


Increased Risk for Atrial Alternans in Rabbit Heart Failure: The Role of Ca2+/Calmodulin-Dependent Kinase II and Inositol-1,4,5-trisphosphate Signaling



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Heart Failure Model
2.3. Myocyte Isolation
2.4. Patch Clamp Experiments
2.5. [Ca2+] Measurements
2.6. CaT Alternans
2.7. Drugs
2.8. Data Analysis and Presentation
3. Results
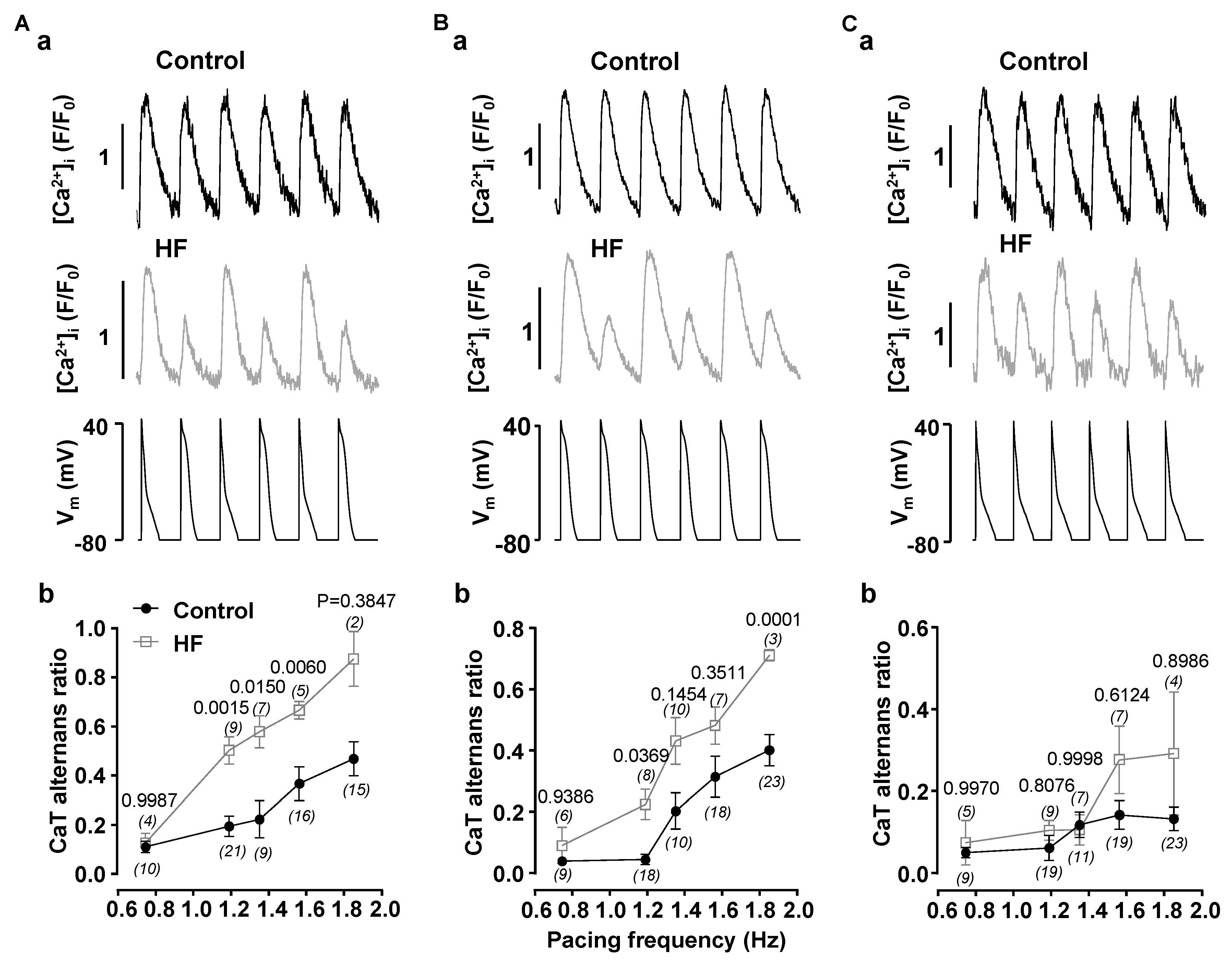
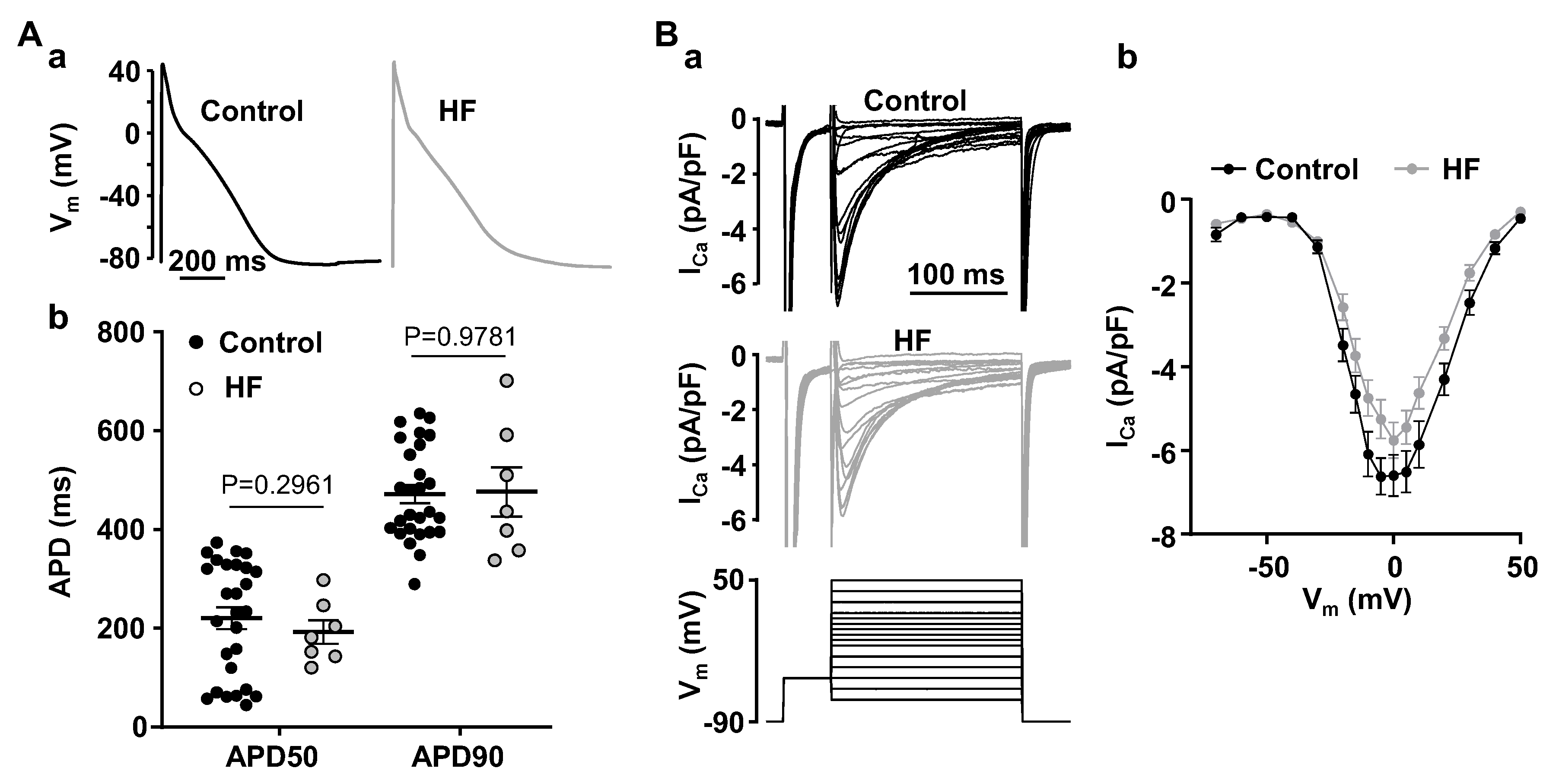
3.1. Alternans Is Enhanced in Atrial Myocytes from Failing Hearts
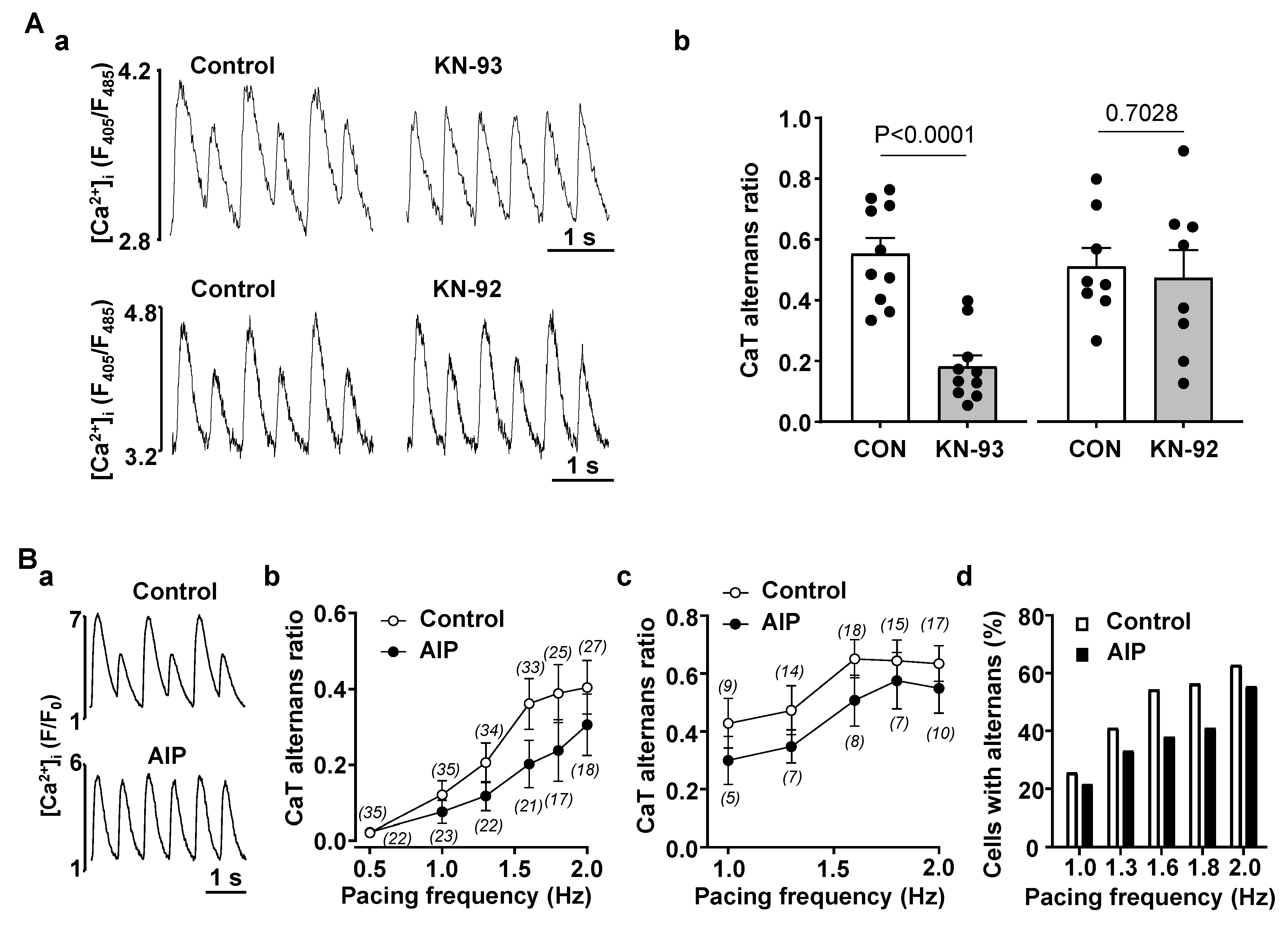
3.2. Enhanced CaMKII Activity Increases Risk for Atrial Alternans
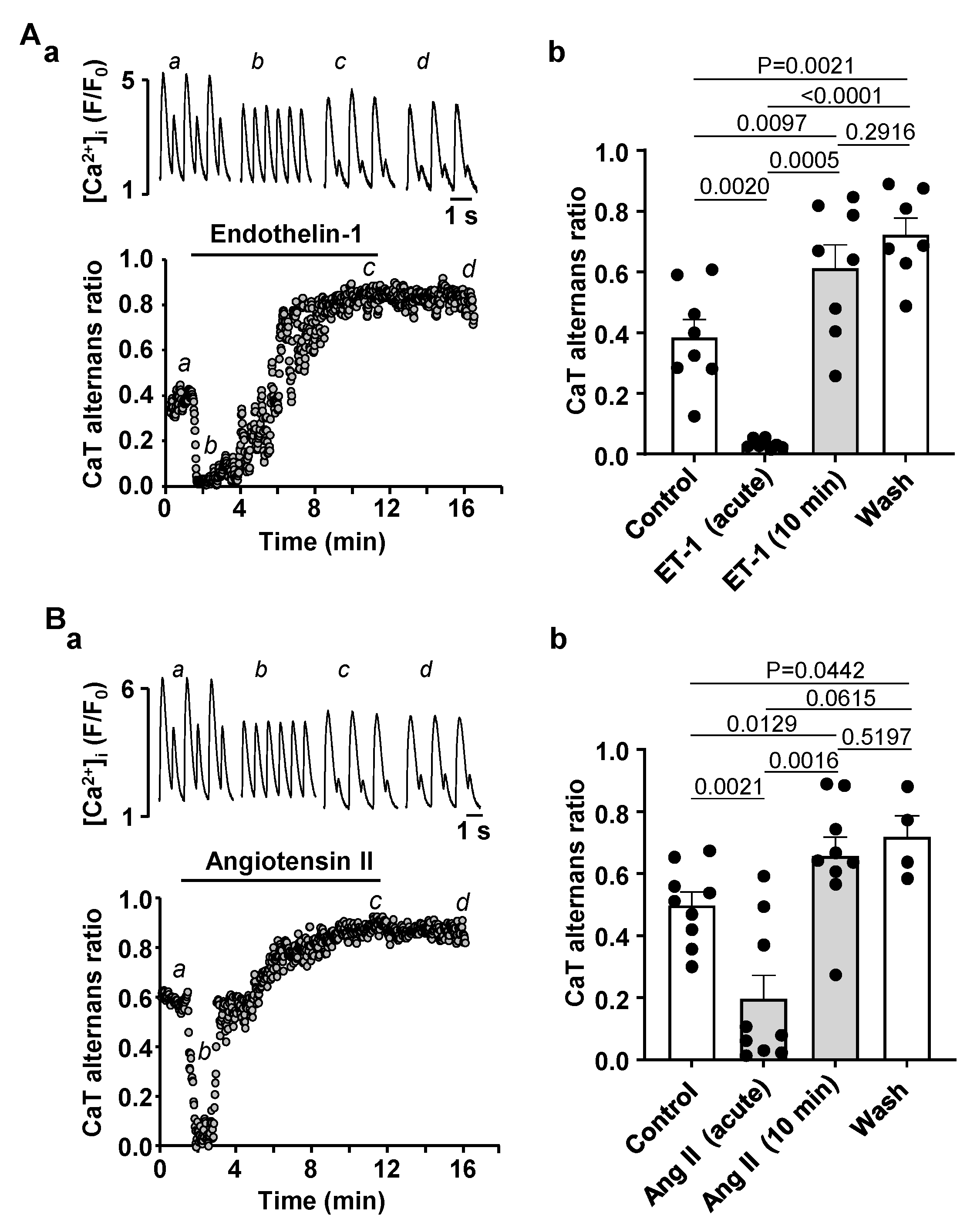
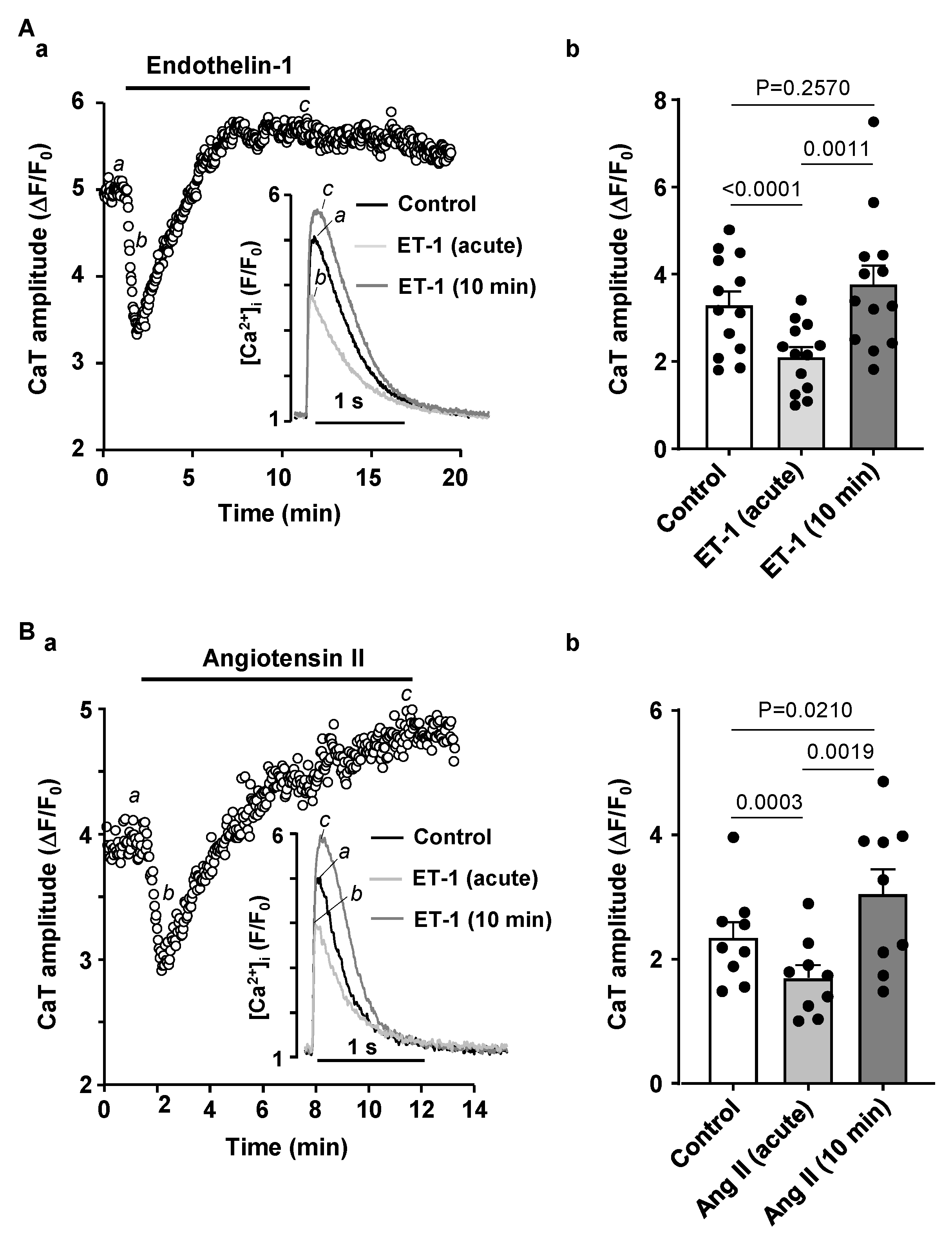
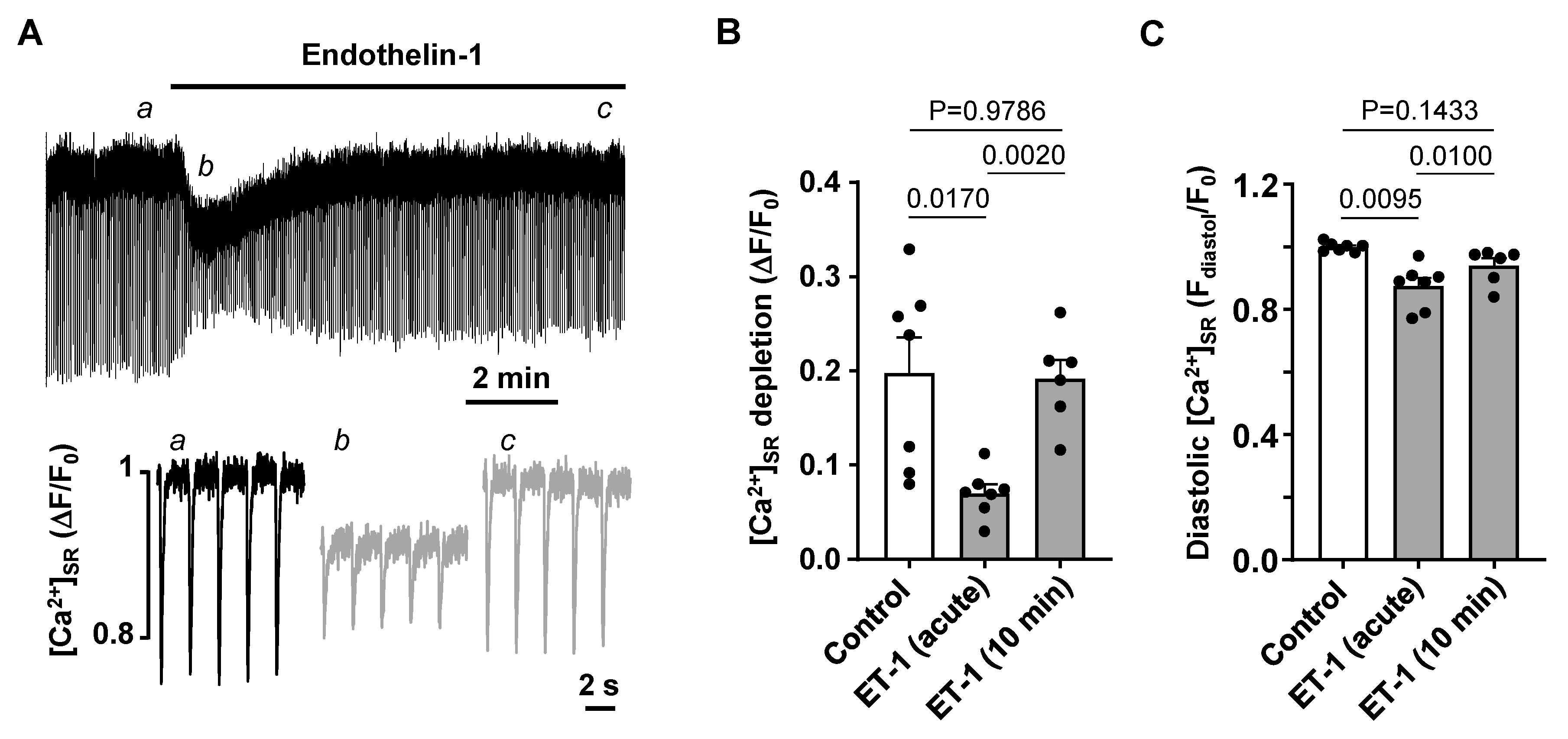
3.3. Activation of IP3 Signaling in Atrial Myocytes Affects Alternans Development and CaT Properties
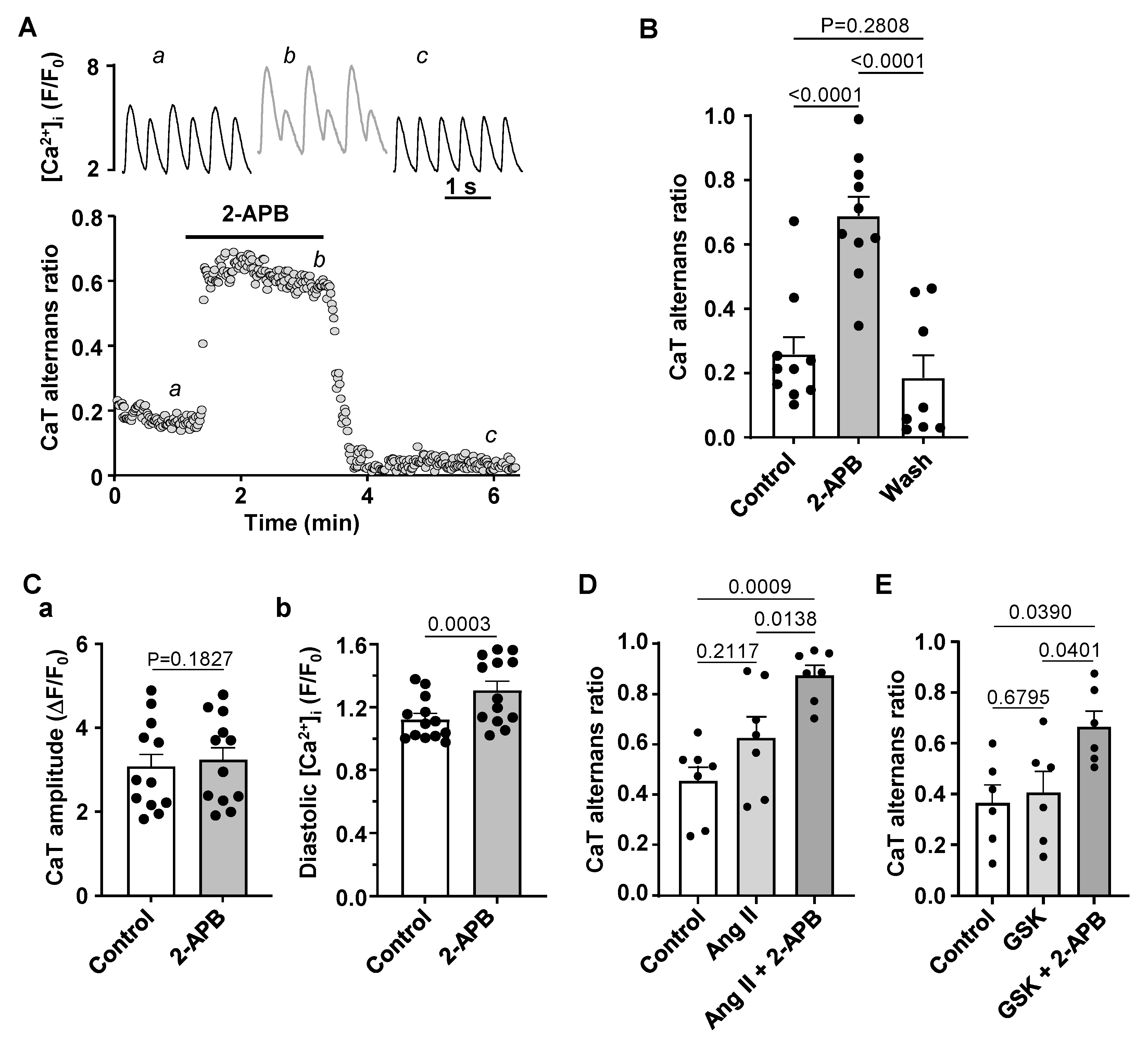
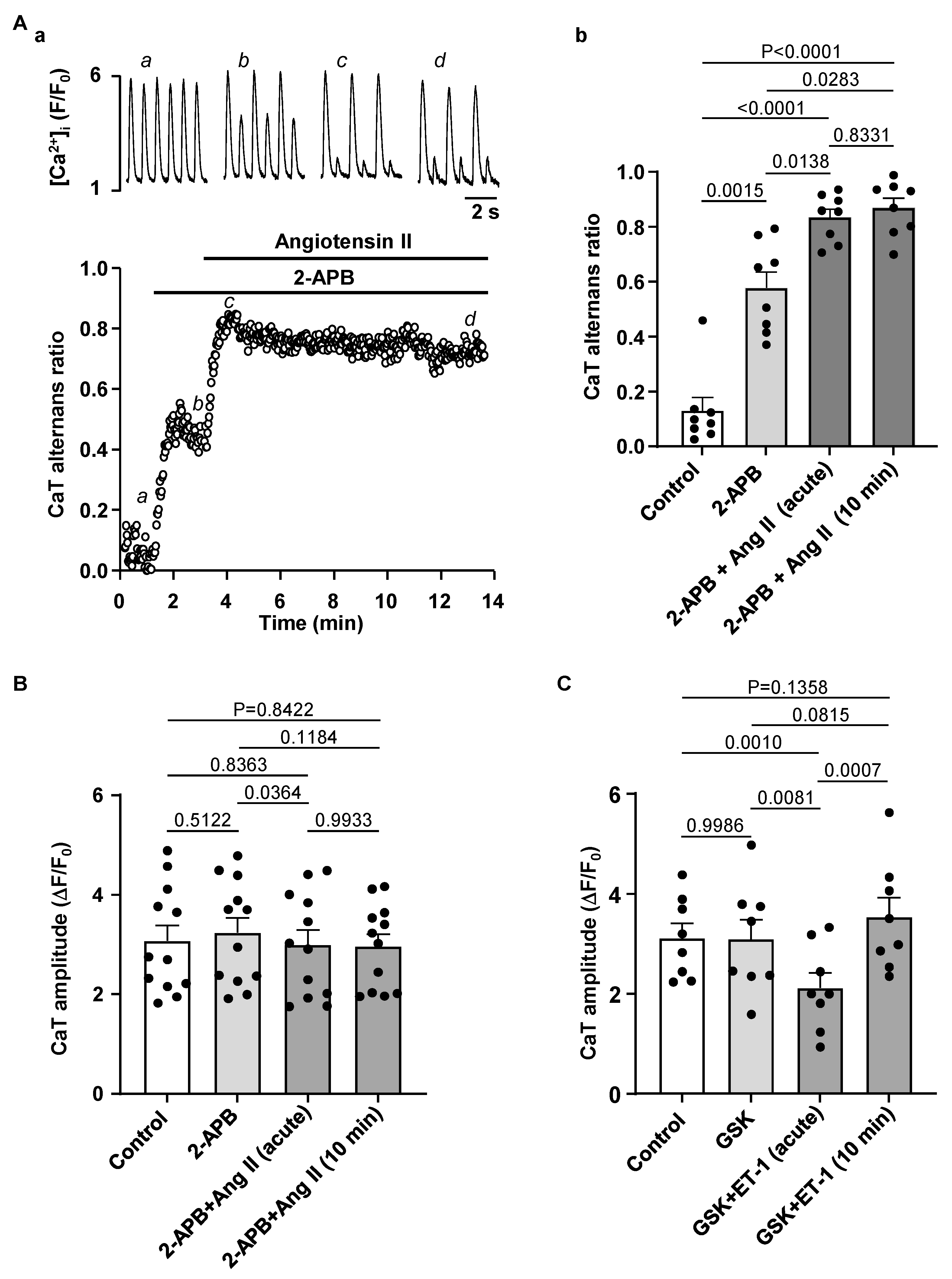
3.4. Effect of IP3R and TRP Channel Inhibition on CaT Alternans
4. Discussion
4.1. HF Remodeling Leads to Higher Risk of Proarrhythmic Alternans
4.2. CaMKII Promotes Atrial Alternans
4.3. Modulation of IP3 Signaling Pathways Affects Development of Atrial Alternans
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mundisugih, J.; Franke, K.B.; Tully, P.J.; Munawar, D.A.; Kumar, S.; Mahajan, R. Prevalence and Prognostic Implication of Atrial Fibrillation in Heart Failure Subtypes: Systematic Review and Meta-Analysis. Heart Lung Circ. 2023, 32, 666–677. [Google Scholar] [CrossRef]
- Thihalolipavan, S.; Morin, D.P. Atrial fibrillation and heart failure: Update 2015. Prog. Cardiovasc. Dis. 2015, 58, 126–135. [Google Scholar] [CrossRef]
