
An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Peptides
2.2. ThT Assay
2.3. SEM
2.4. Cell Culture and Viability
2.5. Phagocytosis Assay
2.6. Animals
2.7. Intraventricular Injection of Aβ25-35 and YS-RD11
2.8. Intranasal Injection of YS-RD11
2.9. Spontaneous Alternation in Y-Maze Test
2.10. Statistical Analysis
3. Results
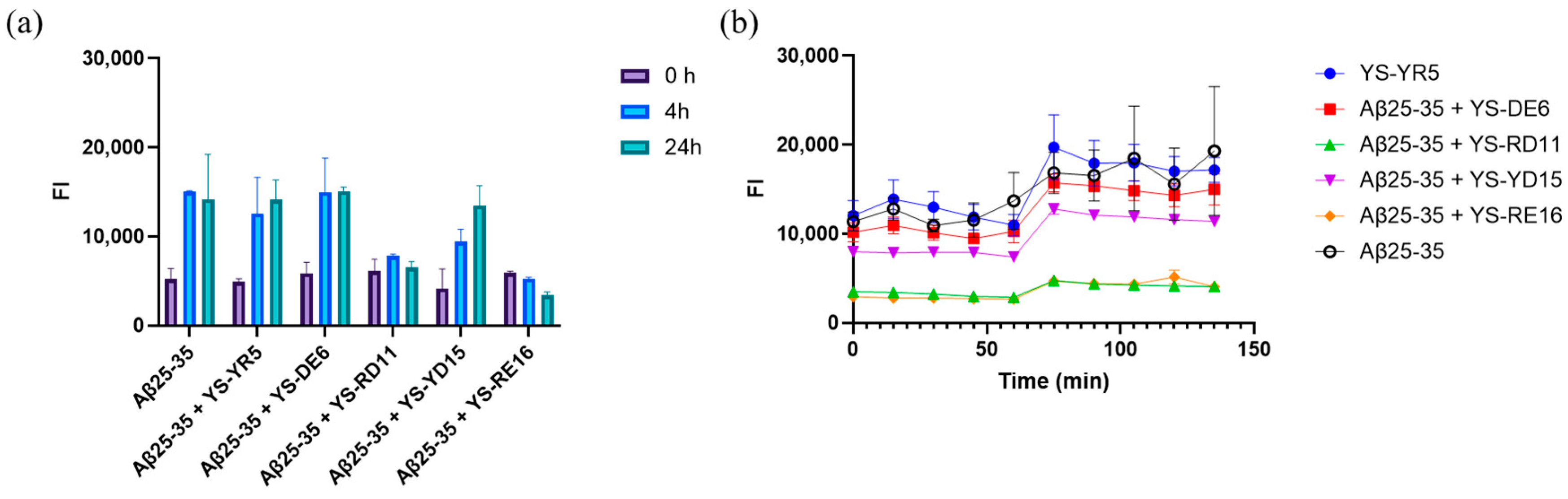
3.1. Screening for Effective Peptides
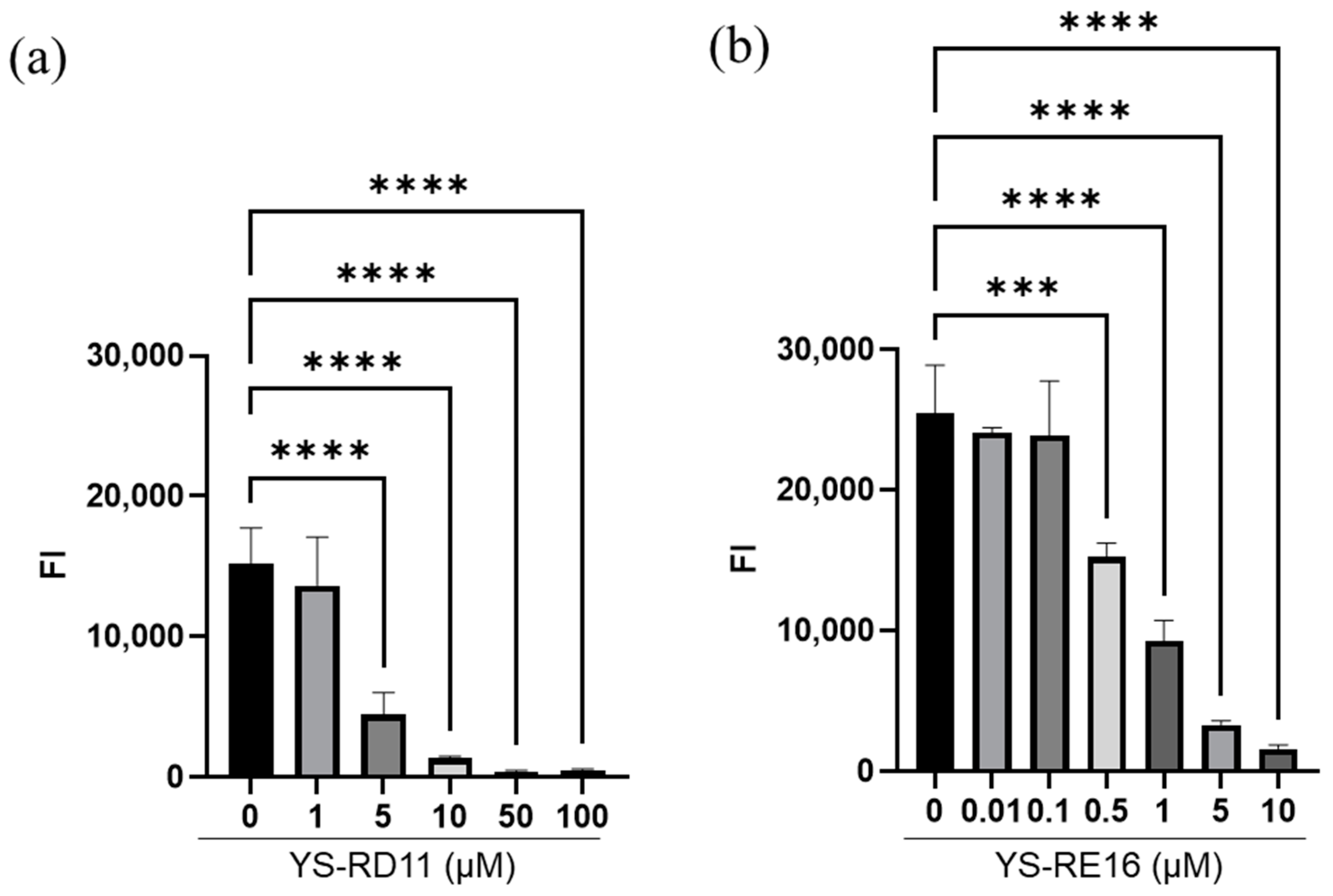
3.2. Inhibitory Effect of YS-RD11 and YS-RE16 on Aβ 25-35 Aggregation
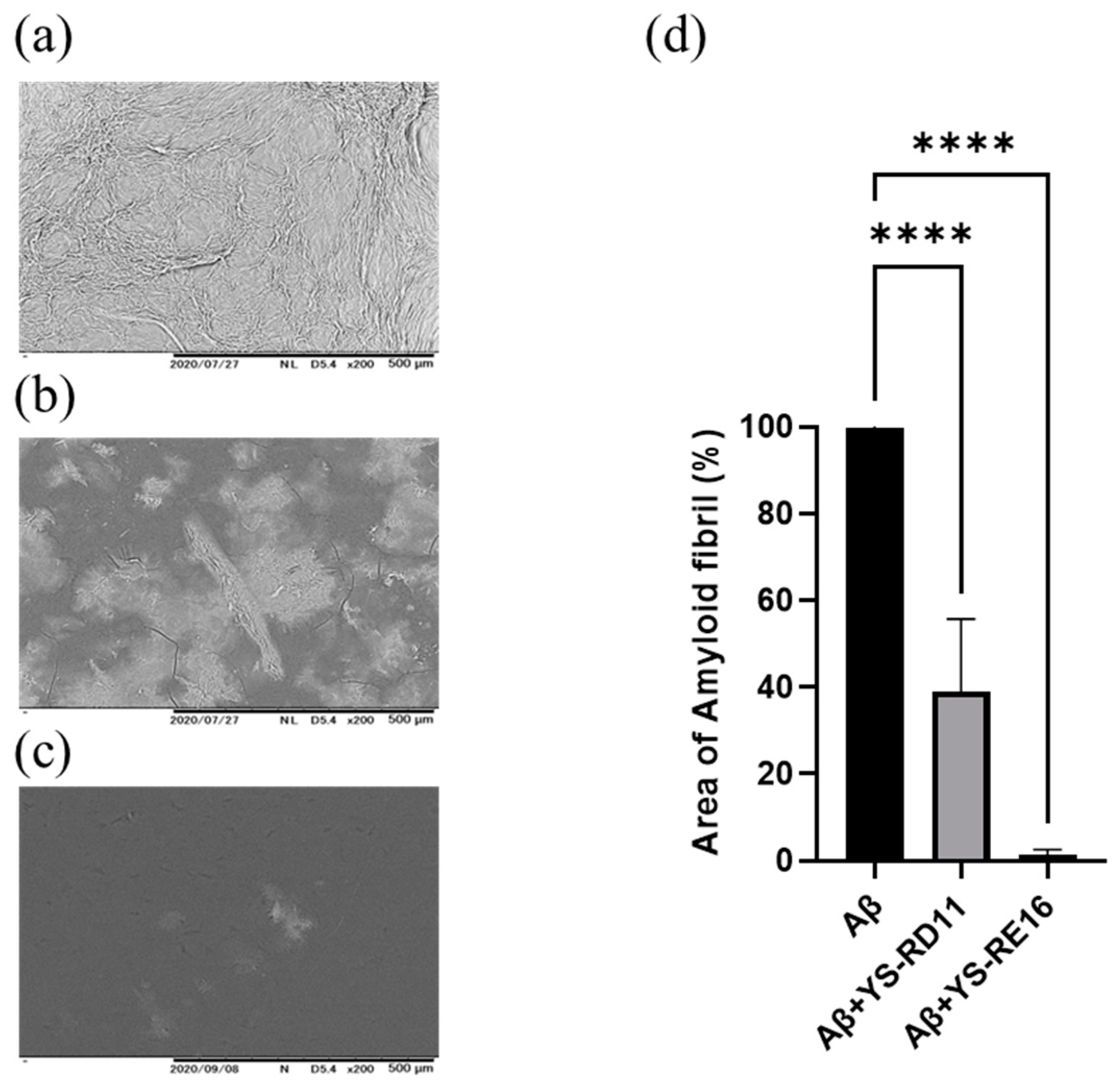
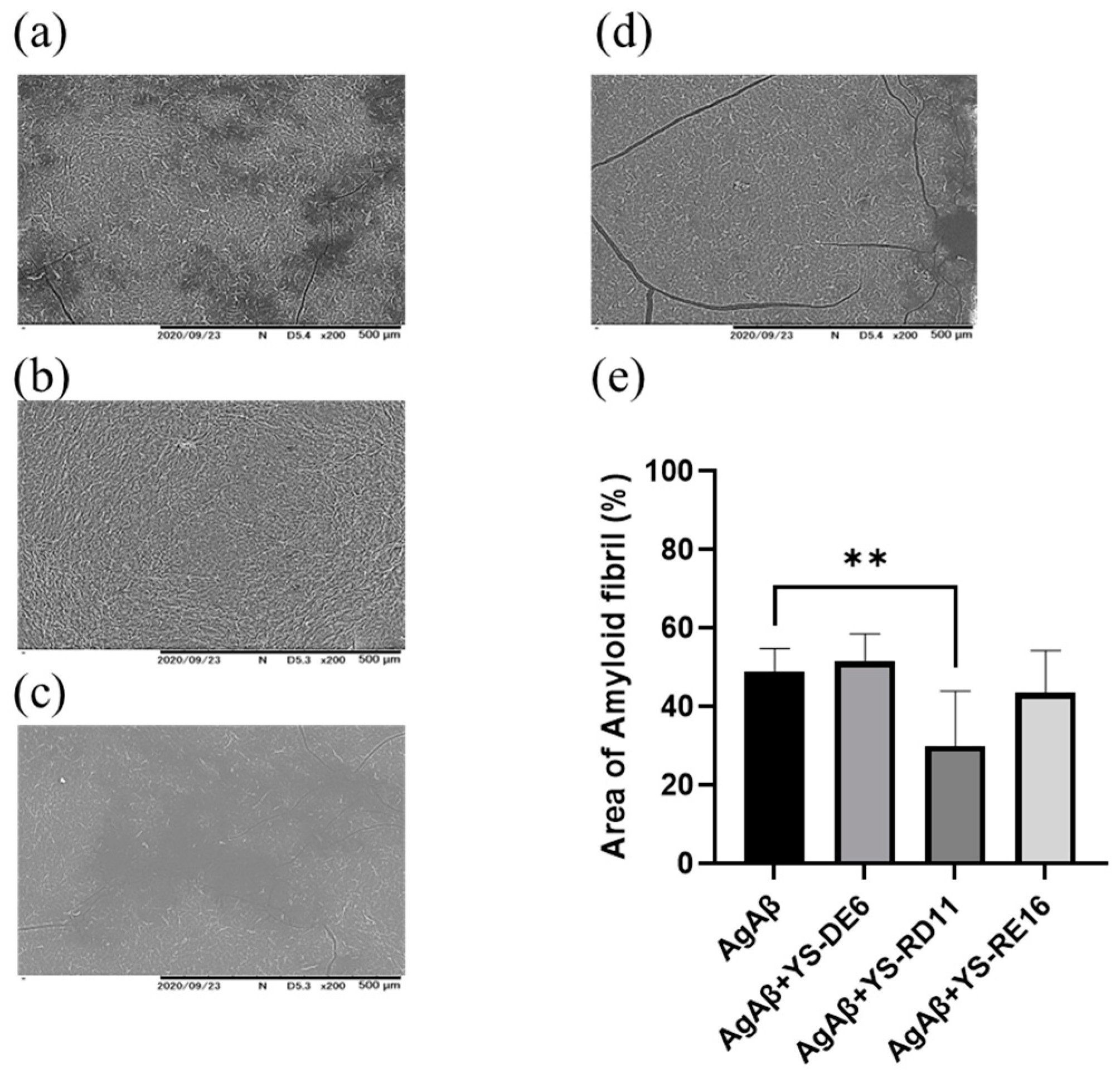
3.3. Resolving Effects on Aggregated Aβ 25-35
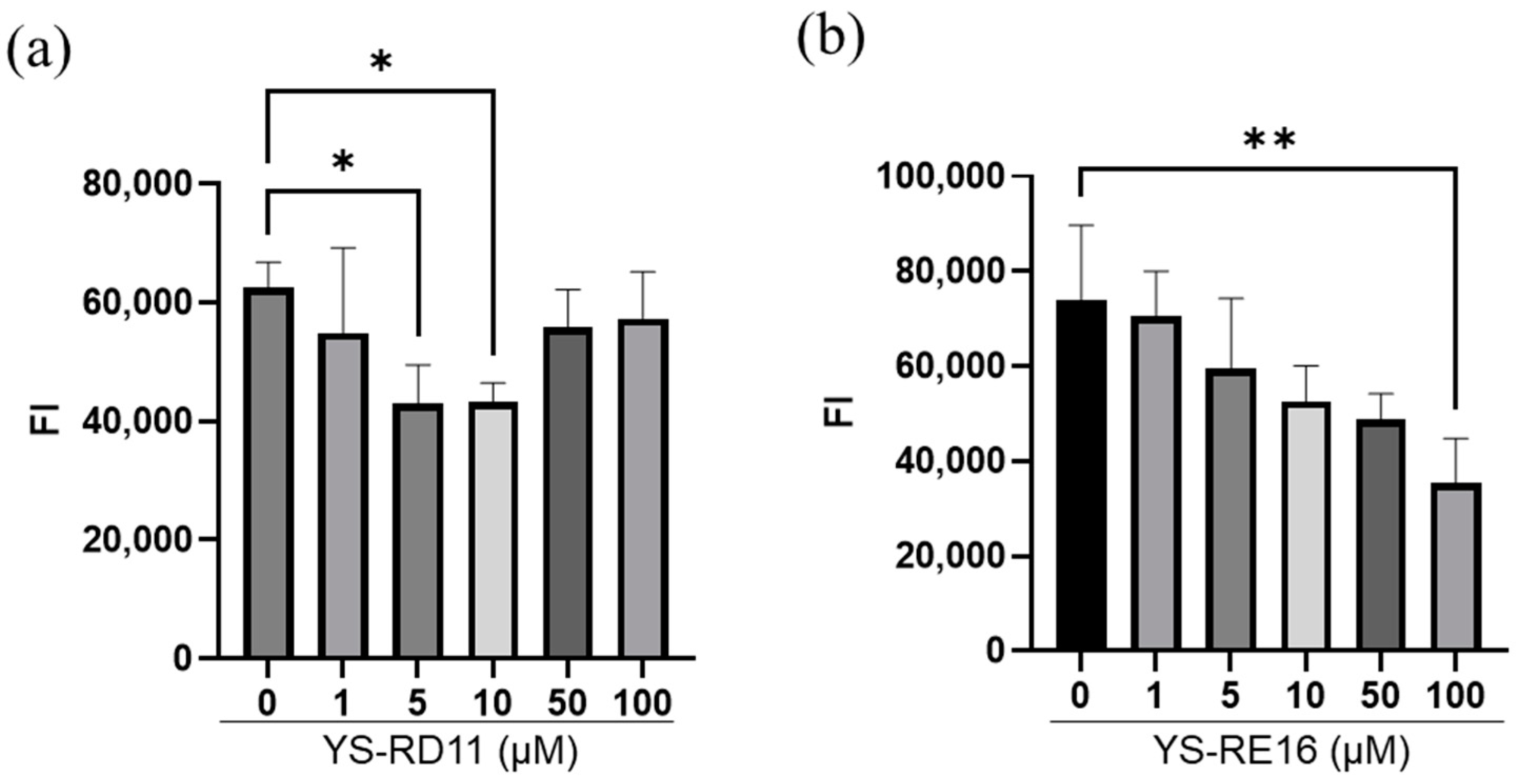
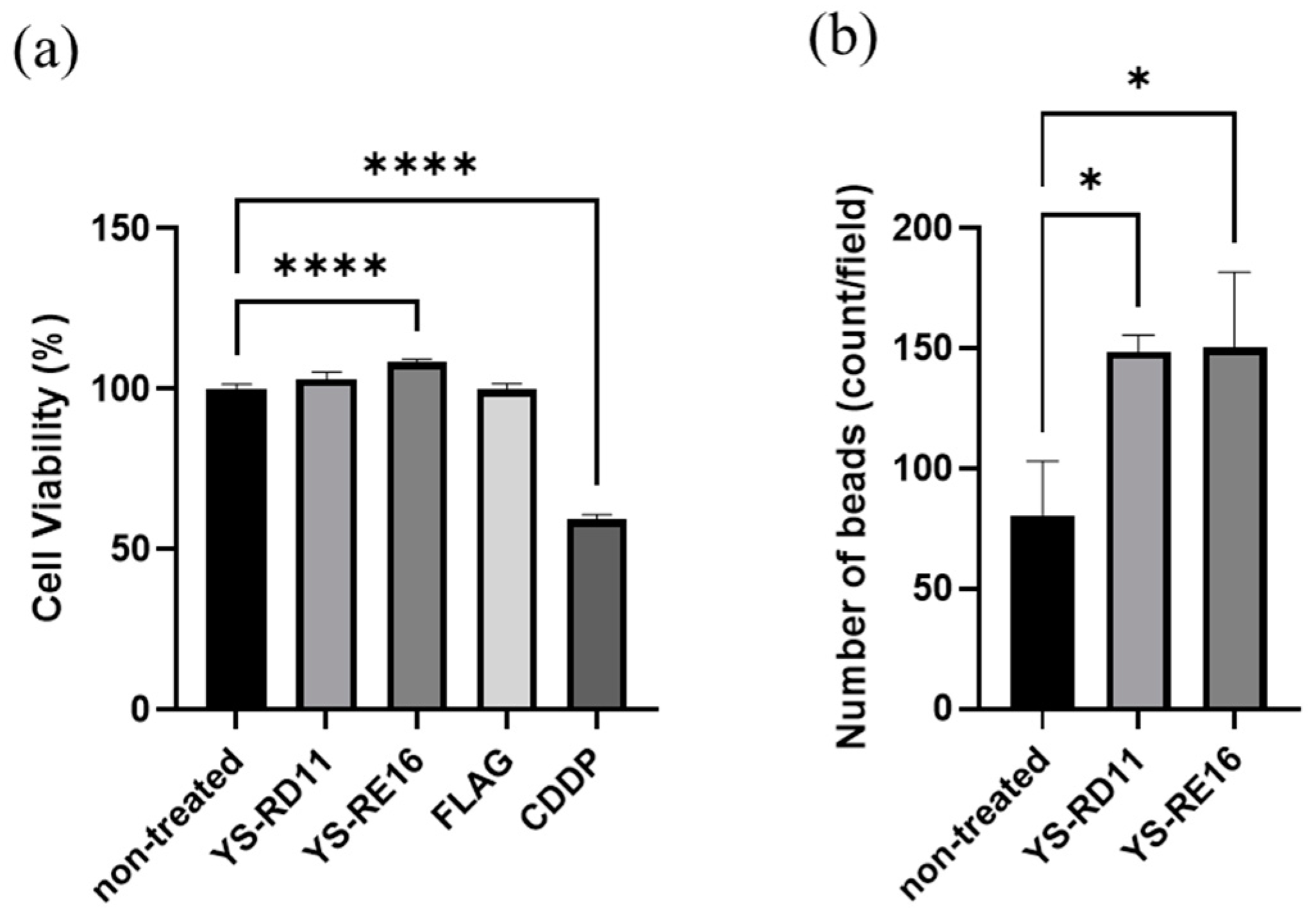
3.4. Cell Experiments
3.5. Animal Experiments
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aishwarya, R.; Abdullah, C.S.; Remex, N.S.; Bhuiyan, M.A.N.; Lu, X.H.; Dhanesha, N.; Stokes, K.Y.; Orr, A.W.; Kevil, C.G.; Bhuiyan, M.S. Diastolic dysfunction in Alzheimer’s disease model mice is associated with Aβ-amyloid aggregate formation and mitochondrial dysfunction. Sci. Rep. 2024, 14, 16715. [Google Scholar] [CrossRef] [PubMed]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I. Amyloid-β Pathology Is the Common Nominator Proteinopathy of the Primate Brain Aging. J. Alzheimers Dis. 2024, 100, S153–S164. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Deshmukh, R. Embelin Mitigates Amyloid-β-Induced Neurotoxicity and Cognitive Impairment in Rats: Potential Therapeutic Implications for Alzheimer’s Disease. Mol. Neurobiol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, K.N.; Manelli, A.M.; Blaine Stine, W.; Baker, L.K.; Krafft, G.A.; Ladu, M.J. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. New precision medicine avenues to the prevention of Alzheimer’s disease from insights into the structure and function of γ-secretases. EMBO J. 2024, 43, 887–903. [Google Scholar] [CrossRef]
- Dehury, B.; Kepp, K.P. Membrane dynamics of γ-secretase with the anterior pharynx-defective 1B subunit. J. Cell. Biochem. 2021, 122, 69–85. [Google Scholar] [CrossRef]
- Fuchino, K.; Mitsuoka, Y.; Masui, M.; Kurose, N.; Yoshida, S.; Komano, K.; Yamamoto, T.; Ogawa, M.; Unemura, C.; Hosono, M.; et al. Rational Design of Novel 1,3-Oxazine Based β-Secretase (BACE1) Inhibitors: Incorporation of a Double Bond To Reduce P-gp Efflux Leading to Robust Aβ Reduction in the Brain. J. Med. Chem. 2018, 61, 5122–5137. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J. Alzheimers Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Lopez, S.; Del Percio, C.; Forloni, G.; Frasca, A.; Drinkenburg, W.H.; Lizio, R.; Noce, G.; Ferri, R.; Soricelli, A.; Stocchi, F.; et al. Chronic BACE-1 Inhibitor Administration in TASTPM Mice (APP KM670/671NL and PSEN1 M146V Mutation): An EEG Study. Int. J. Mol. Sci. 2020, 21, 9072. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Green, B.D. Temporal Effects of Neuron-specific beta-secretase 1 (BACE1) Knock-in on the Mouse Brain Metabolome: Implications for Alzheimer’s Disease. Neuroscience 2019, 397, 138–146. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab—Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401—A clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. Correction: A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 2022, 14, 70. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic soluble amyloid oligomers drive alzheimer’s pathogenesis and represent a clinically validated target for slowing disease progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.X.; Wei, W.; Xu, S.Y. Protective effects of melatonin on cortico-hippocampal neurotoxicity induced by amyloid beta-peptide 25-35. Acta Pharmacol. Sin. 2002, 23, 71–76. [Google Scholar] [PubMed]
- Sun, M.K.; Alkon, D.L. Impairment of hippocampal CA1 heterosynaptic transformation and spatial memory by β-amyloid25–35. J. Neurophysiol. 2002, 87, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, T.; Li, G.; Zhou, L.; Sha, S.; Chen, L. Simvastatin prevents β-amyloid25-35-impaired neurogenesis in hippocampal dentate gyrus through α7nAChR-dependent cascading PI3K-Akt and increasing BDNF via reduction of farnesyl pyrophosphate. Neuropharmacology 2015, 97, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and Biological Activities of Amyloid Beta Peptide 25-35. Curr. Protein Pept. Sci. 2009, 999, 54–67. [Google Scholar] [CrossRef]
