
Altered TRPM7-Dependent Calcium Influx in Natural Killer Cells of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Peripheral Blood Mononuclear Cell and Natural Killer Cell Isolation
2.3. Calcium Imaging
2.4. Statistical Analysis
3. Results
3.1. Participant Characteristics
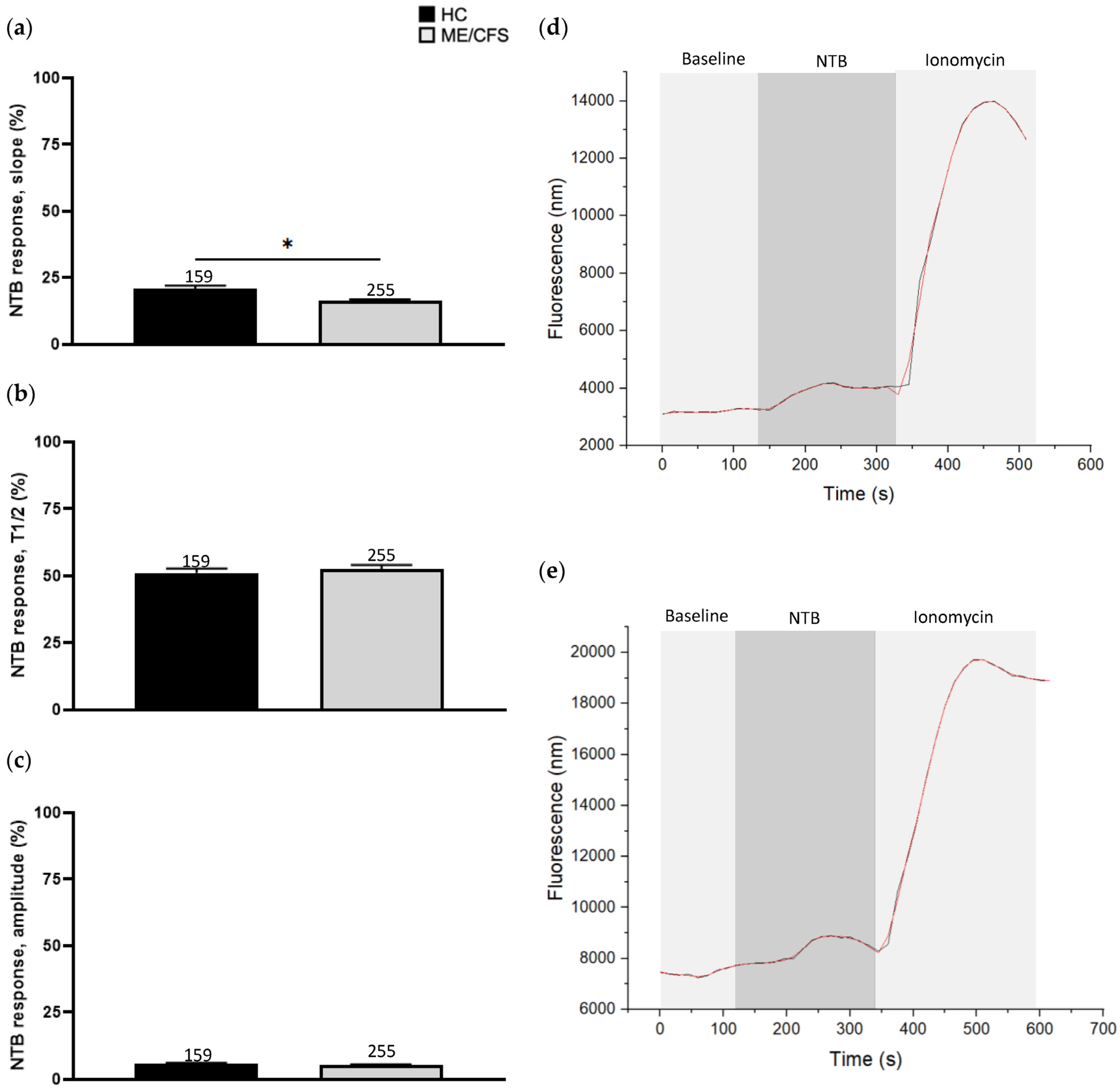
3.2. Effect of Naltriben Treatment on Calcium Influx
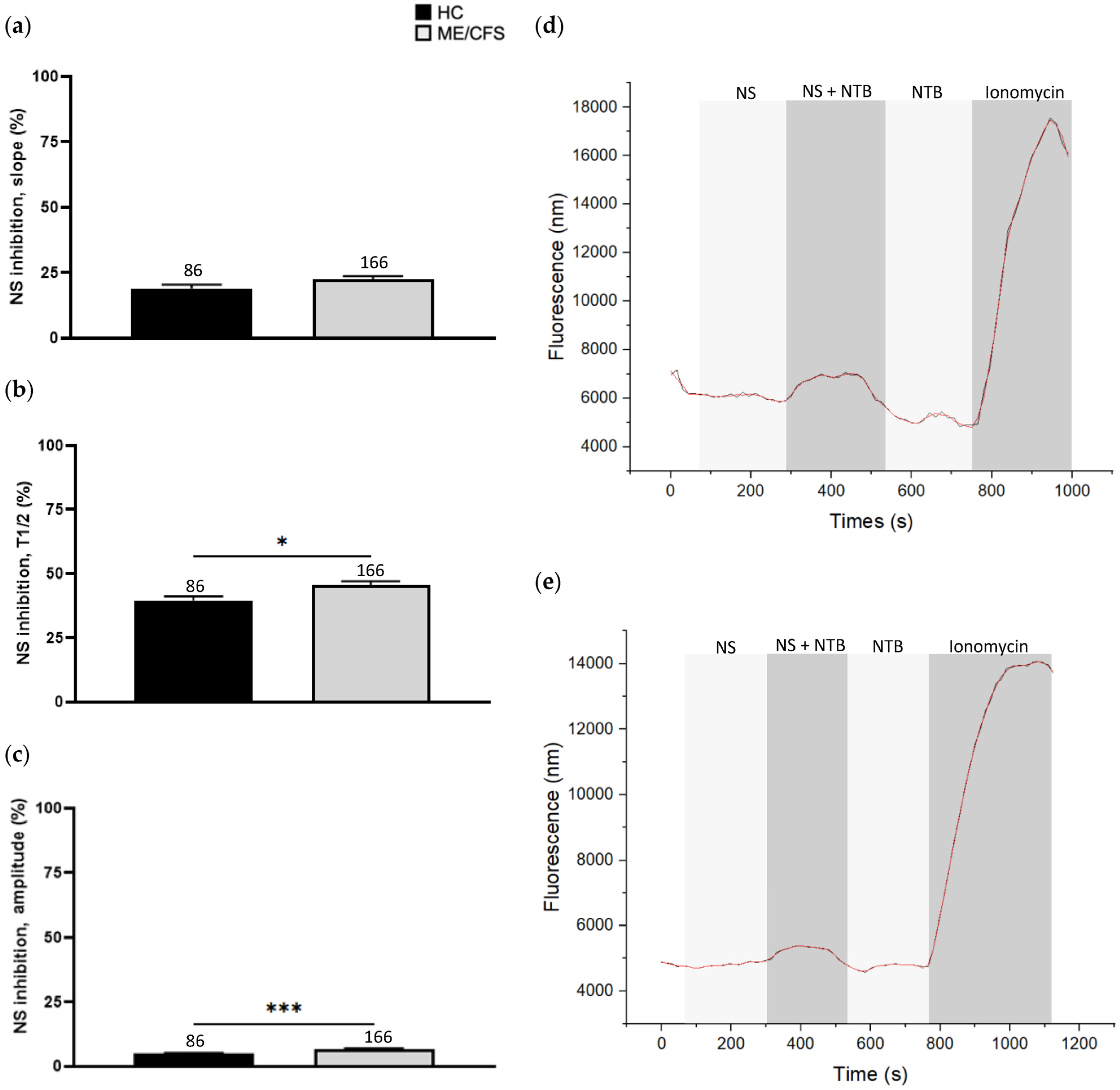
3.3. Effect of NS8593 Treatment on Calcium Influx
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carruthers, B.M.; van de Sande, M.I.; De Meirleir, K.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.P.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International Consensus Criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef]
- Clayton, E.W. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: An IOM Report on Redefining an Illness. JAMA 2015, 313, 1101–1102. [Google Scholar] [CrossRef]
- Abdulla, J.; Torpy, B.D. Chronic Fatigue Syndrome. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Bozzini, S.; Albergati, A.; Capelli, E.; Lorusso, L.; Gazzaruso, C.; Pelissero, G.; Falcone, C. Cardiovascular characteristics of chronic fatigue syndrome. Biomed. Rep. 2018, 8, 26–30. [Google Scholar] [CrossRef]
- Monro, J.A.; Puri, B.K. A Molecular Neurobiological Approach to Understanding the Aetiology of Chronic Fatigue Syndrome (Myalgic Encephalomyelitis or Systemic Exertion Intolerance Disease) with Treatment Implications. Mol. Neurobiol. 2018, 55, 7377–7388. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.-J.; Ahn, Y.-C.; Jang, E.-S.; Lee, S.-W.; Lee, S.-H.; Son, C.-G. Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). J. Transl. Med. 2020, 18, 100. [Google Scholar] [CrossRef]
- Chew-Graham, C.; Dowrick, C.; Wearden, A.; Richardson, V.; Peters, S. Making the diagnosis of Chronic Fatigue Syndrome/Myalgic Encephalitis in primary care: A qualitative study. BMC Fam. Pract. 2010, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.P.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Chronic Fatigue Syndr. 2003, 11, 7–115. [Google Scholar] [CrossRef]
- Eaton-Fitch, N.; du Preez, S.; Cabanas, H.; Staines, D.; Marshall-Gradisnik, S. A systematic review of natural killer cells profile and cytotoxic function in myalgic encephalomyelitis/chronic fatigue syndrome. Syst. Rev. 2019, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Nunès, J.A.; Vély, F. Natural Killer Cell Signaling Pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.; Yagita, H.; Takeda, K.; Van Dommelen, S.L.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef]