- Hohendanner, F.; Walther, S.; Maxwell, J.T.; Kettlewell, S.; Awad, S.; Smith, G.L.; Lonchyna, V.A.; Blatter, L.A. Inositol-1,4,5-trisphosphate induced Ca2+ release and excitation-contraction coupling in atrial myocytes from normal and failing hearts. J. Physiol. 2015, 593, 1459–1477. [Google Scholar] [CrossRef]
- Hohendanner, F.; DeSantiago, J.; Heinzel, F.R.; Blatter, L.A. Dyssynchronous calcium removal in heart failure-induced atrial remodeling. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1352–H1359. [Google Scholar] [CrossRef]
- Comtois, P.; Nattel, S. Atrial repolarization alternans as a path to atrial fibrillation. J. Cardiovasc. Electrophysiol. 2012, 23, 1013–1015. [Google Scholar] [CrossRef]
- Franz, M.R.; Jamal, S.M.; Narayan, S.M. The role of action potential alternans in the initiation of atrial fibrillation in humans: A review and future directions. Europace 2012, 14 (Suppl. S5), v58–v64. [Google Scholar] [CrossRef]
- Walker, M.L.; Rosenbaum, D.S. Repolarization alternans: Implications for the mechanism and prevention of sudden cardiac death. Cardiovasc. Res. 2003, 57, 599–614. [Google Scholar] [CrossRef]
- Lipp, P.; Laine, M.; Tovey, S.C.; Burrell, K.M.; Berridge, M.J.; Li, W.; Bootman, M.D. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr. Biol. 2000, 10, 939–942. [Google Scholar] [CrossRef]
- Domeier, T.L.; Zima, A.V.; Maxwell, J.T.; Huke, S.; Mignery, G.A.; Blatter, L.A. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H596–H604. [Google Scholar] [CrossRef]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Ljubojevic, S.; Radulovic, S.; Leitinger, G.; Sedej, S.; Sacherer, M.; Holzer, M.; Winkler, C.; Pritz, E.; Mittler, T.; Schmidt, A.; et al. Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure. Circulation 2014, 130, 244–255. [Google Scholar] [CrossRef]
- Yamda, J.; Ohkusa, T.; Nao, T.; Ueyama, T.; Yano, M.; Kobayashi, S.; Hamano, K.; Esato, K.; Matsuzaki, M. Up-regulation of inositol 1,4,5 trisphosphate receptor expression in atrial tissue in patients with chronic atrial fibrillation. J. Am. Coll. Cardiol. 2001, 37, 1111–1119. [Google Scholar] [CrossRef]
