
Integrated Proteomics and Metabolomics of Safflower Petal Wilting and Seed Development



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
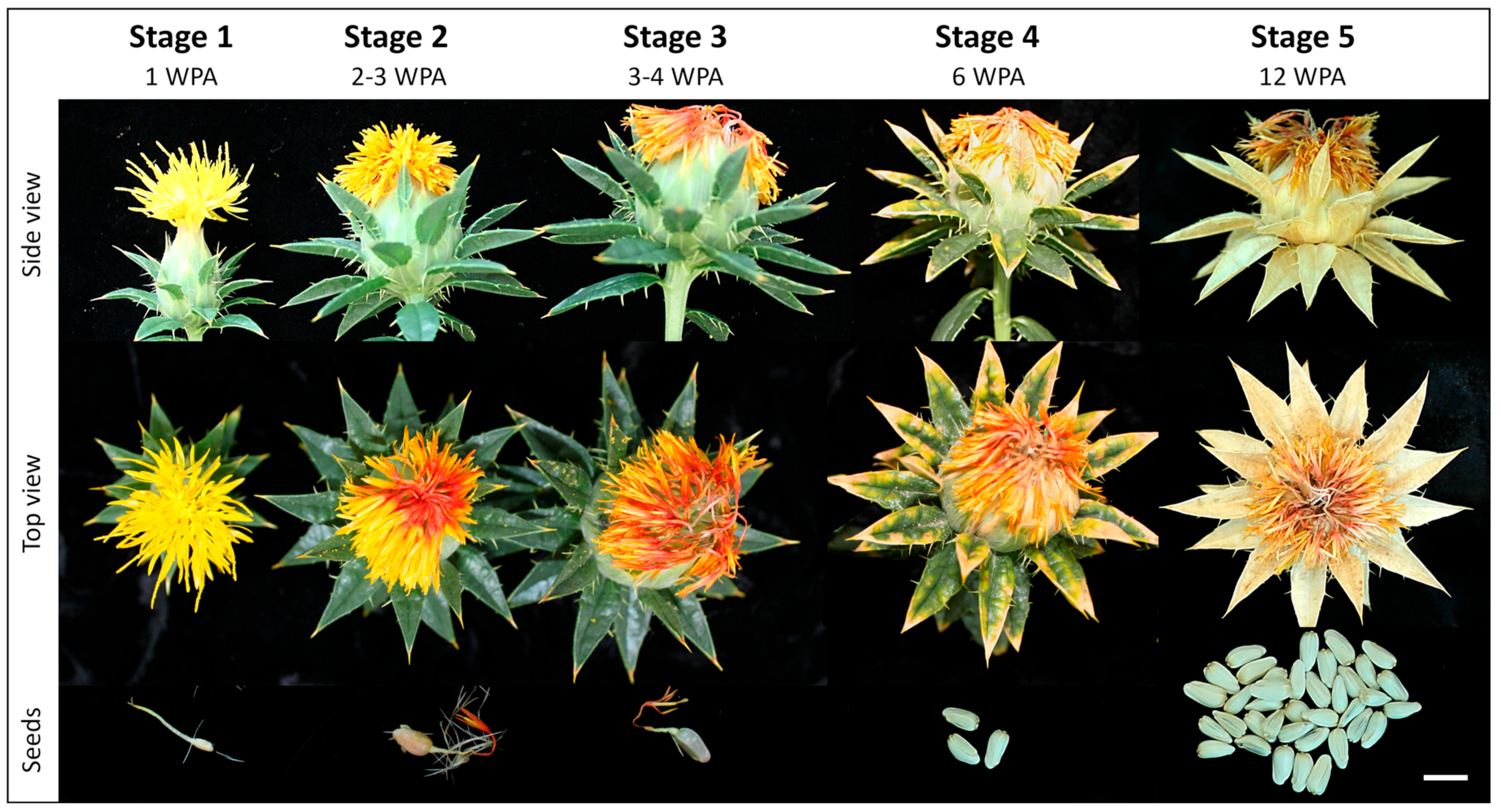
2.1. Safflower Cultivation, Sampling, and Storage
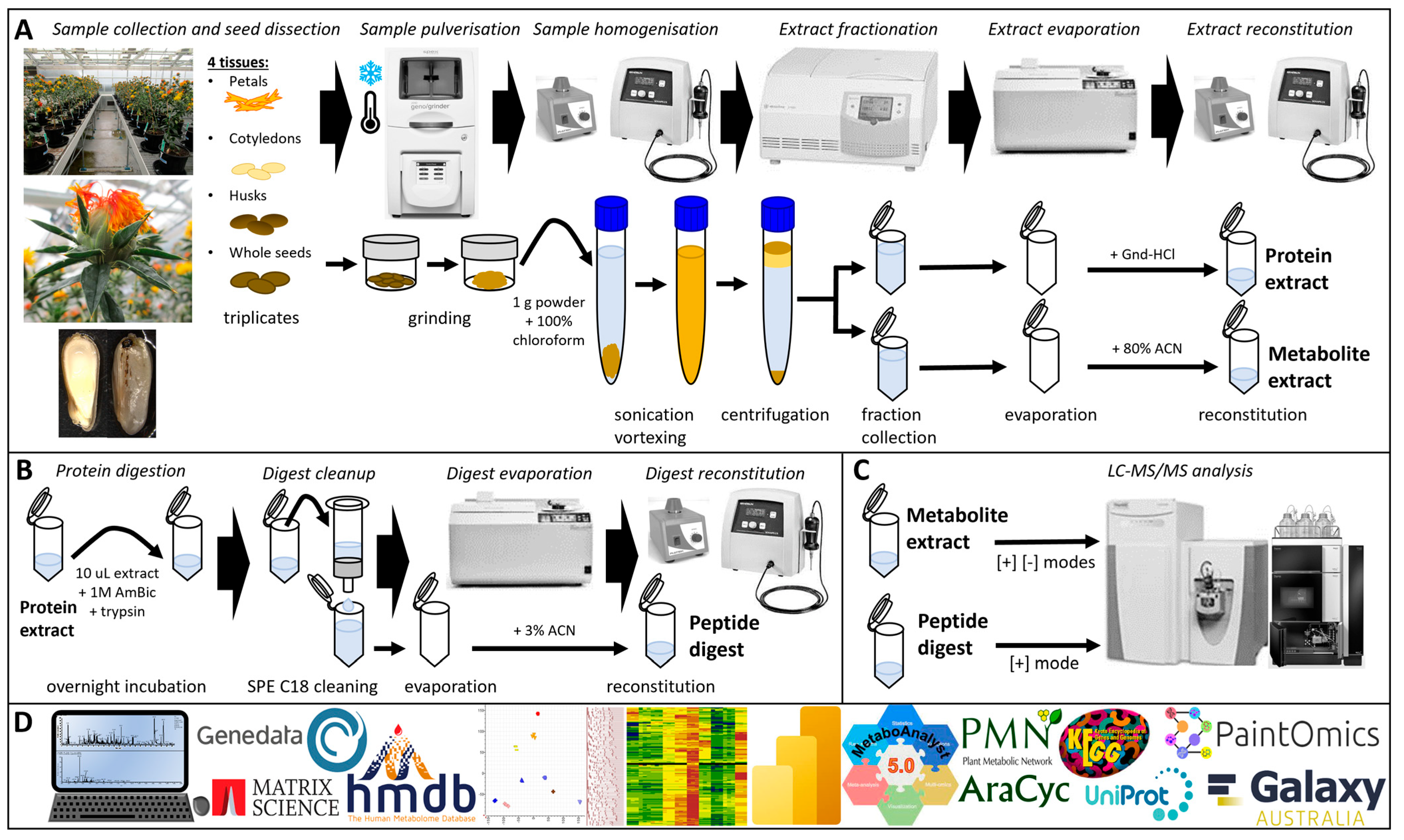
2.2. Sample Preparation
2.3. Bottom-up Proteomics
2.3.1. Protein Extraction and Digestion
2.3.2. Liquid Chromatography (LC) and Mass Spectrometry (MS) of Peptides
2.3.3. Protein Database and Mascot Identification
2.4. Metabolomics
2.4.1. Metabolite Extraction
2.4.2. LC-MS Analysis of Metabolites
2.4.3. Metabolite Identification
2.5. Quantitation and Statistical Analyses
2.6. Data Visualisation, Data Mining, and Bioinformatics
3. Results and Discussion
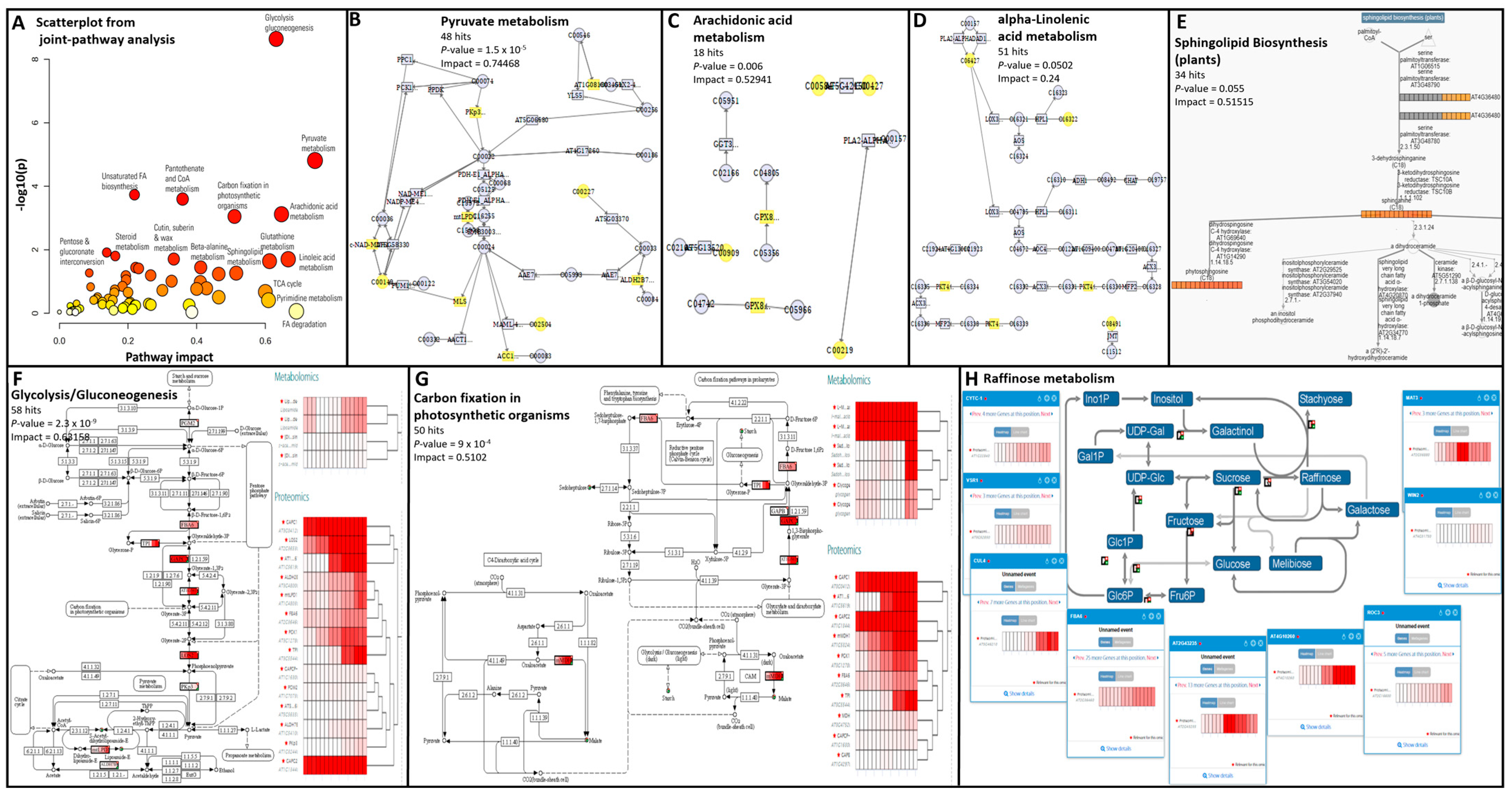
3.1. Proteomics and Metabolomics Successfully Discriminated Safflower Organs and Developmental Stages
3.2. Comparison of Cotyledon and Husk Reveals the Complementarity of Metabolomics and Proteomics
3.3. Petal Molecular Signature Shifts during Colour Transition and Wilting
3.4. Safflower Seed Growth and Maturation Are Driven by a Complex Tapestry of Biochemical Mechanisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xian, B.; Wang, R.; Jiang, H.; Zhou, Y.; Yan, J.; Huang, X.; Chen, J.; Wu, Q.; Chen, C.; Xi, Z.; et al. Comprehensive Review of Two Groups of Flavonoids in Carthamus tinctorius L. Biomed. Pharmacother. 2022, 153, 113462. [Google Scholar] [CrossRef]
- Khalid, N.; Khan, R.S.; Hussain, M.I.; Farooq, M.; Ahmad, A.; Ahmed, I. A Comprehensive Characterisation of Safflower Oil for Its Potential Applications as a Bioactive Food Ingredient—A Review. Trends Food Sci. Technol. 2017, 66, 176–186. [Google Scholar] [CrossRef]
- Mani, V.; Lee, S.-K.; Yeo, Y.; Hahn, B.-S. A Metabolic Perspective and Opportunities in Pharmacologically Important Safflower. Metabolites 2020, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.J. (Ed.) Genetic Resources, Chromosome Engineering, and Crop Improvement: Oilseed Crops; Genetic Resources, Chromosome Engineering, and Crop Improvement Series; CRC Press: Boca Raton, FL, USA, 2007; ISBN 978-0-8493-3639-3. [Google Scholar]