- Naldi, M.; Fiori, J.; Pistolozzi, M.; Drake, A.F.; Bertucci, C.; Wu, R.; Mlynarczyk, K.; Filipek, S.; De Simone, A.; Andrisano, V. Amyloid β-peptide 25-35 self-assembly and its inhibition: A model undecapeptide system to gain atomistic and secondary structure details of the Alzheimers disease process and treatment. ACS Chem. Neurosci. 2012, 3, 952–962. [Google Scholar] [CrossRef]
- Nakamura, R.; Konishi, M.; Taniguchi, M.; Hatakawa, Y.; Akizawa, T. The discovery of shorter synthetic proteolytic peptides derived from Tob1 protein. Peptides 2019, 116, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hatakawa, Y.; Nakamura, R.; Konishi, M.; Sakane, T.; Saito, M.; Akizawa, T. Catalytides derived from the Box A region in the ANA/BTG3 protein cleave amyloid-β fragment peptide. Heliyon 2019, 5, e02454. [Google Scholar] [CrossRef]
- Hatakawa, Y.; Nakamura, R.; Konishi, M.; Sakane, T.; Tanaka, A.; Matsuda, A.; Saito, M.; Akizawa, T. Amyloid beta cleavage by ANA-TA9, a synthetic peptide from the ANA/BTG3 Box A region. Alzheimers Dement. Transl. Res. Clin. Interv. 2021, 7, e12146. [Google Scholar] [CrossRef]
- Nakamura, R.; Konishi, M.; Hatakawa, Y.; Saito, M.; Akizawa, T. The Novel Catalytic Peptide, A Synthetic Nona-Peptide (JAL-TA9) Derived from Tob1 Protein, Digests the Amyloid-β Peptide. J. R. Sci. 2019, 1, 30–35. [Google Scholar]
