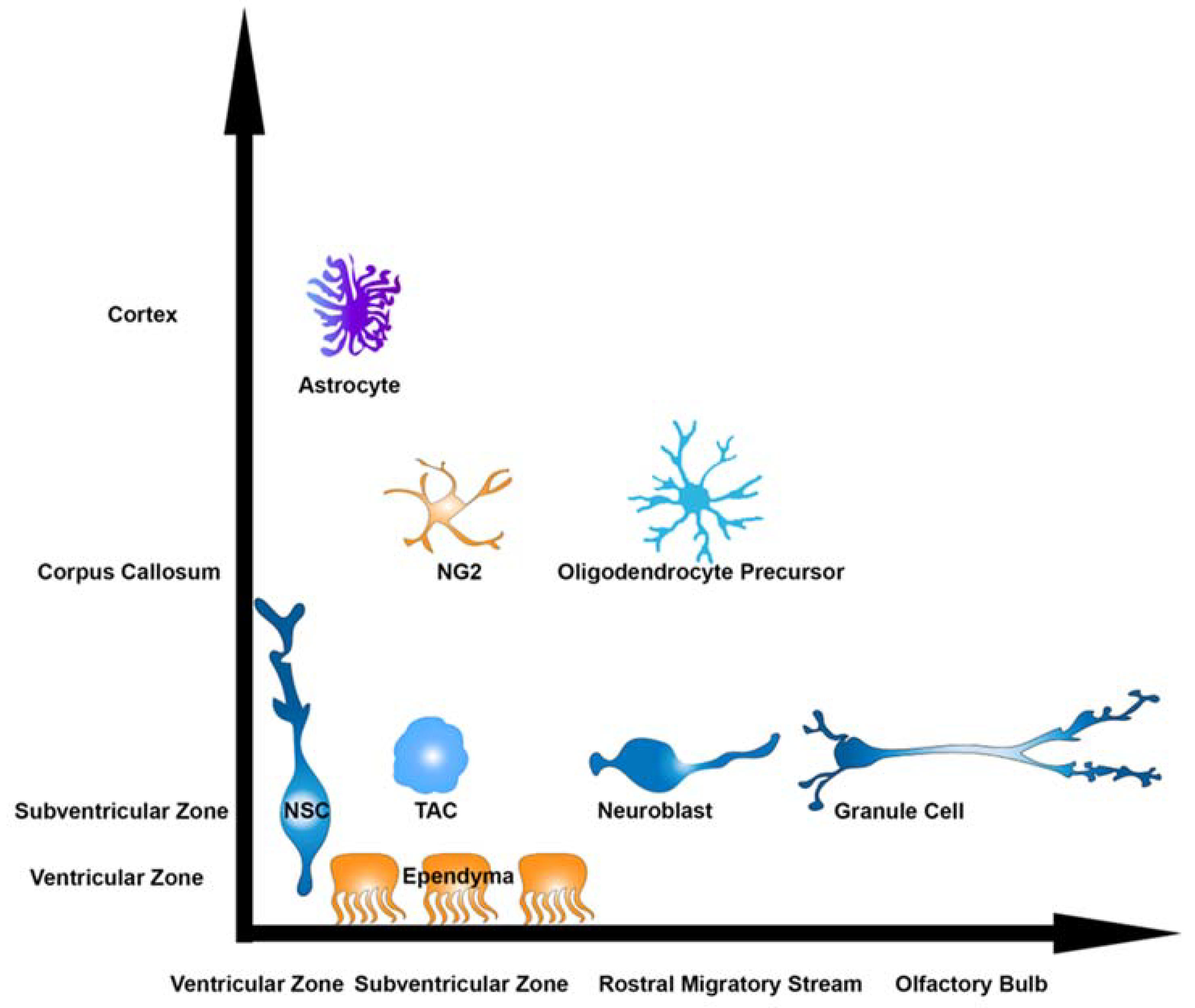
2. Heterogeneity in the V-SVZ
3. Targets of Postnatal Neurogenesis
4. Techniques for Studying V-SVZ NSCs
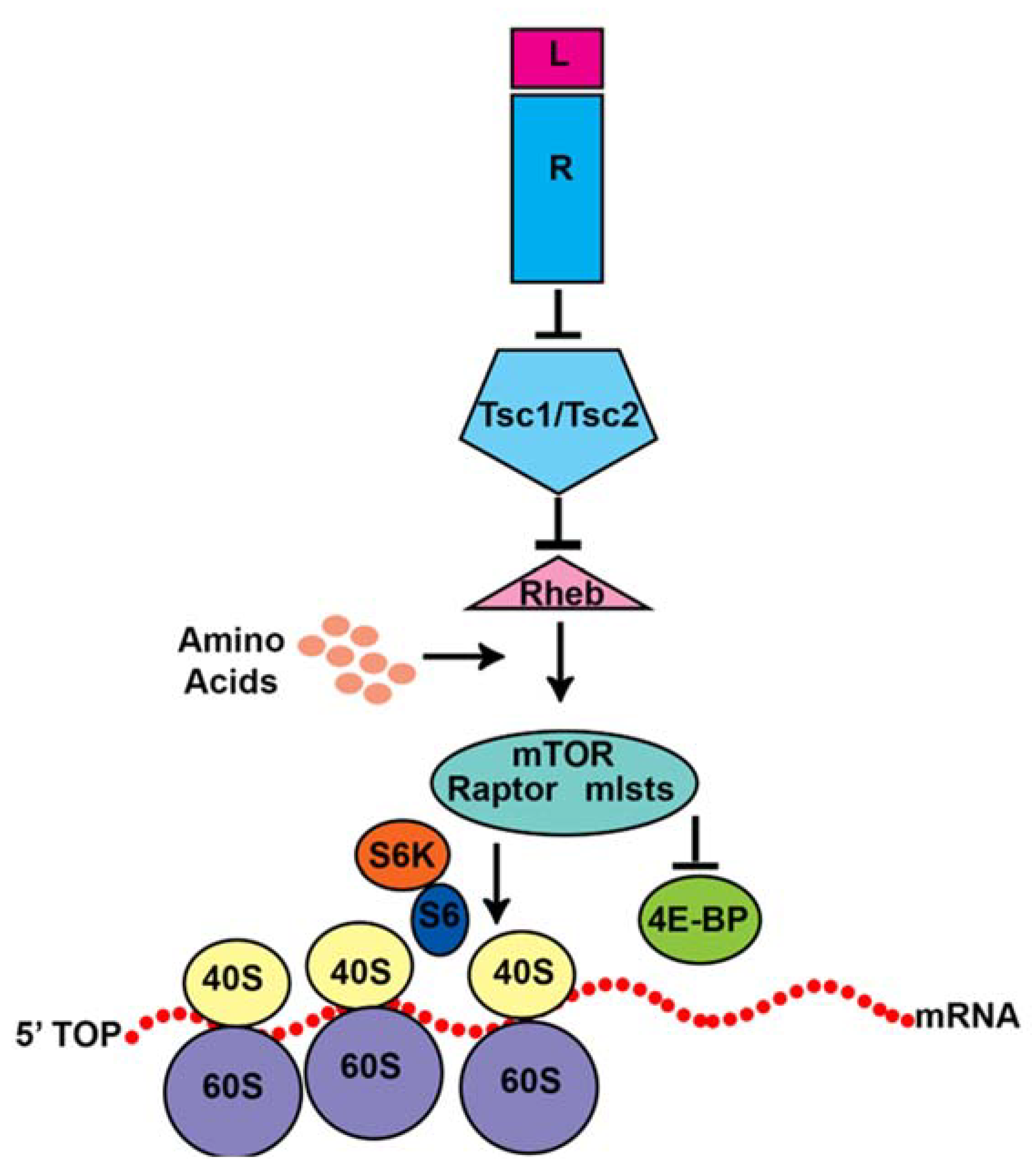
5. The mTOR Pathway
6. Tuberous Sclerosis Complex (TSC)
7. The TSC-mTORC1 Pathway in V-SVZ Neurogenesis
8. Therapeutic Implications for TSC Patients
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Altman, J.; Das, G.D. Post-natal origin of microneurones in the rat brain. Nature 1965, 207, 953–956. [Google Scholar] [CrossRef]
- Altman, J. Autoradiographic Study of Degenerative and Regenerative Proliferation of Neuroglia Cells with Tritiated Thymidine. Exp. Neurol. 1962, 5, 302–318. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.; Das, G.D. Postnatal neurogenesis in the guinea-pig. Nature 1967, 214, 1098–1101. [Google Scholar] [CrossRef]
- Altman, J. Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior fore-brain, with special reference to persisting neurogenesis in the olfactory bulb. J. Comp. Neurol. 1969, 137, 433–457. [Google Scholar] [CrossRef] [PubMed]
- Doetsch, F.; García-Verdugo, J.M.; Alvarez-Buylla, A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J. Neurosci. 1997, 17, 5046–5061. [Google Scholar] [CrossRef]
- Luskin, M.B. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron 1993, 11, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Ihrie, R.A.; Álvarez-Buylla, A. Lake-Front Property: A Unique Germinal Niche by the Lateral Ventricles of the Adult Brain. Neuron 2011, 70, 674–686. [Google Scholar] [CrossRef]
- Casingal, C.R.; Descant, K.D.; Anton, E.S. Coordinating cerebral cortical construction and connectivity: Unifying influence of radial progenitors. Neuron 2022, 110, 1100–1115. [Google Scholar] [CrossRef]
- Kwan, K.Y.; Šestan, N.; Anton, E.S. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 2012, 139, 1535–1546. [Google Scholar] [CrossRef]
- Marshall, C.A.; Suzuki, S.O.; Goldman, J.E. Gliogenic and neurogenic progenitors of the subventricular zone: Who are they, where did they come from, and where are they going? Glia 2003, 43, 52–61. [Google Scholar] [CrossRef]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed]
- de Chevigny, A.; Core, N.; Follert, P.; Wild, S.; Bosio, A.; Yoshikawa, K.; Cremer, H.; Beclin, C. Dynamic expression of the pro-dopaminergic transcription factors Pax6 and Dlx2 during postnatal olfactory bulb neurogenesis. Front. Cell. Neurosci. 2012, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Winpenny, E.; Lebel-Potter, M.; Fernandez, M.E.; Brill, M.S.; Götz, M.; Guillemot, F.; Raineteau, O. Sequential generation of olfactory bulb glutamatergic neurons by Neurog2-expressing precursor cells. Neural Dev. 2011, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Merkle, F.T.; Mirzadeh, Z.; Alvarez-Buylla, A. Mosaic organization of neural stem cells in the adult brain. Science 2007, 317, 381–384. [Google Scholar] [CrossRef]