- Thoday-Kennedy, E.; Banerjee, B.; Panozzo, J.; Maharjan, P.; Hudson, D.; Spangenberg, G.; Hayden, M.; Kant, S. Dissecting Physiological and Agronomic Diversity in Safflower Populations Using Proximal Phenotyping. Agriculture 2023, 13, 620. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Tian, K.; Tang, Z.-H.; Chen, X.-J.; Bian, Z.-X.; Wang, Y.-T.; Lu, J.-J. Phytochemistry and Pharmacology of Carthamus tinctorius L. Am. J. Chin. Med. 2016, 44, 197–226. [Google Scholar] [CrossRef] [PubMed]
- Hegazi, N.M.; Khattab, A.R.; Frolov, A.; Wessjohann, L.A.; Farag, M.A. Authentication of Saffron Spice Accessions from Its Common Substitutes via a Multiplex Approach of UV/VIS Fingerprints and UPLC/MS Using Molecular Networking and Chemometrics. Food Chem. 2022, 367, 130739. [Google Scholar] [CrossRef]
- Ryparova Kvirencova, J.; Navratilova, K.; Hrbek, V.; Hajslova, J. Detection of Botanical Adulterants in Saffron Powder. Anal. Bioanal. Chem. 2023, 415, 5723–5734. [Google Scholar] [CrossRef]
- Paredi, G.; Raboni, S.; Marchesani, F.; Ordoudi, S.A.; Tsimidou, M.Z.; Mozzarelli, A. Insight of Saffron Proteome by Gel-Electrophoresis. Molecules 2016, 21, 167. [Google Scholar] [CrossRef] [PubMed]
- Adamska, I.; Biernacka, P. Bioactive Substances in Safflower Flowers and Their Applicability in Medicine and Health-Promoting Foods. Int. J. Food Sci. 2021, 2021, 6657639. [Google Scholar] [CrossRef]
- Sato, S.; Kusakari, T.; Suda, T.; Kasai, T.; Kumazawa, T.; Onodera, J.; Obara, H. Efficient Synthesis of Analogs of Safflower Yellow B, Carthamin, and Its Precursor: Two Yellow and One Red Dimeric Pigments in Safflower Petals. Tetrahedron 2005, 61, 9630–9636. [Google Scholar] [CrossRef]
- Ji, Y.; Guo, S.; Wang, B.; Yu, M. Extraction and Determination of Flavonoids in Carthamus tinctorius. Open Chem. 2018, 16, 1129–1133. [Google Scholar] [CrossRef]