- Qi, X.Y.; Vahdati Hassani, F.; Hoffmann, D.; Xiao, J.; Xiong, F.; Villeneuve, L.R.; Ljubojevic-Holzer, S.; Kamler, M.; Abu-Taha, I.; Heijman, J.; et al. Inositol Trisphosphate Receptors and Nuclear Calcium in Atrial Fibrillation. Circ. Res. 2021, 128, 619–635. [Google Scholar] [CrossRef]
- Anderson, M.E.; Brown, J.H.; Bers, D.M. CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 468–473. [Google Scholar] [CrossRef]
- Hoeker, G.S.; Hanafy, M.A.; Oster, R.A.; Bers, D.M.; Pogwizd, S.M. Reduced Arrhythmia Inducibility with Calcium/Calmodulin-dependent Protein Kinase II Inhibition in Heart Failure Rabbits. J. Cardiovasc. Pharmacol. 2016, 67, 260–265. [Google Scholar] [CrossRef]
- Heijman, J.; Muna, A.P.; Veleva, T.; Molina, C.E.; Sutanto, H.; Tekook, M.; Wang, Q.; Abu-Taha, I.H.; Gorka, M.; Kunzel, S.; et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ. Res. 2020, 127, 1036–1055. [Google Scholar] [CrossRef]
- Zima, A.V.; Blatter, L.A. Inositol-1,4,5-trisphosphate-dependent Ca2+ signalling in cat atrial excitation-contraction coupling and arrhythmias. J. Physiol. 2004, 555, 607–615. [Google Scholar] [CrossRef]
- Pogwizd, S.M. Nonreentrant mechanisms underlying spontaneous ventricular arrhythmias in a model of nonischemic heart failure in rabbits. Circulation 1995, 92, 1034–1048. [Google Scholar] [CrossRef]
- Bers, D.M.; Pogwizd, S.M.; Schlotthauer, K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic. Res. Cardiol. 2002, 97 (Suppl. S1), I36–I42. [Google Scholar] [CrossRef] [PubMed]