- Merkle, F.T.; Fuentealba, L.C.; Sanders, T.A.; Magno, L.; Kessaris, N.; Alvarez-Buylla, A. Adult neural stem cells in distinct microdomains generate previously unknown interneuron types. Nat. Neurosci. 2014, 17, 207–214. [Google Scholar] [CrossRef]
- Delgado, R.N.; Lim, D.A. Embryonic Nkx2.1-expressing neural precursor cells contribute to the regional heterogeneity of adult V-SVZ neural stem cells. Dev. Biol. 2015, 407, 265–274. [Google Scholar] [CrossRef]
- Stenman, J.; Toresson, H.; Campbell, K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: Implications for striatal and olfactory bulb neurogenesis. J. Neurosci. 2003, 23, 167–174. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, Y.; Dimos, J.T.; Fasano, C.A.; Phoenix, T.N.; Lemischka, I.R.; Ivanova, N.B.; Stifani, S.; Morrisey, E.E.; Temple, S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat. Neurosci. 2006, 9, 743–751. [Google Scholar] [CrossRef]
- Delgado, A.C.; Maldonado-Soto, A.R.; Silva-Vargas, V.; Mizrak, D.; von Känel, T.; Tan, K.R.; Paul, A.; Madar, A.; Cuervo, H.; Kitajewski, J.; et al. Release of stem cells from quiescence reveals gliogenic domains in the adult mouse brain. Science 2021, 372, 1205–1209. [Google Scholar] [CrossRef]
- Lois, C.; Alvarez-Buylla, A. Long-distance neuronal migration in the adult mammalian brain. Science 1994, 264, 1145–1148. [Google Scholar] [CrossRef]
- Doetsch, F.; Alvarez-Buylla, A. Network of tangential pathways for neuronal migration in adult mammalian brain. Proc. Natl. Acad. Sci. USA 1996, 93, 14895–14900. [Google Scholar] [CrossRef] [PubMed]
- Whitman, M.C.; Greer, C.A. Synaptic integration of adult-generated olfactory bulb granule cells: Basal axodendritic centrifugal input precedes apical dendrodendritic local circuits. J. Neurosci. 2007, 27, 9951–9961. [Google Scholar] [CrossRef] [PubMed]
- Belluzzi, O.; Benedusi, M.; Ackman, J.; LoTurco, J.J. Electrophysiological Differentiation of New Neurons in the Olfactory Bulb. J. Neurosci. 2003, 23, 10411–10418. [Google Scholar] [CrossRef]
- Sailor, K.A.; Valley, M.T.; Wiechert, M.T.; Riecke, H.; Sun, G.J.; Adams, W.; Dennis, J.C.; Sharafi, S.; Ming, G.-L.; Song, H.; et al. Persistent Structural Plasticity Optimizes Sensory Information Processing in the Olfactory Bulb. Neuron 2016, 91, 384–396. [Google Scholar] [CrossRef]
- Kelsch, W.; Lin, C.W.; Lois, C. Sequential development of synapses in dendritic domains during adult neurogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 16803–16808. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, G.M.; Chen, W.R.; Willhite, D.; Migliore, M.; Greer, C.A. The olfactory granule cell: From classical enigma to central role in olfactory processing. Brain Res. Rev. 2007, 55, 373–382. [Google Scholar] [CrossRef]
- Rall, W.; Shepherd, G.M.; Reese, T.S.; Brightman, M.W. Dendrodendritic synaptic pathway for inhibition in the olfactory bulb. Exp. Neurol. 1966, 14, 44–56. [Google Scholar] [CrossRef]