- Hatakawa, Y.; Tanaka, A.; Furubayashi, T.; Nakamura, R.; Konishi, M.; Akizawa, T.; Sakane, T. Direct Delivery of ANA-TA9, a Peptide Capable of Aβ Hydrolysis, to the Brain by Intranasal Administration. Pharmaceutics 2021, 13, 1673. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Higaki, K.; Ninomiya, H.; Luan, Z.; Iida, M.; Ogawa, S.; Suzuki, Y.; Ohno, K.; Nanba, E. Chemical chaperone therapy: Luciferase assay for screening of β-galactosidase mutations. Mol. Genet. Metab. 2010, 101, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Quan, S.; Bardwell, J.C. Chaperone discovery. Bioessays 2012, 34, 973–981. [Google Scholar] [CrossRef]
- Sharma, L.; Sharma, A. Influence of cyclodextrin ring substituents on folding-related aggregation of bovine carbonic anhydrase. Eur. J. Biochem. 2001, 268, 2456–2463. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Kuboi, R. Oxidative refolding of Denatured/Reduced lysozyme utilizing the chaperone-like function of liposomes and immobilized liposome chromatography. Biotechnol. Prog. 1999, 15, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Khanna, R.; Schilling, A.; Flanagan, J.J.; Pellegrino, L.J.; Brignol, N.; Lun, Y.; Guillen, D.; Ranes, B.E.; Frascella, M.; et al. Co-administration with the pharmacological chaperone AT1001 increases recombinant human α-galactosidase A tissue uptake and improves substrate reduction in Fabry mice. Mol. Ther. 2012, 20, 717–726. [Google Scholar] [CrossRef]