- Zhou, L.; Lu, L.; Chen, C.; Zhou, T.; Wu, Q.; Wen, F.; Chen, J.; Pritchard, H.W.; Peng, C.; Pei, J.; et al. Comparative Changes in Sugars and Lipids Show Evidence of a Critical Node for Regeneration in Safflower Seeds during Aging. Front. Plant Sci. 2022, 13, 1020478. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.C.; Okada, S.; Taylor, M.C.; Menon, A.; Mathew, A.; Cullerne, D.; Stephen, S.J.; Allen, R.S.; Zhou, X.; Liu, Q.; et al. Seed-specific RNAi in Safflower Generates a Superhigh Oleic Oil with Extended Oxidative Stability. Plant Biotechnol. J. 2018, 16, 1788–1796. [Google Scholar] [CrossRef] [PubMed]
- Sardouei-Nasab, S.; Nemati, Z.; Mohammadi-Nejad, G.; Haghi, R.; Blattner, F.R. Phylogenomic Investigation of Safflower (Carthamus tinctorius) and Related Species Using Genotyping-by-Sequencing (GBS). Sci. Rep. 2023, 13, 6212. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, H.; Zhan, W.; Yu, Z.; Qin, E.; Liu, S.; Yang, T.; Xiang, N.; Kudrna, D.; Chen, Y.; et al. The Chromosome-Scale Reference Genome of Safflower (Carthamus tinctorius) Provides Insights into Linoleic Acid and Flavonoid Biosynthesis. Plant Biotechnol. J. 2021, 19, 1725–1742. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, Y.; Yang, J.; Liu, X.; Wang, Y.; Yao, N.; Guan, L.; Wang, N.; Wu, J.; Li, X. De Novo Transcriptome of Safflower and the Identification of Putative Genes for Oleosin and the Biosynthesis of Flavonoids. PLoS ONE 2012, 7, e30987. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, D.; Reyes-Chin-Wo, S.; Acquadro, A.; Froenicke, L.; Portis, E.; Beitel, C.; Tirone, M.; Mauro, R.; Lo Monaco, A.; Mauromicale, G.; et al. The Genome Sequence of the Outbreeding Globe Artichoke Constructed de Novo Incorporating a Phase-Aware Low-Pass Sequencing Strategy of F1 Progeny. Sci. Rep. 2016, 6, 19427. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Qin, Y.; Hu, X.; Xu, J.; Ye, Q.; Zhang, C.; Ding, Y.; Li, G.; Chen, Y.; Liu, J.; et al. Identification of Genes Associated with Fatty Acid Biosynthesis Based on 214 Safflower Core Germplasm. BMC Genom. 2023, 24, 763. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhou, X.-R.; Wood, C.C.; Green, A.G.; Singh, S.P.; Liu, L.; Liu, Q. A Large and Functionally Diverse Family of Fad2 Genes in Safflower (Carthamus tinctorius L.). BMC Plant Biol. 2013, 13, 5. [Google Scholar] [CrossRef]