- Lepousez, G.; Valley, M.T.; Lledo, P.-M. The impact of adult neurogenesis on olfactory bulb circuits and computations. Annu. Rev. Physiol. 2013, 75, 339–363. [Google Scholar] [CrossRef]
- Moreno, M.M.; Linster, C.; Escanilla, O.; Sacquet, J.; Didier, A.; Mandairon, N. Olfactory perceptual learning requires adult neurogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 17980–17985. [Google Scholar] [CrossRef]
- Ernst, A.; Alkass, K.; Bernard, S.; Salehpour, M.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; Frisén, J. Neurogenesis in the striatum of the adult human brain. Cell 2014, 156, 1072–1083. [Google Scholar] [CrossRef]
- Schlösser, B.; Klausa, G.; Prime, G.; Bruggencate, G.T. Postnatal development of calretinin- and parvalbumin-positive in-terneurons in the rat neostriatum: An immunohistochemical study. J. Comp. Neurol. 1999, 405, 185–198. [Google Scholar] [CrossRef]
- García-González, D.; Dumitru, I.; Zuccotti, A.; Yen, T.-Y.; Herranz-Pérez, V.; Tan, L.L.; Neitz, A.; García-Verdugo, J.M.; Kuner, R.; Alfonso, J.; et al. Neurogenesis of medium spiny neurons in the nucleus accumbens continues into adulthood and is enhanced by pathological pain. Mol. Psychiatry 2021, 26, 4616–4632. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ninomiya, M.; Acosta, P.H.; García-Verdugo, J.M.; Sunabori, T.; Sakaguchi, M.; Adachi, K.; Kojima, T.; Hirota, Y.; Kawase, T.; et al. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 2006, 26, 6627–6636. [Google Scholar] [CrossRef]
- Sanai, N.; Nguyen, T.; Ihrie, R.A.; Mirzadeh, Z.; Tsai, H.-H.; Wong, M.; Gupta, N.; Berger, M.S.; Huang, E.; Garcia-Verdugo, J.-M.; et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature 2011, 478, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Alvarez-Buylla, A. The adult ventricular–subventricular zone (V-SVZ) and olfactory bulb (OB) neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8, a018820. [Google Scholar] [CrossRef] [PubMed]
- Inta, D.; Alfonso, J.; von Engelhardt, J.; Kreuzberg, M.M.; Meyer, A.H.; van Hooft, J.A.; Monyer, H. Neurogenesis and widespread forebrain migration of distinct GABAergic neurons from the postnatal subven-tricular zone. Proc. Natl. Acad. Sci. USA 2008, 105, 20994–20999. [Google Scholar] [CrossRef]
- Ge, W.P.; Miyawaki, A.; Gage, F.H.; Jan, Y.N.; Jan, L.Y. Local generation of glia is a major astrocyte source in postnatal cortex. Nature 2012, 484, 376–380. [Google Scholar] [CrossRef]
- Mirzadeh, Z.; Merkle, F.T.; Soriano-Navarro, M.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Neural Stem Cells Confer Unique Pinwheel Architecture to the Ventricular Surface in Neurogenic Regions of the Adult Brain. Cell Stem Cell 2008, 3, 265–278. [Google Scholar] [CrossRef]
- Kuhn, H.G.; Winkler, J.; Kempermann, G.; Thal, L.J.; Gage, F.H. Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J. Neurosci. 1997, 17, 5820–5829. [Google Scholar] [CrossRef]
