



Conformational Analysis of Charged Homo-Polypeptides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
All-Atom Molecular Dynamics Simulations
3. Results
3.1. D/E Repeats Are More Common Than K/R Repeats
3.2. Dimensions of Polyelectrolytic Polypeptides
3.3. Conformational Ensemble of Polyelectrolytic Polypeptides
3.4. Flory Exponents and Relaxation Times
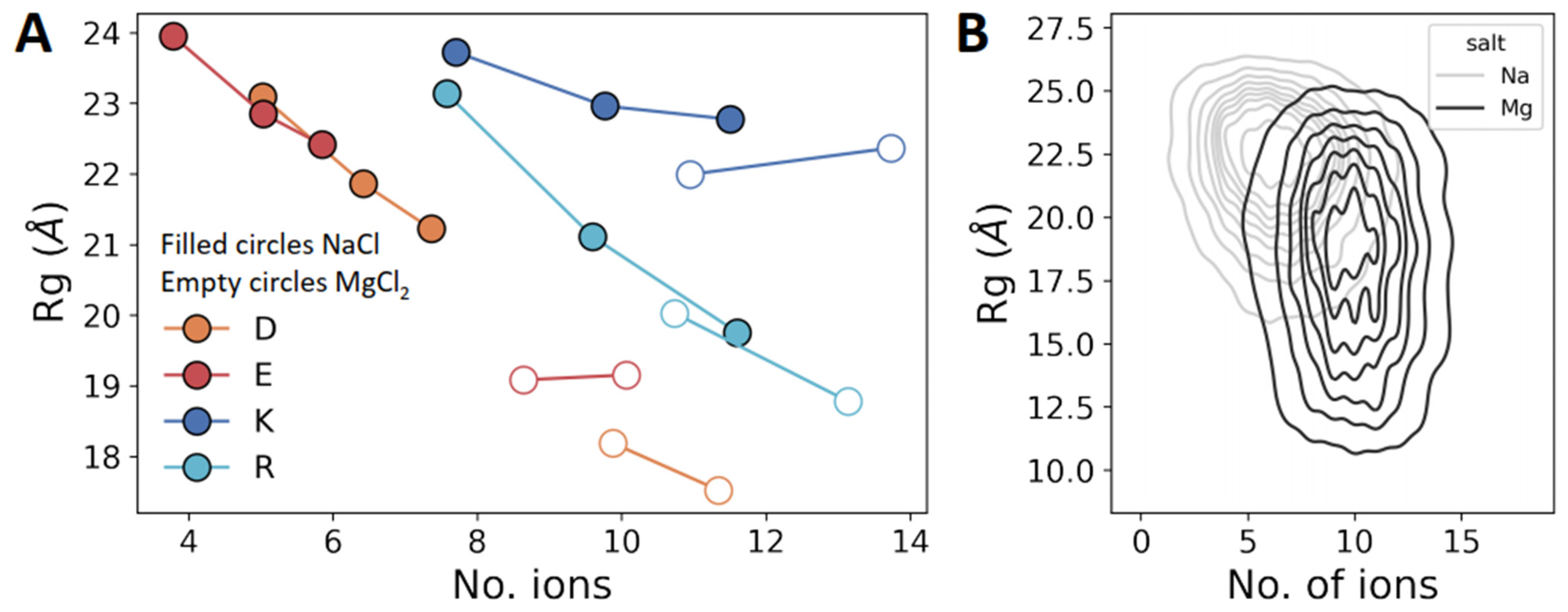
3.5. Sensitivity to Cation Valency Is Greater for D/E Repeats Than for K/R Repeats
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The most important thing is the tail: Multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013, 587, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Muller-Spath, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Ruegger, S.; Reymond, L.; Nettels, D.; Schuler, B. From the Cover: Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614. [Google Scholar] [CrossRef]
- Hofmann, H.; Soranno, A.; Borgia, A.; Gast, K.; Nettels, D.; Schuler, B. Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 16155–16160. [Google Scholar] [CrossRef]
- Bianchi, G.; Longhi, S.; Grandori, R.; Brocca, S. Relevance of Electrostatic Charges in Compactness, Aggregation, and Phase Separation of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2020, 21, 6208. [Google Scholar] [CrossRef]
- Vuzman, D.; Azia, A.; Levy, Y. Searching DNA via a “Monkey Bar” Mechanism: The Significance of Disordered Tails. J. Mol. Biol. 2010, 396, 674–684. [Google Scholar] [CrossRef]
- Vuzman, D.; Levy, Y. DNA search efficiency is modulated by charge composition and distribution in the intrinsically disordered tail. Proc. Natl. Acad. Sci. USA 2010, 107, 21004–21009. [Google Scholar] [CrossRef]
- Vuzman, D.; Levy, Y. Intrinsically disordered regions as affinity tuners in protein–DNA interactions. Mol. Biosyst. 2011, 8, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Bigman, L.S.; Greenblatt, H.M.; Levy, Y. What Are the Molecular Requirements for Protein Sliding along DNA? J. Phys. Chem. B 2021, 125, 3119–3131. [Google Scholar] [CrossRef] [PubMed]
- Hazra, M.K.; Levy, Y. Charge pattern affects the structure and dynamics of polyampholyte condensates. Phys. Chem. Chem. Phys. 2020, 22, 19368–19375. [Google Scholar] [CrossRef] [PubMed]
- Hazra, M.K.; Levy, Y. Biophysics of Phase Separation of Disordered Proteins Is Governed by Balance between Short- And Long-Range Interactions. J. Phys. Chem. B 2021, 125, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Hazra, M.K.; Levy, Y. Affinity of disordered protein complexes is modulated by entropy–energy reinforcement. Proc. Natl. Acad. Sci. USA 2022, 119, e2120456119. [Google Scholar] [CrossRef] [PubMed]
- Bigman, L.S.; Iwahara, J.; Levy, Y. Negatively Charged Disordered Regions are Prevalent and Functionally Important Across Proteomes. J. Mol. Biol. 2022, 434, 167660. [Google Scholar] [CrossRef]