- Sim, G.C.; Radvanyi, L.G. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.M.; Tsun, A.; Stinchcombe, J.C. The immunological synapse: A focal point for endocytosis and exocytosis. J. Cell Biol. 2010, 189, 399–406. [Google Scholar] [CrossRef]
- Krzewski, K.; Coligan, J.E. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 335. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, C.A.; Jankowska, K.I.; Burkhardt, J.K.; Lewis, R.S. Calcium influx through CRAC channels controls actin organization and dynamics at the immune synapse. eLife 2016, 5, e14850. [Google Scholar] [CrossRef] [PubMed]
- Peacock, M. Calcium Metabolism in Health and Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, S23–S30. [Google Scholar] [CrossRef]
- Piste, P.; Sayaji, D.; Avinash, M. Calcium and its Role in Human Body. Int. J. Res. Pharm. Biomed. Sci. 2012, 4, 2229–3701. [Google Scholar]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Nguyen, T.; Staines, D.; Nilius, B.; Smith, P.; Marshall-Gradisnik, S. Novel identification and characterisation of Transient receptor potential melastatin 3 ion channels on Natural Killer cells and B lymphocytes: Effects on cell signalling in Chronic fatigue syndrome/Myalgic encephalomyelitis patients. Biol. Res. 2016, 49, 27. [Google Scholar] [CrossRef]
- Nguyen, T.; Johnston, S.; Clarke, L.; Smith, P.; Staines, D.; Marshall-Gradisnik, S. Impaired calcium mobilization in natural killer cells from chronic fatigue syndrome/myalgic encephalomyelitis patients is associated with transient receptor potential melastatin 3 ion channels. Clin. Exp. Immunol. 2017, 187, 284–293. [Google Scholar] [CrossRef]
- Cabanas, H.; Muraki, K.; Eaton, N.; Balinas, C.; Staines, D.; Marshall-Gradisnik, S. Loss of Transient Receptor Potential Melastatin 3 ion channel function in natural killer cells from Chronic Fatigue Syndrome/Myalgic Encephalomyelitis patients. Mol. Med. 2018, 24, 44. [Google Scholar] [CrossRef]
- Balinas, C.; Cabanas, H.; Staines, D.; Marshall-Gradisnik, S. Transient receptor potential melastatin 2 channels are overexpressed in myalgic encephalomyelitis/chronic fatigue syndrome patients. J. Transl. Med. 2019, 17, 401. [Google Scholar] [CrossRef]