- Doetsch, F.; Caille, I.; Lim, D.A.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef]
- Morton, M.C.; Neckles, V.N.; Seluzicki, C.M.; Holmberg, J.C.; Feliciano, D.M. Neonatal Subventricular Zone Neural Stem Cells Release Extracellular Vesicles that Act as a Microglial Morphogen. Cell Rep. 2018, 23, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Peitz, M.; Pfannkuche, K.; Rajewsky, K.; Edenhofer, F. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: A tool for efficient genetic engineering of mammalian genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Spatazza, J.; Lee, H.H.; Di Nardo, A.A.; Tibaldi, L.; Joliot, A.; Hensch, T.K.; Prochiantz, A. Choroid-Plexus-Derived Otx2 Homeoprotein Constrains Adult Cortical Plasticity. Cell Rep. 2013, 3, 1815–1823. [Google Scholar] [CrossRef]
- Boutin, C.; Diestel, S.; Desoeuvre, A.; Tiveron, M.C.; Cremer, H. Efficient in vivo electroporation of the postnatal rodent fore-brain. PLoS ONE 2008, 3, e1883. [Google Scholar] [CrossRef]
- Feliciano, D.M.; Lafourcade, C.A.; Bordey, A. Neonatal subventricular zone electroporation. J. Vis. Exp. 2013, 72, e50197. [Google Scholar] [CrossRef]
- Hartman, N.W.; Lin, T.V.; Zhang, L.; Paquelet, G.E.; Feliciano, D.M.; Bordey, A. MTORC1 Targets the Translational Repressor 4E-BP2, but Not S6 Kinase 1/2, to Regulate Neural Stem Cell Self-Renewal In Vivo. Cell Rep. 2013, 5, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, J.C.; Riley, V.A.; Sokolov, A.M.; Mukherjee, S.; Feliciano, D.M. Protocol for electroporating and isolating murine (sub)ventricular zone cells for single-nuclei omics. STAR Protoc. 2024, 5, 103095. [Google Scholar] [CrossRef]
- Feliciano, D.M.; Su, T.; Lopez, J.; Platel, J.C.; Bordey, A. Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Investig. 2011, 121, 1596–1607. [Google Scholar] [CrossRef]
- Platel, J.C.; Dave, K.A.; Gordon, V.; Lacar, B.; Rubio, M.E.; Bordey, A. NMDA Receptors Activated by Subventricular Zone Astrocytic Glutamate Are Critical for Neuroblast Survival Prior to Entering a Synaptic Network. Neuron 2010, 65, 859–872. [Google Scholar] [CrossRef]
- Siddiqi, F.; Chen, F.; Aron, A.W.; Fiondella, C.G.; Patel, K.; LoTurco, J.J. Fate mapping by piggybac transposase reveals that neocortical glast+ progenitors generate more astrocytes than nestin+ progenitors in rat neocortex. Cereb. Cortex 2012, 24, 508–520. [Google Scholar] [CrossRef]
- Chen, F.; Rosiene, J.; Che, A.; Becker, A.; Loturco, J. Tracking and transforming neocortical progenitors by crispr/cas9 gene targeting and piggybac transposase lineage labeling. Development 2015, 142, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Riley, V.A.; Holmberg, J.C.; Sokolov, A.M.; Feliciano, D.M. Tsc2 shapes olfactory bulb granule cell molecular and morphological characteristics. Front. Mol. Neurosci. 2022, 15, 19. [Google Scholar] [CrossRef]