- Li, D.; Wang, Q.; Xu, X.; Yu, J.; Chen, Z.; Wei, B.; Wu, W. Temporal Transcriptome Profiling of Developing Seeds Reveals Candidate Genes Involved in Oil Accumulation in Safflower (Carthamus tinctorius L.). BMC Plant Biol. 2021, 21, 181. [Google Scholar] [CrossRef]
- Çulha Erdal, Ş.; Eyidoğan, F.; Ekmekçi, Y. Comparative Physiological and Proteomic Analysis of Cultivated and Wild Safflower Response to Drought Stress and Re-Watering. Physiol. Mol. Biol. Plants 2021, 27, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Shaki, F.; Ebrahimzadeh Maboud, H.; Niknam, V. Differential Proteomics: Effect of Growth Regulators on Salt Stress Responses in Safflower Seedlings. Pestic. Biochem. Physiol. 2020, 164, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Bárta, J.; Bártová, V.; Jarošová, M.; Švajner, J.; Smetana, P.; Kadlec, J.; Filip, V.; Kyselka, J.; Berčíková, M.; Zdráhal, Z.; et al. Oilseed Cake Flour Composition, Functional Properties and Antioxidant Potential as Effects of Sieving and Species Differences. Foods 2021, 10, 2766. [Google Scholar] [CrossRef]
- Pu, Z.; Zhang, S.; Tang, Y.; Shi, X.; Tao, H.; Yan, H.; Chen, J.; Yue, S.; Chen, Y.; Zhu, Z.; et al. Study on Changes in Pigment Composition during the Blooming Period of Safflower Based on Plant Metabolomics and Semi-quantitative Analysis. J. Sep. Sci. 2021, 44, 4082–4091. [Google Scholar] [CrossRef]
- Kim, N.S.; Kim, J.K.; Sathasivam, R.; Park, H.W.; Nguyen, B.V.; Kim, M.C.; Cuong, D.M.; Chung, Y.S.; Park, S.U. Impact of Betaine Under Salinity on Accumulation of Phenolic Compounds in Safflower (Carthamus tinctorius L.) Sprouts. Nat. Product. Commun. 2021, 16, 1934578X2110150. [Google Scholar] [CrossRef]
- Qiang, T.; Liu, J.; Dong, Y.; Ma, Y.; Zhang, B.; Wei, X.; Liu, H.; Xiao, P. Transcriptome Sequencing and Chemical Analysis Reveal the Formation Mechanism of White Florets in Carthamus tinctorius L. Plants 2020, 9, 847. [Google Scholar] [CrossRef]
