
Intact Transition Epitope Mapping—Force Differences between Original and Unusual Residues (ITEM-FOUR)
 , and
, and
Abstract

1. Introduction
2. Materials and Methods
2.1. Preparation of Antibody, Peptide, and Immune Complex Solutions
2.2. Offline Nanoesi-MS Instrument Settings and Data Acquisition
2.3. Offline NanoESI-MS Data Analysis
2.4. ITEM-FOUR Analysis of Apparent Kinetic and Apparent Thermodynamic Values
2.5. Isothermal Titration Calorimetry in Solution
2.6. Atomistic Molecular Dynamics Simulations
3. Results
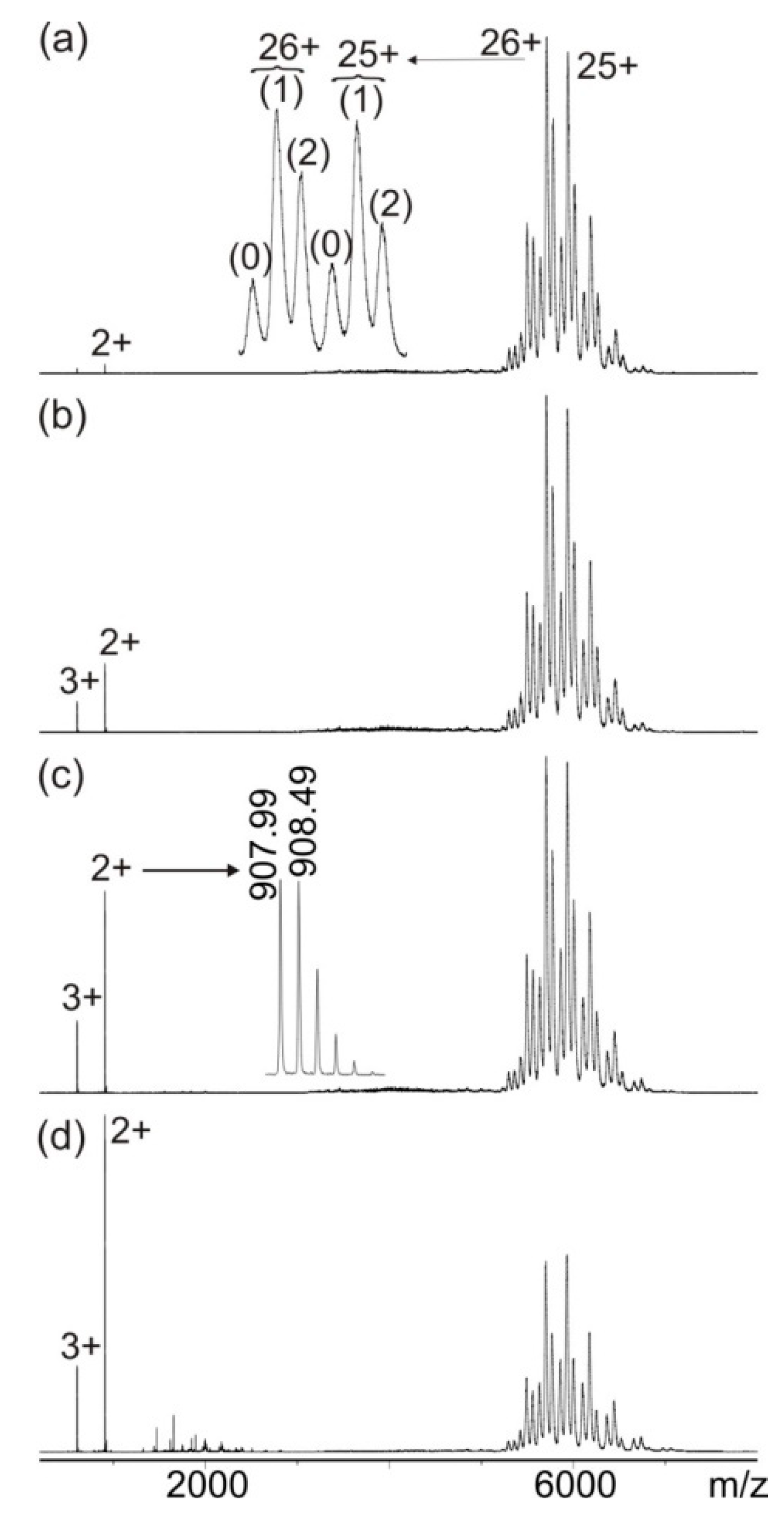
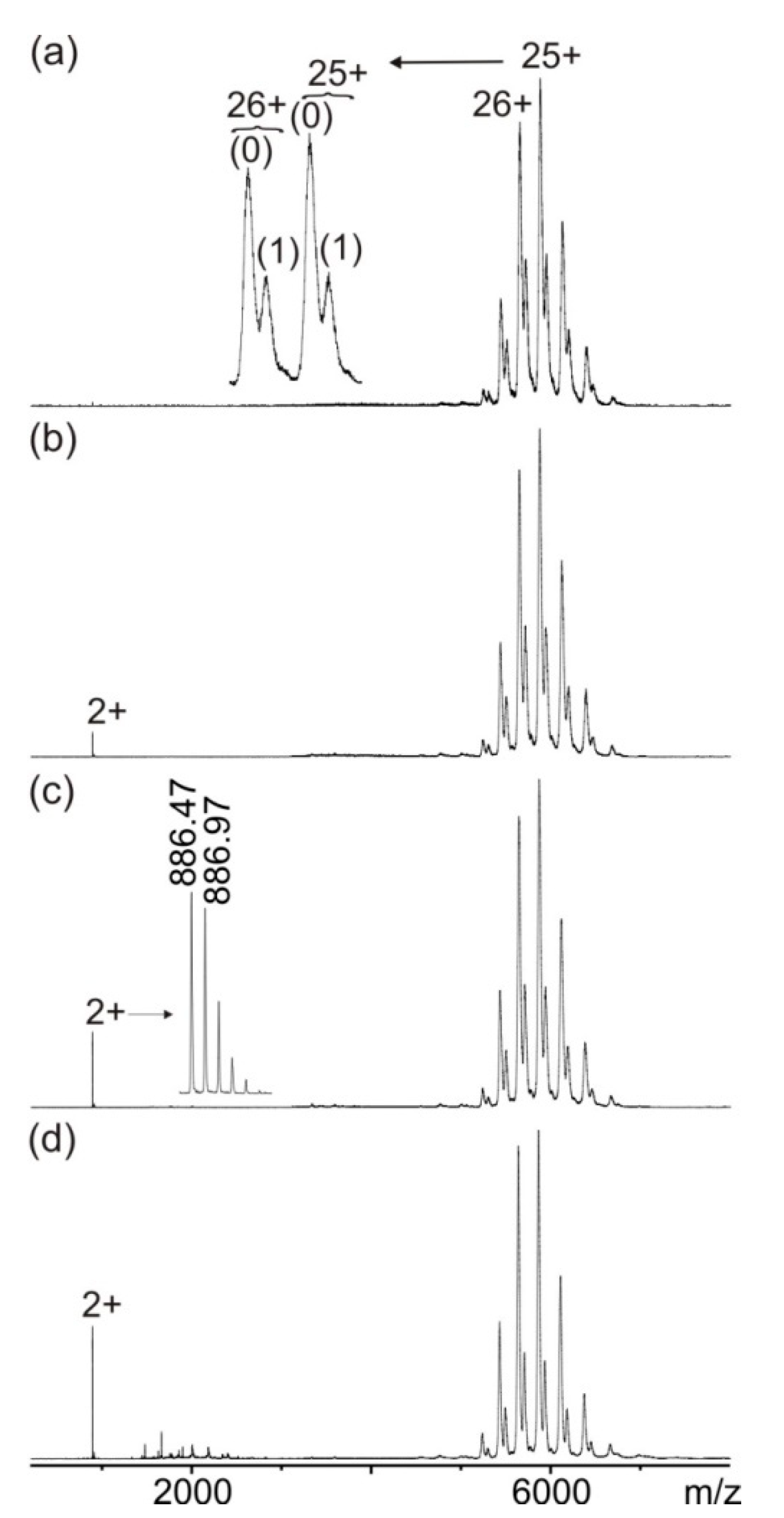
3.1. Mass Spectrometric Characterization of hcTn I Epitope Peptides and Anti-hcTn I Antibody

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide No. (wt or SAP) (a) | Amino Acid Sequence (wt or SAP) (a) | SNP Entry (b) | SNP (b) | Cardiomyopathy Association (b) | Calc. m/z (Charge) | Exp. m/z (Charge) |
|---|---|---|---|---|---|---|
| 1 (wt) | ENREVGDWRKNIDAL | n.a. | n.a. | n.a. | 605.64 (3+) | 605.63 (3+) |