- Hattori, Y.; Miyata, T. Embryonic neocortical microglia express toll-like receptor 9 and respond to plasmid DNA injected into the ventricle: Technical considerations regarding microglial distribution in electroporated brain walls. eNeuro 2018, 5, ENEURO.0312-18.2018. [Google Scholar] [CrossRef] [PubMed]
- Lagace, D.C.; Whitman, M.C.; Noonan, M.A.; Ables, J.L.; DeCarolis, N.A.; Arguello, A.A.; Donovan, M.H.; Fischer, S.J.; Farnbauch, L.A.; Beech, R.D.; et al. Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J. Neurosci. 2007, 27, 12623–12629. [Google Scholar] [CrossRef]
- Levison, S.W.; Goldman, J.E. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron 1993, 10, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22, 989) a new antifungal antibiotic. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits MTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the Human mTOR Complex I and Its Implications for Rapamycin Inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef]
- Aylett, C.H.S.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mTOR complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.-I.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Price, D.J.; Grove, J.R.; Calvo, V.; Avruch, J.; Bierer, B.E. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 1992, 257, 973–977. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTpase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin Activation of Rheb, a Mediator of mTOR/S6K/4E-BP Signaling, Is Inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Hasbani, D.M.; Crino, P.B. Tuberous sclerosis complex. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 148, pp. 813–822. [Google Scholar]
- Chan, D.L.; Calder, T.; Lawson, J.A.; Mowat, D.; Kennedy, S.E. The natural history of subependymal giant cell astrocytomas in tuberous sclerosis complex: A review. Rev. Neurosci. 2018, 29, 295–301. [Google Scholar] [CrossRef]
- Feliciano, D.M. The Neurodevelopmental Pathogenesis of Tuberous Sclerosis Complex (TSC). Front. Neuroanat. 2020, 14, 39. [Google Scholar] [CrossRef]
- Van Slegtenhorst, M.; Hoogt, R.D.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; Ouweland, A.V.D.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef]
- The European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993, 75, 1305–1315. [Google Scholar] [CrossRef]
- Henske, E.P.; Jóźwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Prim. 2016, 2, 16035. [Google Scholar] [CrossRef]
- Feliciano, D.M.; Lin, T.V.; Hartman, N.W.; Bartley, C.M.; Kubera, C.; Hsieh, L.; Lafourcade, C.; O’Keefe, R.A.; Bordey, A. A circuitry and biochemical basis for tuberous sclerosis symptoms: From epilepsy to neurocognitive deficits. Int. J. Dev. Neurosci. 2013, 31, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Zywitza, V.; Misios, A.; Bunatyan, L.; Willnow, T.E.; Rajewsky, N. Single-Cell Transcriptomics Characterizes Cell Types in the Subventricular Zone and Uncovers Molecular Defects Impairing Adult Neurogenesis. Cell Rep. 2018, 25, 2457–2469. [Google Scholar] [CrossRef] [PubMed]