- Ren, C.; Chen, C.; Dong, S.; Wang, R.; Xian, B.; Liu, T.; Xi, Z.; Pei, J.; Chen, J. Integrated Metabolomics and Transcriptome Analysis on Flavonoid Biosynthesis in Flowers of Safflower (Carthamus tinctorius L.) during Colour-Transition. PeerJ 2022, 10, e13591. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ren, C.; Dong, S.; Chen, C.; Xian, B.; Wu, Q.; Wang, J.; Pei, J.; Chen, J. Integrated Metabolomics and Transcriptome Analysis of Flavonoid Biosynthesis in Safflower (Carthamus tinctorius L.) with Different Colors. Front. Plant Sci. 2021, 12, 712038. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Wang, R.; Xian, B.; Ren, C.; Liu, Q.; Wu, Q.; Pei, J. Integrated Metabolomics and Transcriptome Analysis on Flavonoid Biosynthesis in Safflower (Carthamus tinctorius L.) under MeJA Treatment. BMC Plant Biol. 2020, 20, 353. [Google Scholar] [CrossRef]
- Chen, C.; Wang, R.; Dong, S.; Wang, J.; Ren, C.; Chen, C.; Yan, J.; Zhou, T.; Wu, Q.; Pei, J.; et al. Integrated Proteome and Lipidome Analysis of Naturally Aged Safflower Seeds Varying in Vitality. Plant Biol. J. 2022, 24, 266–277. [Google Scholar] [CrossRef]
- Vincent, D.; Bui, A.; Ram, D.; Ezernieks, V.; Bedon, F.; Panozzo, J.; Maharjan, P.; Rochfort, S.; Daetwyler, H.; Hayden, M. Mining the Wheat Grain Proteome. Int. J. Mol. Sci. 2022, 23, 713. [Google Scholar] [CrossRef]
- Vincent, D.; Bui, A.; Ezernieks, V.; Shahinfar, S.; Luke, T.; Ram, D.; Rigas, N.; Panozzo, J.; Rochfort, S.; Daetwyler, H.; et al. A Community Resource to Mass Explore the Wheat Grain Proteome and Its Application to the Late-Maturity Alpha-Amylase (LMA) Problem. GigaScience 2022, 12, giad084. [Google Scholar] [CrossRef]
- Holman, J.D.; Tabb, D.L.; Mallick, P. Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Curr. Protoc. Bioinform. 2014, 46, 13.24.1–13.24.9. [Google Scholar] [CrossRef] [PubMed]
- Creasy, D.M.; Cottrell, J.S. Error Tolerant Searching of Uninterpreted Tandem Mass Spectrometry Data. Proteomics 2002, 2, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Reddy, P.; Plozza, T.; Ezernieks, V.; Stefanelli, D.; Scalisi, A.; Goodwin, I.; Rochfort, S. Metabolic Pathways for Observed Impacts of Crop Load on Floral Induction in Apple. Int. J. Mol. Sci. 2022, 23, 6019. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Chilton, J.M.; Grüning, B.; Johnson, J.E.; Soranzo, N. NCBI BLAST+ Integrated into Galaxy. GigaScience 2015, 4, 39. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for Inferring Cellular Functions from Protein Sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG Mapping Tools for Uncovering Hidden Features in Biological Data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Salguero, P.; Petek, M.; Martinez-Mira, C.; Balzano-Nogueira, L.; Ramšak, Ž.; McIntyre, L.; Gruden, K.; Tarazona, S.; Conesa, A. PaintOmics 4: New Tools for the Integrative Analysis of Multi-Omics Datasets Supported by Multiple Pathway Databases. Nucleic Acids Res. 2022, 50, W551–W559. [Google Scholar] [CrossRef] [PubMed]
- Plant Metabolics Network (PMN). 2023. Available online: https://pmn.plantcyc.org/Organism-Summary?object=ARA (accessed on 22 November 2023).
- Ahmadi, S.; Winter, D. Identification of Unexpected Protein Modifications by Mass Spectrometry-Based Proteomics. In Functional Proteomics; Wang, X., Kuruc, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1871, pp. 225–251. ISBN 978-1-4939-8813-6. [Google Scholar]
- Bockisch, M. Vegetable Fats and Oils. In Fats and Oils Handbook; Elsevier: Amsterdam, The Netherlands, 1998; pp. 174–344. ISBN 978-0-9818936-0-0. [Google Scholar]
- Zhang, Q.; Hu, N.; Li, W.; Ding, C.; Ma, T.; Bai, B.; Wang, H.; Suo, Y.; Wang, X.; Ding, C. Preparative Separation of N-Feruloyl Serotonin and N-(p-Coumaroyl) Serotonin from Safflower Seed Meal Using High-Speed Counter-Current Chromatography. J. Chromatogr. Sci. 2015, 53, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Nazeam, J.A.; El-Hefnawy, H.M.; Omran, G.; Singab, A.-N. Chemical Profile and Antihyperlipidemic Effect of Portulaca oleracea L. Seeds in Streptozotocin-Induced Diabetic Rats. Nat. Prod. Res. 2018, 32, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, B.-C.; Xu, Y.; Zhu, Y.-X. Systematic Studies of 12S Seed Storage Protein Accumulation and Degradation Patterns during Arabidopsis Seed Maturation and Early Seedling Germination Stages. J. Biochem. Mol. Biol. 2007, 40, 373–381. [Google Scholar] [CrossRef]