- Xie, L.; Jakob, U. Inorganic polyphosphate, a multifunctional polyanionic protein scaffold. J. Biol. Chem. 2019, 294, 2180–2190. [Google Scholar] [CrossRef]
- Yoshida-Moriguchi, T.; Campbell, K.P. Matriglycan: A novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology 2015, 25, 702–713. [Google Scholar] [CrossRef]
- Hosaka, M.; Nagahama, M.; Kim, W.; Watanabe, T.; Hatsuzawa, K.; Ikemizu, J.; Murakami, K.; Nakayama, K. Arg-X-Lys/Arg-Arg motif as a signal for precursor cleavage catalyzed by furin within the constitutive secretory pathway. J. Biol. Chem. 1991, 266, 12127–12130. [Google Scholar] [CrossRef]
- Leininger, S.E.; Rodriguez, J.; Vu, Q.V.; Jiang, Y.; Li, M.S.; Deutsch, C.; O’Brien, E.P. Ribosome Elongation Kinetics of Consecutively Charged Residues Are Coupled to Electrostatic Force. Biochemistry 2021, 60, 3223–3235. [Google Scholar] [CrossRef]
- Lu, J.; Deutsch, C. Electrostatics in the Ribosomal Tunnel Modulate Chain Elongation Rates. J. Mol. Biol. 2008, 384, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Phillips, R. Cell Biology by the Numbers; Garland Science: New York City, NY, USA, 2015. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Vitalis, A.; Wang, X.; Pappu, R.V. Quantitative Characterization of Intrinsic Disorder in Polyglutamine: Insights from Analysis Based on Polymer Theories. Biophys. J. 2007, 93, 1923–1937. [Google Scholar] [CrossRef]
- Wang, X.; Greenblatt, H.M.; Bigman, L.S.; Yu, B.; Pletka, C.C.; Levy, Y.; Iwahara, J. Dynamic Autoinhibition of the HMGB1 Protein via Electrostatic Fuzzy Interactions of Intrinsically Disordered Regions. J. Mol. Biol. 2021, 433, 167122. [Google Scholar] [CrossRef]
- Wang, X.; Bigman, L.; Greenblatt, H.; Yu, B.; Levy, Y.; Iwahara, J. Negatively charged, intrinsically disordered regions can accelerate target search by DNA-binding proteins. Nucleic Acid Res. 2023, gkad045. [Google Scholar] [CrossRef]
- Fossat, M.J.; Zeng, X.; Pappu, R.V. Uncovering Differences in Hydration Free Energies and Structures for Model Compound Mimics of Charged Side Chains of Amino Acids. J. Phys. Chem. B 2021, 125, 4148–4161. [Google Scholar] [CrossRef]
- Lemke, T.; Edte, M.; Gebauer, D.; Peter, C. Three Reasons Why Aspartic Acid and Glutamic Acid Sequences Have a Surprisingly Different Influence on Mineralization. J. Phys. Chem. B 2021, 125, 10335–10343. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bigman, L.S.; Levy, Y. Conformational Analysis of Charged Homo-Polypeptides. Biomolecules 2023, 13, 363. https://doi.org/10.3390/biom13020363
Bigman LS, Levy Y. Conformational Analysis of Charged Homo-Polypeptides. Biomolecules. 2023; 13(2):363. https://doi.org/10.3390/biom13020363
Chicago/Turabian StyleBigman, Lavi S., and Yaakov Levy. 2023. "Conformational Analysis of Charged Homo-Polypeptides" Biomolecules 13, no. 2: 363. https://doi.org/10.3390/biom13020363
APA StyleBigman, L. S., & Levy, Y. (2023). Conformational Analysis of Charged Homo-Polypeptides. Biomolecules, 13(2), 363. https://doi.org/10.3390/biom13020363