- Cabanas, H.; Muraki, K.; Balinas, C.; Eaton-Fitch, N.; Staines, D.; Marshall-Gradisnik, S. Validation of impaired Transient Receptor Potential Melastatin 3 ion channel activity in natural killer cells from Chronic Fatigue Syndrome/Myalgic Encephalomyelitis patients. Mol. Med. 2019, 25, 14. [Google Scholar] [CrossRef] [PubMed]
- Cabanas, H.; Muraki, K.; Staines, D.; Marshall-Gradisnik, S. Naltrexone Restores Impaired Transient Receptor Potential Melastatin 3 Ion Channel Function in Natural Killer Cells From Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Front. Immunol. 2019, 10, 2545. [Google Scholar] [CrossRef]
- Eaton-Fitch, N.; Cabanas, H.; du Preez, S.; Staines, D.; Marshall-Gradisnik, S. The effect of IL-2 stimulation and treatment of TRPM3 on channel co-localisation with PIP2 and NK cell function in myalgic encephalomyelitis/chronic fatigue syndrome patients. J. Transl. Med. 2021, 19, 306. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef]
- Gees, M.; Owsianik, G.; Nilius, B.; Voets, T. TRP Channels. Compr. Physiol. Am. Cancer Soc. 2012, 2, 563–608. [Google Scholar] [CrossRef]
- Marshall-Gradisnik, S.M.; Smith, P.; Brenu, E.W.; Nilius, B.; Ramos, S.B.; Staines, D.R. Examination of Single Nucleotide Polymorphisms (SNPs) in Transient Receptor Potential (TRP) Ion Channels in Chronic Fatigue Syndrome Patients. Immunol. Immunogenet. Insights 2015, 7, III-S25147. [Google Scholar] [CrossRef]
- Du Preez, S.; Eaton-Fitch, N.; Cabanas, H.; Staines, D.; Marshall-Gradisnik, S. Characterization of IL-2 Stimulation and TRPM7 Pharmacomodulation in NK Cell Cytotoxicity and Channel Co-Localization with PIP2 in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Int. J. Environ. Res. Public. Health 2021, 18, 11879. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Kilch, T.; Horgen, F.D.; Fleig, A.; Penner, R. The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. 2017, 595, 3165–3180. [Google Scholar] [CrossRef]
- Chubanov, V.; Schäfer, S.; Ferioli, S.; Gudermann, T. Natural and Synthetic Modulators of the TRPM7 Channel. Cells 2014, 3, 1089–1101. [Google Scholar] [CrossRef]
- Hofmann, T.; Schäfer, S.; Linseisen, M.; Sytik, L.; Gudermann, T.; Chubanov, V. Activation of TRPM7 channels by small molecules under physiological conditions. Pflug. Arch. Eur. J. Physiol. 2014, 466, 2177–2189. [Google Scholar] [CrossRef]
- Won, J.; Vang, H.; Kim, J.H.; Lee, P.R.; Kang, Y.; Oh, S.B. TRPM7 Mediates Mechanosensitivity in Adult Rat Odontoblasts. J. Dent. Res. 2018, 97, 1039–1046. [Google Scholar] [CrossRef]