- Basak, O.; Krieger, T.G.; Muraro, M.J.; Wiebrands, K.; Stange, D.E.; Frias-Aldeguer, J.; Rivron, N.C.; van de Wetering, M.; van Es, J.H.; van Oudenaarden, A.; et al. Troy+ brain stem cells cycle through quiescence and regulate their number by sensing niche occupancy. Proc. Natl. Acad. Sci. USA 2018, 115, E610–E619. [Google Scholar] [CrossRef] [PubMed]
- Cebrian-Silla, A.; Nascimento, M.A.; Redmond, S.A.; Mansky, B.; Wu, D.; Obernier, K.; Rodriguez, R.R.; Gonzalez-Granero, S.; García-Verdugo, J.M.; Lim, D.A.; et al. Single-cell analysis of the ventricular-subventricular zone reveals signatures of dorsal and ventral adult neurogenic lineages. eLife 2021, 10, e67436. [Google Scholar] [CrossRef]
- Blair, J.D.; Hockemeyer, D.; Doudna, J.A.; Bateup, H.S.; Floor, S.N. Widespread Translational Remodeling during Human Neuronal Differentiation. Cell Rep. 2017, 21, 2005–2016. [Google Scholar] [CrossRef]
- Baser, A.; Skabkin, M.; Kleber, S.; Dang, Y.; Balta, G.S.G.; Kalamakis, G.; Göpferich, M.; Ibañez, D.C.; Schefzik, R.; Lopez, A.S.; et al. Onset of differentiation is post-transcriptionally controlled in adult neural stem cells. Nature 2019, 566, 100–104. [Google Scholar] [CrossRef]
- Paliouras, G.N.; Hamilton, L.K.; Aumont, A.; Joppé, S.E.; Barnabé-Heider, F.; Fernandes, K.J.L. Mammalian target of rapamycin signaling is a key regulator of the transit-amplifying progenitor pool in the adult and aging forebrain. J. Neurosci. 2012, 32, 15012–15026. [Google Scholar] [CrossRef]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. MTORC1 Phosphorylation Sites Encode Their Sensitivity to Starvation and Rapamycin. Science 2013, 341, 364–373. [Google Scholar] [CrossRef]
- Cochard, L.M.; Levros, L.-C.; Joppé, S.E.; Pratesi, F.; Aumont, A.; Fernandes, K.J.L. Manipulation of EGFR-Induced Signaling for the Recruitment of Quiescent Neural Stem Cells in the Adult Mouse Forebrain. Front. Neurosci. 2021, 15, 621076. [Google Scholar] [CrossRef]
- Holland, E.C.; Hively, W.P.; DePinho, R.A.; Varmus, H.E. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998, 12, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
- Romero-Pozuelo, J.; Figlia, G.; Kaya, O.; Martin-Villalba, A.; Teleman, A.A. Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep. 2020, 31, 107504. [Google Scholar] [CrossRef]
- Zhou, J.; Shrikhande, G.; Xu, J.; McKay, R.M.; Burns, D.K.; Johnson, J.E.; Parada, L.F. Tsc1 mutant neural stem/progenitor cells exhibit migration deficits and give rise to subependymal lesions in the lateral ventricle. Genes Dev. 2011, 25, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, C.; Feliciano, D.M.; Bordey, A.; Hartman, N.W. Switching on mTORC1 induces neurogenesis but not proliferation in neural stem cells of young mice. Neurosci. Lett. 2016, 614, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, D.M.; Quon, J.L.; Su, T.; Taylor, M.M.; Bordey, A. Postnatal neurogenesis generates heterotopias, olfactory mi-cronodules and cortical infiltration following single-cell TSC1 deletion. Hum. Mol. Genet. 2012, 21, 799–810. [Google Scholar] [CrossRef]