- D’Agostino, N.; Buonanno, M.; Ayoub, J.; Barone, A.; Monti, S.M.; Rigano, M.M. Identification of Non-Specific Lipid Transfer Protein Gene Family Members in Solanum Lycopersicum and Insights into the Features of Sola l 3 Protein. Sci. Rep. 2019, 9, 1607. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, X.; Lu, C.; Zeng, X.; Li, Y.; Fu, D.; Wu, G. Non-Specific Lipid Transfer Proteins in Plants: Presenting New Advances and an Integrated Functional Analysis. J. Exp. Bot. 2015, 66, 5663–5681. [Google Scholar] [CrossRef]
- Lees, J.A.; Reinisch, K.M. Inter-Organelle Lipid Transfer: A Channel Model for Vps13 and Chorein-N Motif Proteins. Curr. Opin. Cell Biol. 2020, 65, 66–71. [Google Scholar] [CrossRef]
- Boatright, J.; Negre, F.; Chen, X.; Kish, C.M.; Wood, B.; Peel, G.; Orlova, I.; Gang, D.; Rhodes, D.; Dudareva, N. Understanding in Vivo Benzenoid Metabolism in Petunia Petal Tissue. Plant Physiol. 2004, 135, 1993–2011. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, S.; Wang, Y.; Zeng, W.; Jin, B. Floral Scents and Fruit Aromas: Functions, Compositions, Biosynthesis, and Regulation. Front. Plant Sci. 2022, 13, 860157. [Google Scholar] [CrossRef] [PubMed]
- Paniagua, C.; Bilkova, A.; Jackson, P.; Dabravolski, S.; Riber, W.; Didi, V.; Houser, J.; Gigli-Bisceglia, N.; Wimmerova, M.; Budínská, E.; et al. Dirigent Proteins in Plants: Modulating Cell Wall Metabolism during Abiotic and Biotic Stress Exposure. J. Exp. Bot. 2017, 68, 3287–3301. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, K.; Huang, A.H.C. Lipid-Rich Tapetosomes in Brassica Tapetum Are Composed of Oleosin-Coated Oil Droplets and Vesicles, Both Assembled in and Then Detached from the Endoplasmic Reticulum. Plant J. 2005, 43, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Bannenberg, G.; Martínez, M.; Hamberg, M.; Castresana, C. Diversity of the Enzymatic Activity in the Lipoxygenase Gene Family of Arabidopsis Thaliana. Lipids 2009, 44, 85–95. [Google Scholar] [CrossRef]