- Carvacho, I.; Ardestani, G.; Lee, H.C.; Mcgarvey, K.; Fissore, R.A.; Lykke-Hartmann, K. TRPM7-like channels are functionally expressed in oocytes and modulate post-fertilization embryo development in mouse. Sci. Rep. Nat. Publ. Group. Lond. 2016, 6, 34236. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, M.; Inoue, H.; Konishi, M. Effects of an Activator of TRPM7, Naltriben, on Magnesium Influx of Rat Ventricular Myocytes. Biophys. J. 2017, 112, 409a. [Google Scholar] [CrossRef]
- Schilling, T.; Miralles, F.; Eder, C. TRPM7 regulates proliferation and polarisation of macrophages. J. Cell Sci. 2014, 127, 4561–4566. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, S.; Ferioli, S.; Hofmann, T.; Zierler, S.; Gudermann, T.; Chubanov, V. Mibefradil represents a new class of benzimidazole TRPM7 channel agonists. Pflüg Arch. Eur. J. Physiol. 2016, 468, 623–634. [Google Scholar] [CrossRef]
- Zierler, S.; Sumoza-Toledo, A.; Suzuki, S.; Dúill, F.Ó.; Ryazanova, L.V.; Penner, R.; Ryazanov, A.G.; Fleig, A. TRPM7 kinase activity regulates murine mast cell degranulation: TRPM7 kinase in mast cell degranulation. J. Physiol. 2016, 594, 2957–2970. [Google Scholar] [CrossRef] [PubMed]
- Nadezhdin, K.D.; Correia, L.; Narangoda, C.; Patel, D.S.; Neuberger, A.; Gudermann, T.; Kurnikova, M.G.; Chubanov, V.; Sobolevsky, A.I. Structural mechanisms of TRPM7 activation and inhibition. Nat. Commun. 2023, 14, 2639. [Google Scholar] [CrossRef] [PubMed]
- Cabanas, H.; Harnois, T.; Magaud, C.; Cousin, L.; Constantin, B.; Bourmeyster, N.; Déliot, N. Deregulation of calcium homeostasis in Bcr-Abl-dependent chronic myeloid leukemia. Oncotarget 2018, 9, 26309. [Google Scholar] [CrossRef]
- Qin, F. Hill Coefficients of a Polymodal Monod-Wyman-Changeux Model for Ion Channel Gating. Biophys. J. 2010, 99, L29–L31. [Google Scholar] [CrossRef]
- Hong, C.; Choi, S.H.; Kwak, M.; Jeong, B.; Ko, J.; Park, H.J.; Choi, S.; Jun, J.Y.; So, I. TRPC5 channel instability induced by depalmitoylation protects striatal neurons against oxidative stress in Huntington’s disease. Biochim. Biophys. Acta (BBA) Mol. Cell. Res. 2020, 1867, 118620. [Google Scholar] [CrossRef]