- Rushing, G.V.; Brockman, A.A.; Bollig, M.K.; Leelatian, N.; Mobley, B.C.; Irish, J.M.; Ess, K.C.; Fu, C.; Ihrie, R.A. Location-dependent maintenance of intrinsic susceptibility to mTORC1-driven tumorigenesis. Life Sci. Alliance 2019, 2, e201800218. [Google Scholar] [CrossRef]
- Gorski, J.A.; Talley, T.; Qiu, M.; Puelles, L.; Rubenstein, J.L.R.; Jones, K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 2002, 22, 6309–6314. [Google Scholar] [CrossRef]
- Kohwi, M.; Petryniak, M.A.; Long, J.E.; Ekker, M.; Obata, K.; Yanagawa, Y.; Rubenstein, J.L.; Alvarez-Buylla, A. A subpopulation of olfactory bulb GABAergic interneurons is derived from Emx1- and Dlx5/6-expressing progenitors. J. Neurosci. 2007, 27, 6878–6891. [Google Scholar] [CrossRef]
- Magri, L.; Cambiaghi, M.; Cominelli, M.; Alfaro-Cervello, C.; Cursi, M.; Pala, M.; Bulfone, A.; Garcìa-Verdugo, J.M.; Leocani, L.; Minicucci, F.; et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 2011, 9, 447–462. [Google Scholar] [CrossRef]
- Riley, V.A.; Shankar, V.; Holmberg, J.C.; Sokolov, A.M.; Neckles, V.N.; Williams, K.; Lyman, R.; Mackay, T.F.; Feliciano, D.M. Tsc2 coordinates neuroprogenitor differentiation. iScience 2023, 26, 108442. [Google Scholar] [CrossRef]
- Crino, P.B.; Trojanowski, J.Q.; Dichter, M.A.; Eberwine, J. Embryonic neuronal markers in tuberous sclerosis: Single-cell mo-lecular pathology. Proc. Natl. Acad. Sci. USA 1996, 93, 14152–14157. [Google Scholar] [CrossRef]
- Zordan, P.; Cominelli, M.; Cascino, F.; Tratta, E.; Poliani, P.L.; Galli, R. Tuberous sclerosis complex-associated CNS abnormalities depend on hyperactivation of mTORC1 and Akt. J. Clin. Investig. 2018, 128, 1688–1706. [Google Scholar] [CrossRef] [PubMed]
- Lafourcade, C.A.; Lin, T.V.; Feliciano, D.M.; Zhang, L.; Hsieh, L.S.; Bordey, A. Rheb activation in subventricular zone progenitors leads to heterotopia, ectopic neuronal differentiation, and rapamycin-sensitive olfactory micronodules and dendrite hypertrophy of newborn neurons. J. Neurosci. 2013, 33, 2419–2431. [Google Scholar] [CrossRef]
- Magini, A.; Polchi, A.; Di Meo, D.; Mariucci, G.; Sagini, K.; De Marco, F.; Cassano, T.; Giovagnoli, S.; Dolcetta, D.; Emiliani, C. TFEB activation restores migration ability to Tsc1-deficient adult neural stem/progenitor cells. Hum. Mol. Genet. 2017, 26, 3303–3312. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, A.M.; Holmberg, J.C.; Feliciano, D.M. The amino acid transporter Slc7a5 regulates the mTOR pathway and is required for granule cell development. Hum. Mol. Genet. 2020, 29, 3003–3013. [Google Scholar] [CrossRef]