- Mogk, A.; Tomoyasu, T.; Goloubinoff, P.; Rüdiger, S.; Röder, D.; Langen, H.; Bukau, B. Identification of thermolabile Escherichia coli proteins: Prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 1999, 18, 6934–6949. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Ishikawa, T. A nuclear localization signal is essential for stress-induced dimer-to-trimer transition of heat shock transcription factor 3. J. Biol. Chem. 2000, 275, 34665–34671. [Google Scholar] [CrossRef]
- Pike, C.J.; Walencewicz-Wasserman, A.J.; Kosmoski, J.; Cribbs, D.H.; Glabe, C.G.; Cotman, C.W. Structure-activity analyses of beta-amyloid peptides: Contributions of the beta 25-35 region to aggregation and neurotoxicity. J. Neurochem. 1995, 64, 253–265. [Google Scholar] [CrossRef]
- Nakamura, R.; Konishi, M.; Higashi, Y.; Saito, M.; Akizawa, T. Five-mer peptides prevent short-term spatial memory deficits in Aβ25-35-induced Alzheimer’s model mouse by suppressing Aβ25-35 aggregation and resolving its aggregate form. Alzheimers Res. Ther. 2023, 15, 83. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Akizawa, T.; Konishi, M. Structure–Activity Relationship of 5-mer Catalytides, GSGYR and RYGSG. Biomolecules 2022, 12, 1766. [Google Scholar] [CrossRef]
- Hatakawa, Y.; Nakamura, R.; Akizawa, T.; Konishi, M.; Matsuda, A.; Oe, T.; Saito, M.; Ito, F. SKGQA, a Peptide Derived from the ANA/BTG3 Protein, Cleaves Amyloid-β with Proteolytic Activity. Biomolecules 2024, 14, 586. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kamatsuka, Y.; Ohishi, A.; Nishida, K.; Nagasawa, K. P2X7 receptors regulate engulfing activity of non-stimulated resting astrocytes. Biochem. Biophys. Res. Commun. 