- Du Preez, S.; Cabanas, H.; Staines, D.; Marshall-Gradisnik, S. Potential Implications of Mammalian Transient Receptor Potential Melastatin 7 in the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Review. Int. J. Environ. Res. Public Health 2021, 18, 10708. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, C.; Perraud, A.L.; Johnson, C.O.; Inabe, K.; Smith, M.K.; Penner, R.; Kurosaki, T.; Fleig, A. Regulation of Vertebrate Cellular Mg2+ Homeostasis by TRPM7. Cell 2003, 114, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Eaton-Fitch, N.; Du Preez, S.; Cabanas, H.; Muraki, K.; Staines, D.; Marshall-Gradisnik, S. Impaired TRPM3-dependent calcium influx and restoration using Naltrexone in natural killer cells of myalgic encephalomyelitis/chronic fatigue syndrome patients. J. Transl. Med. 2022, 20, 94. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rubil, S.; Lesch, A.; Guethlein, L.A.; Rössler, O.G. Transient receptor potential TRPM3 channels: Pharmacology, signaling, and biological functions. Pharmacol. Res. 2017, 124, 92–99. [Google Scholar] [CrossRef]
- Nicotera, P.; Orrenius, S. The role of calcium in apoptosis. Cell Calcium 1998, 23, 173–180. [Google Scholar] [CrossRef]
- Jahn, R.; Scheller, R.H. SNAREs—Engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef]
- Law, R.H.; Lukoyanova, N.; Voskoboinik, I.; Caradoc-Davies, T.T.; Baran, K.; Dunstone, M.A.; D’Angelo, M.E.; Orlova, E.V.; Coulibaly, F.; Verschoor, S.; et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 2010, 468, 447–451. [Google Scholar] [CrossRef]
- Broad, L.M.; Braun, F.-J.; Lievremont, J.-P.; Bird, G.S.J.; Kurosaki, T.; Putney, J.W. Role of the Phospholipase C-Inositol 1,4,5-Trisphosphate Pathway in Calcium Release-activated Calcium Current and Capacitative Calcium Entry. J. Biol. Chem. 2001, 276, 15945–15952. [Google Scholar] [CrossRef]
- Robert, V.; Triffaux, E.; Savignac, M.; Pelletier, L. Calcium signalling in T-lymphocytes. Biochimie 2011, 93, 2087–2094. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V. Calcium signalling and calcium channels: Evolution and general principles. Eur. J. Pharmacol. 2014, 739, 1–3. [Google Scholar] [CrossRef]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef]
- Trebak, M.; Kinet, J.-P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef]
- Kaschek, L.; Zöphel, S.; Knörck, A.; Hoth, M. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. Semin. Cell Dev. Biol. 2021, 596, 2681–2698. [Google Scholar] [CrossRef]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Chuderland, D.; Seger, R. Calcium regulates ERK signaling by modulating its protein-protein interactions. Commun. Integr. Biol. 2008, 1, 4–5. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Leibson, P.J. Signal Transduction During Natural Killer Cell Activation. Curr. Protoc. Immunol. 2000, 35, 11.9B.1–11.9B.13. [Google Scholar] [CrossRef]