- Shannon, T.R.; Pogwizd, S.M.; Bers, D.M. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res. 2003, 93, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Kanaporis, G.; Blatter, L.A. Membrane potential determines calcium alternans through modulation of SR Ca2+ load and L-type Ca2+ current. J. Mol. Cell. Cardiol. 2017, 105, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Kanaporis, G.; Blatter, L.A. The mechanisms of calcium cycling and action potential dynamics in cardiac alternans. Circ. Res. 2015, 116, 846–856. [Google Scholar] [CrossRef]
- Kanaporis, G.; Blatter, L.A. Activation of small conductance Ca2+-activated K+ channels suppresses Ca2+ transient and action potential alternans in ventricular myocytes. J. Physiol. 2023, 601, 51–67. [Google Scholar] [CrossRef]
- Kanaporis, G.; Kalik, Z.M.; Blatter, L.A. Action potential shortening rescues atrial calcium alternans. J. Physiol. 2019, 597, 723–740. [Google Scholar] [CrossRef]
- Kanaporis, G.; Blatter, L.A. Calcium-activated chloride current determines action potential morphology during calcium alternans in atrial myocytes. J. Physiol. 2016, 594, 699–714. [Google Scholar] [CrossRef]
- Glukhov, A.V.; Balycheva, M.; Sanchez-Alonso, J.L.; Ilkan, Z.; Alvarez-Laviada, A.; Bhogal, N.; Diakonov, I.; Schobesberger, S.; Sikkel, M.B.; Bhargava, A.; et al. Direct Evidence for Microdomain-Specific Localization and Remodeling of Functional L-Type Calcium Channels in Rat and Human Atrial Myocytes. Circulation 2015, 132, 2372–2384. [Google Scholar] [CrossRef]
- Clarke, J.D.; Caldwell, J.L.; Horn, M.A.; Bode, E.F.; Richards, M.A.; Hall, M.C.; Graham, H.K.; Briston, S.J.; Greensmith, D.J.; Eisner, D.A.; et al. Perturbed atrial calcium handling in an ovine model of heart failure: Potential roles for reductions in the L-type calcium current. J. Mol. Cell. Cardiol. 2015, 79, 169–179. [Google Scholar] [CrossRef]
- Li, D.; Melnyk, P.; Feng, J.; Wang, Z.; Petrecca, K.; Shrier, A.; Nattel, S. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation 2000, 101, 2631–2638. [Google Scholar] [CrossRef] [PubMed]