2013, 439, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Aratake, T.; Shimizu, S.; Shimizu, T.; Nakamura, K.; Tsuda, M.; Yawata, T.; Ueba, T.; Saito, M. Influence of extracellular zinc on M1 microglial activation. Sci. Rep. 2017, 7, 43778. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-Β25-35-induced learning and memory impairment in mice, an experimental model of alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Namsechi, R.; Sim, V.L. Structure-based peptide design to modulate amyloid beta aggregation and reduce cytotoxicity. PLoS ONE 2015, 10, e0129087. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.K.; Samuel, D.; Jayaraman, G.; Srimathi, T.; Yu, C. The role of proline in the prevention of aggregation during protein folding in vitro. Biochem. Mol. Biol. Int. 1998, 46, 509–517. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Li, Q.; Wu, Y.; Chen, J.; Xuan, A.; Wang, X. Microglia and immunotherapy in Alzheimer’s disease. Acta Neurol. Scand. 2022, 145, 273–278. [Google Scholar] [CrossRef]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Colonna, M. Microglia in Alzheimer’s disease: A target for immunotherapy. J. Leukoc. Biol. 2019, 106, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takayama, K.; Furubayashi, T.; Mori, K.; Takemura, Y.; Amano, M.; Maeda, C.; Inoue, D.; Kimura, S.; Kiriyama, A.; et al. Transnasal Delivery of the Peptide Agonist Specific to Neuromedin-U Receptor 2 to the Brain for the Treatment of Obesity. Mol. Pharm. 2020, 17, 32–39. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | |
|---|---|
| YS-YE20 | YKNMRETLVYLTHLDYDDTE |
| YS-YR5 | YKNMR |
| YS-DE6 | DYDDTE |
| YS-RD11 | RETLVYLTHLD |
| YS-YD15 | YKNMRETLVYLTHLD |
| YS-RE16 | RETLVYLTHLDYDDTE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, R.; Matsuda, A.; Higashi, Y.; Hayashi, Y.; Konishi, M.; Saito, M.; Akizawa, T. An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model. Biomolecules 2024, 14, 1234. https://doi.org/10.3390/biom14101234
Nakamura R, Matsuda A, Higashi Y, Hayashi Y, Konishi M, Saito M, Akizawa T. An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model. Biomolecules. 2024; 14(10):1234. https://doi.org/10.3390/biom14101234
Chicago/Turabian StyleNakamura, Rina, Akira Matsuda, Youichirou Higashi, Yoshihiro Hayashi, Motomi Konishi, Motoaki Saito, and Toshifumi Akizawa. 2024. "An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model" Biomolecules 14, no. 10: 1234. https://doi.org/10.3390/biom14101234
APA StyleNakamura, R., Matsuda, A., Higashi, Y., Hayashi, Y., Konishi, M., Saito, M., & Akizawa, T. (2024). An 11-mer Synthetic Peptide Suppressing Aggregation of Aβ25-35 and Resolving Its Aggregated Form Improves Test Performance in an Aβ25-35-Induced Alzheimer’s Mouse Model. Biomolecules, 14(10), 1234. https://doi.org/10.3390/biom14101234