- Watzl, C.; Long, E.O. Signal Transduction During Activation and Inhibition of Natural Killer Cells. Curr. Protoc. Immunol. 2010, 90, 11–19. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Davidson, D.; Pérez-Quintero, L.A.; Kurosaki, T.; Swat, W.; Veillette, A. The Adaptor SAP Controls NK Cell Activation by Regulating the Enzymes Vav-1 and SHIP-1 and by Enhancing Conjugates with Target Cells. Immunity 2012, 36, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Huth, T.K.; Staines, D.; Marshall-Gradisnik, S. ERK1/2, MEK1/2 and p38 downstream signalling molecules impaired in CD56dimCD16+ and CD56brightCD16dim/− natural killer cells in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis patients. J. Transl. Med. 2016, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-G.; Rios, F.J.; Montezano, A.C.; Touyz, R.M. TRPM7, Magnesium, and Signaling. Int. J. Mol. Sci. 2019, 20, 1877. [Google Scholar] [CrossRef]
- Staines, D.; Preez, S.D.; Cabanas, H.; Balinas, C.; Eaton, N.; Passmore, R.; Maksoud, R.; Redmayne, J.; Marshall-Gradisnik, S. Transient Receptor Potential Ion Channels in the Etiology and Pathomechanism of Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. Int. J. Clin. Med. 2018, 9, 445. [Google Scholar] [CrossRef]
- Sato-Kasai, M.; Kato, T.A.; Ohgidani, M.; Mizoguchi, Y.; Sagata, N.; Inamine, S.; Horikawa, H.; Hayakawa, K.; Shimokawa, N.; Kyuragi, S.; et al. Aripiprazole inhibits polyI:C-induced microglial activation possibly via TRPM7. Schizophr. Res. 2016, 178, 35–43. [Google Scholar] [CrossRef]
- Crosby, L.D.; Kalanidhi, S.; Bonilla, A.; Subramanian, A.; Ballon, J.S.; Bonilla, H. Off label use of Aripiprazole shows promise as a treatment for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A retrospective study of 101 patients treated with a low dose of Aripiprazole. J. Transl. Med. 2021, 19, 50. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Category | Item | HC | ME/CFS | p-Value |
|---|---|---|---|---|
| General demographics | Age (years) | 39.8 ± 4.0 | 40.3 ± 4.5 | 0.7263 |
| Gender | ||||
| Male (%, n) | 0, 0 | 0, 0 | ||
| Female (%, n) | 100, 9 | 100, 9 | ||
| BMI (kg/m2) | 22.0 ± 0.7 | 23.7 ± 1.1 | 0.3789 | |
| WHODAS | Understanding and communication | 8.3 ± 2.5 | 59.7 ± 8.4 | *** |
| Mobility | 2.8 ± 2.8 | 61.1 ± 7.3 | *** | |