- Skalecka, A.; Liszewska, E.; Bilinski, R.; Gkogkas, C.; Khoutorsky, A.; Malik, A.R.; Sonenberg, N.; Jaworski, J. mTOR kinase is needed for the development and stabilization of dendritic arbors in newly born olfactory bulb neurons. Dev. Neurobiol. 2016, 76, 1308–1327. [Google Scholar] [CrossRef]
- Cox, R.L.; de Anda, F.C.; Mangoubi, T.; Yoshii, A. Multiple critical periods for rapamycin treatment to correct structural defects in Tsc-1-suppressed brain. Front. Mol. Neurosci. 2018, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional Transport of Amino Acids Regulates mTOR and Autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically Engineered Human Cortical Spheroid Models of Tuberous Sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef]
- Franz, D.N.; Belousova, E.; Sparagana, S.; Bebin, E.M.; Frost, M.; Kuperman, R.; Witt, O.; Kohrman, M.H.; Flamini, J.R.; Wu, J.Y.; et al. Efficacy and Safety of Everolimus for Subependymal Giant Cell Astrocytomas Associated with Tuberous Sclerosis Complex (EXIST-1): A Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet 2013, 381, 125–132. [Google Scholar] [CrossRef]
- Kaplan, B.; Qazi, Y.; Wellen, J.R. Strategies for the Management of Adverse Events Associated with MTOR Inhibitors. Transplant. Rev. 2014, 28. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-Competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-Resistant Functions of MTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Francipane, M.G.; Lagasse, E. Selective Targeting of Human Colon Cancer Stem-like Cells by the MTOR Inhibitor Torin-1. Oncotarget 2013, 4. [Google Scholar] [CrossRef]
- Pollizzi, K.; Malinowska-Kolodziej, I.; Stumm, M.; Lane, H.; Kwiatkowski, D. Equivalent Benefit of MTORC1 Blockade and Combined PI3K-MTOR Blockade in a Mouse Model of Tuberous Sclerosis. Mol. Cancer 2009, 8. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; De Stanchina, E.; et al. Overcoming MTOR Resistance Mutations with a New-Generation MTOR Inhibitor. Nature 2016, 534. [Google Scholar] [CrossRef]
- Mukherjee, S.; Wolan, M.J.; Scott, M.K.; Riley, V.A.; Feliciano, D.M. A Bitopic MTORC Inhibitor Reverses Phenotypes in a Tuberous Sclerosis Complex Model 2025. bioRxiv 2025. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feliciano, D.M.; Bordey, A. TSC-mTORC1 Pathway in Postnatal V-SVZ Neurodevelopment. Biomolecules 2025, 15, 573. https://doi.org/10.3390/biom15040573
Feliciano DM, Bordey A. TSC-mTORC1 Pathway in Postnatal V-SVZ Neurodevelopment. Biomolecules. 2025; 15(4):573. https://doi.org/10.3390/biom15040573
Chicago/Turabian StyleFeliciano, David M., and Angelique Bordey. 2025. "TSC-mTORC1 Pathway in Postnatal V-SVZ Neurodevelopment" Biomolecules 15, no. 4: 573. https://doi.org/10.3390/biom15040573
APA StyleFeliciano, D. M., & Bordey, A. (2025). TSC-mTORC1 Pathway in Postnatal V-SVZ Neurodevelopment. Biomolecules, 15(4), 573. https://doi.org/10.3390/biom15040573