- Hoch, B.; Meyer, R.; Hetzer, R.; Krause, E.G.; Karczewski, P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 1999, 84, 713–721. [Google Scholar] [CrossRef]
- Liu, Z.; Finet, J.E.; Wolfram, J.A.; Anderson, M.E.; Ai, X.; Donahue, J.K. Calcium/calmodulin-dependent protein kinase II causes atrial structural remodeling associated with atrial fibrillation and heart failure. Heart Rhythm. 2019, 16, 1080–1088. [Google Scholar] [CrossRef]
- Yoo, S.; Aistrup, G.; Shiferaw, Y.; Ng, J.; Mohler, P.J.; Hund, T.J.; Waugh, T.; Browne, S.; Gussak, G.; Gilani, M.; et al. Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight 2018, 3, e120728. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Mori, Y.; Hara, Y.; Uchida, K.; Zhou, H.; Mikoshiba, K. 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Recept. Channels 2001, 7, 429–439. [Google Scholar] [PubMed]
- Zhang, Y.H.; Wu, H.J.; Che, H.; Sun, H.Y.; Cheng, L.C.; Li, X.; Au, W.K.; Tse, H.F.; Li, G.R. Functional transient receptor potential canonical type 1 channels in human atrial myocytes. Pflugers Arch. 2013, 465, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Rainer, P.P.; Shalkey Hahn, V.; Lee, D.I.; Jo, S.H.; Andersen, A.; Liu, T.; Xu, X.; Willette, R.N.; Lepore, J.J.; et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- McManus, D.D.; Saczynski, J.S.; Lessard, D.; Kinno, M.; Pidikiti, R.; Esa, N.; Harrington, J.; Goldberg, R.J. Recent trends in the incidence, treatment, and prognosis of patients with heart failure and atrial fibrillation (the Worcester Heart Failure Study). Am. J. Cardiol. 2013, 111, 1460–1465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shinagawa, K.; Shi, Y.F.; Tardif, J.C.; Leung, T.K.; Nattel, S. Dynamic nature of atrial fibrillation substrate during development and reversal of heart failure in dogs. Circulation 2002, 105, 2672–2678. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.; Morton, J.B.; Davidson, N.C.; Spence, S.J.; Vohra, J.K.; Sparks, P.B.; Kalman, J.M. Electrical remodeling of the atria in congestive heart failure: Electrophysiological and electroanatomic mapping in humans. Circulation 2003, 108, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Chen, Y.C.; Chen, Y.A.; Yeh, Y.H.; Chen, S.A.; Chen, Y.J. B-Type Natriuretic Peptide Modulates Pulmonary Vein Arrhythmogenesis: A Novel Potential Contributor to the Genesis of Atrial Tachyarrhythmia in Heart Failure. J. Cardiovasc. Electrophysiol. 2016, 27, 1462–1471. [Google Scholar] [CrossRef]
- Denham, N.C.; Pearman, C.M.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front. Physiol. 2018, 9, 1380. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Qi, M.; Yuan, W.; Samarel, A.M.; Bers, D.M. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 1999, 85, 1009–1019. [Google Scholar] [CrossRef]
- Lalani, G.G.; Schricker, A.A.; Clopton, P.; Krummen, D.E.; Narayan, S.M. Frequency analysis of atrial action potential alternans: A sensitive clinical index of individual propensity to atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2013, 6, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.M.; Franz, M.R.; Clopton, P.; Pruvot, E.J.; Krummen, D.E. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 2011, 123, 2922–2930. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.H.; Wakili, R.; Qi, X.Y.; Chartier, D.; Boknik, P.; Kaab, S.; Ravens, U.; Coutu, P.; Dobrev, D.; Nattel, S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ. Arrhythm. Electrophysiol. 2008, 1, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, A.; Nishijima, Y.; Terentyev, D.; Khan, M.; Terentyeva, R.; Hamlin, R.L.; Nakayama, T.; Gyorke, S.; Cardounel, A.J.; Carnes, C.A. Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc. Res. 2009, 84, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Molina, C.E.; Abu-Taha, I.H.; Wang, Q.; Rosello-Diez, E.; Kamler, M.; Nattel, S.; Ravens, U.; Wehrens, X.H.T.; Hove-Madsen, L.; Heijman, J.; et al. Profibrotic, Electrical, and Calcium-Handling Remodeling of the Atria in Heart Failure Patients with and without Atrial Fibrillation. Front. Physiol. 2018, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Chelu, M.G.; Sarma, S.; Sood, S.; Wang, S.; van Oort, R.J.; Skapura, D.G.; Li, N.; Santonastasi, M.; Muller, F.U.; Schmitz, W.; et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Investig. 2009, 119, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- van Oort, R.J.; McCauley, M.D.; Dixit, S.S.; Pereira, L.; Yang, Y.; Respress, J.L.; Wang, Q.; De Almeida, A.C.; Skapura, D.G.; Anderson, M.E.; et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 2010, 122, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Livshitz, L.M.; Rudy, Y. Regulation of Ca2+ and electrical alternans in cardiac myocytes: Role of CAMKII and repolarizing currents. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2854–H2866. [Google Scholar] [CrossRef]
- Sadredini, M.; Haugsten Hansen, M.; Frisk, M.; Louch, W.E.; Lehnart, S.E.; Sjaastad, I.; Stokke, M.K. CaMKII inhibition has dual effects on spontaneous Ca2+ release and Ca2+ alternans in ventricular cardiomyocytes from mice with a gain-of-function RyR2 mutation. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H446–H460. [Google Scholar] [CrossRef]
- Sag, C.M.; Mallwitz, A.; Wagner, S.; Hartmann, N.; Schotola, H.; Fischer, T.H.; Ungeheuer, N.; Herting, J.; Shah, A.M.; Maier, L.S.; et al. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J. Mol. Cell. Cardiol. 2014, 76, 94–105. [Google Scholar] [CrossRef]