| Self-care | 0.0 ± 0.0 | 36.1 ± 9.3 | ** | |
| Interpersonal relationships | 8.3 ± 3.6 | 47.2 ± 13.0 | * | |
| Life activities | 7.6 ± 3.7 | 73.6 ± 7.0 | *** | |
| Participation in work/school | 4.9 ± 2.5 | 73.4 ± 10.3 | *** | |
| Participation in society | 2.1 ± 0.7 | 64.2 ± 8.4 | *** | |
| Illness demographic (SF-36) | Pain (%) | 90.5 ± 3.8 | 50 ± 9.5 | ** |
| Physical functioning (%) | 98.9 ± 1.1 | 27.8 ± 7.7 | *** | |
| Role physical (%) | 98.6 ± 1.4 | 15.3 ± 5.2 | *** | |
| General health (%) | 72.9 ± 4.5 | 31.9 ± 5.2 | * | |
| Social functioning (%) | 94.4 ± 3.0 | 22.2 ± 8.0 | *** | |
| Role emotional (%) | 92.6 ± 3.2 | 56.5 ± 12.3 | * | |
| Wellbeing (%) | 71.1 ± 3.2 | 57.2 ± 7.9 | 0.1868 | |
| Vitality (%) | 66.7 ± 3.9 | 6.9 ± 3.2 | *** | |
| Full blood count | White blood cells | 5.9 ± 0.4 | 6.4 ± 0.5 | 0.5353 |
| Lymphocytes | 1.68 ± 0.12 | 1.92 ± 0.15 | 0.2005 | |
| Neutrophils | 3.59 ± 0.24 | 3.84 ± 0.41 | 0.5353 | |
| Monocytes | 0.46 ± 0.03 | 0.46 ± 0.03 | 0.8259 | |
| Eosinophils | 0.19 ± 0.04 | 0.14 ± 0.02 | 0.5687 | |
| Basophils | 0.03 ± 0.01 | 0.04 ± 0.01 | 0.5093 | |
| Platelets | 266 ± 13 | 250 ± 13 | 0.5961 | |
| Red blood cells | 4.34 ± 0.09 | 4.44 ± 0.10 | 0.7263 | |
| Haematocrit | 0.39 ± 0.01 | 0.40 ± 0.01 | 0.4009 | |
| Haemoglobin | 131 ± 3 | 135 ± 2 | 0.7949 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du Preez, S.; Eaton-Fitch, N.; Smith, P.K.; Marshall-Gradisnik, S. Altered TRPM7-Dependent Calcium Influx in Natural Killer Cells of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Biomolecules 2023, 13, 1039. https://doi.org/10.3390/biom13071039
Du Preez S, Eaton-Fitch N, Smith PK, Marshall-Gradisnik S. Altered TRPM7-Dependent Calcium Influx in Natural Killer Cells of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Biomolecules. 2023; 13(7):1039. https://doi.org/10.3390/biom13071039
Chicago/Turabian StyleDu Preez, Stanley, Natalie Eaton-Fitch, Peter K. Smith, and Sonya Marshall-Gradisnik. 2023. "Altered TRPM7-Dependent Calcium Influx in Natural Killer Cells of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients" Biomolecules 13, no. 7: 1039. https://doi.org/10.3390/biom13071039
APA StyleDu Preez, S., Eaton-Fitch, N., Smith, P. K., & Marshall-Gradisnik, S. (2023). Altered TRPM7-Dependent Calcium Influx in Natural Killer Cells of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Biomolecules, 13(7), 1039. https://doi.org/10.3390/biom13071039