- Mueller, L.A.; Zhang, P.; Rhee, S.Y. AraCyc: A Biochemical Pathway Database for Arabidopsis. Plant Physiol. 2003, 132, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Floerl, S.; Majcherczyk, A.; Possienke, M.; Feussner, K.; Tappe, H.; Gatz, C.; Feussner, I.; Kües, U.; Polle, A. Verticillium Longisporum Infection Affects the Leaf Apoplastic Proteome, Metabolome, and Cell Wall Properties in Arabidopsis Thaliana. PLoS ONE 2012, 7, e31435. [Google Scholar] [CrossRef]
- López-Hidalgo, C.; Guerrero-Sánchez, V.M.; Gómez-Gálvez, I.; Sánchez-Lucas, R.; Castillejo-Sánchez, M.A.; Maldonado-Alconada, A.M.; Valledor, L.; Jorrín-Novo, J.V. A Multi-Omics Analysis Pipeline for the Metabolic Pathway Reconstruction in the Orphan Species Quercus Ilex. Front. Plant Sci. 2018, 9, 935. [Google Scholar] [CrossRef]
- García-Alcalde, F.; García-López, F.; Dopazo, J.; Conesa, A. Paintomics: A Web Based Tool for the Joint Visualization of Transcriptomics and Metabolomics Data. Bioinformatics 2011, 27, 137–139. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Wang, W.-Q.; Liu, S.-J.; Song, S.-Q.; Møller, I.M. Proteomics of Seed Development, Desiccation Tolerance, Germination and Vigor. Plant Physiol. Biochem. 2015, 86, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Domergue, J.-B.; Abadie, C.; Limami, A.; Way, D.; Tcherkez, G. Seed Quality and Carbon Primary Metabolism. Plant Cell Environ. 2019, 42, 2776–2788. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, D.; Reddy, P.; Isenegger, D. Integrated Proteomics and Metabolomics of Safflower Petal Wilting and Seed Development. Biomolecules 2024, 14, 414. https://doi.org/10.3390/biom14040414
Vincent D, Reddy P, Isenegger D. Integrated Proteomics and Metabolomics of Safflower Petal Wilting and Seed Development. Biomolecules. 2024; 14(4):414. https://doi.org/10.3390/biom14040414
Chicago/Turabian StyleVincent, Delphine, Priyanka Reddy, and Daniel Isenegger. 2024. "Integrated Proteomics and Metabolomics of Safflower Petal Wilting and Seed Development" Biomolecules 14, no. 4: 414. https://doi.org/10.3390/biom14040414
APA StyleVincent, D., Reddy, P., & Isenegger, D. (2024). Integrated Proteomics and Metabolomics of Safflower Petal Wilting and Seed Development. Biomolecules, 14(4), 414. https://doi.org/10.3390/biom14040414