- Zang, Y.; Dai, L.; Zhan, H.; Dou, J.; Xia, L.; Zhang, H. Theoretical investigation of the mechanism of heart failure using a canine ventricular cell model: Especially the role of up-regulated CaMKII and SR Ca2+ leak. J. Mol. Cell. Cardiol. 2013, 56, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Blatter, L.A.; Kanaporis, G.; Martinez-Hernandez, E.; Oropeza-Almazan, Y.; Banach, K. Excitation-contraction coupling and calcium release in atrial muscle. Pflugers Arch. 2021, 473, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Demydenko, K.; Ekhteraei-Tousi, S.; Roderick, H.L. Inositol 1,4,5-trisphosphate receptors in cardiomyocyte physiology and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2022, 377, 20210319. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanaporis, G.; Blatter, L.A. Increased Risk for Atrial Alternans in Rabbit Heart Failure: The Role of Ca2+/Calmodulin-Dependent Kinase II and Inositol-1,4,5-trisphosphate Signaling. Biomolecules 2024, 14, 53. https://doi.org/10.3390/biom14010053
Kanaporis G, Blatter LA. Increased Risk for Atrial Alternans in Rabbit Heart Failure: The Role of Ca2+/Calmodulin-Dependent Kinase II and Inositol-1,4,5-trisphosphate Signaling. Biomolecules. 2024; 14(1):53. https://doi.org/10.3390/biom14010053
Chicago/Turabian StyleKanaporis, Giedrius, and Lothar A. Blatter. 2024. "Increased Risk for Atrial Alternans in Rabbit Heart Failure: The Role of Ca2+/Calmodulin-Dependent Kinase II and Inositol-1,4,5-trisphosphate Signaling" Biomolecules 14, no. 1: 53. https://doi.org/10.3390/biom14010053
APA StyleKanaporis, G., & Blatter, L. A. (2024). Increased Risk for Atrial Alternans in Rabbit Heart Failure: The Role of Ca2+/Calmodulin-Dependent Kinase II and Inositol-1,4,5-trisphosphate Signaling. Biomolecules, 14(1), 53. https://doi.org/10.3390/biom14010053