| 2 (R186Q) | ENQEVGDWRKNIDAL | rs397516357 | G > A | hypertrophic | 596.30 (3+) | 596.30 (3+) |
| 3 (R192H) | ENREVGDWHKNIDAL | rs104894729 | G > A | hypertrophic/restrictive | 599.30 (3+) | 599.31 (3+) |
| 4 (R192L) | ENREVGDWLKNIDAL | rs104894729 | G > T | hypertrophic | 591.30 (3+) | 591.32 (3+) |
| 5 (R192C) | ENREVGDWCKNIDAL | rs727503499 | C > T | restrictive/hypertrophic | 587.95 (3+) | 587.96 (3+) |
| 6 (D190G) | ENREVGGWRKNIDAL | rs104894728 | A > G | familial hypertrophic | 586.31 (3+) | 586.32 (3+) |
| 7 (R192P) | ENREVGDWPKNIDAL | rs104894729 | G > C | hypertrophic | 585.96 (3+) | 585.98 (3+) |
| 8(R192P) 8 (K193E) | ENREVGDWPENIDAL | rs104894729 rs730881080 | G > C A > G | hypertrophic hypertrophic | 878.91 (2+) | 878.94 (2+) |
3.2. Mass Spectrometric Analysis of hcTn I Epitope Peptide Binding to the Anti-hcTn I Antibody
| Peptide No. | Amino Acid Sequence (a) | Initial [%] (b,c) | Final [%] (b,d) | ∆CV50 [V] (e) | dx [V] (e) | Slope [%/V] (e) | R2 (e) |
|---|---|---|---|---|---|---|---|
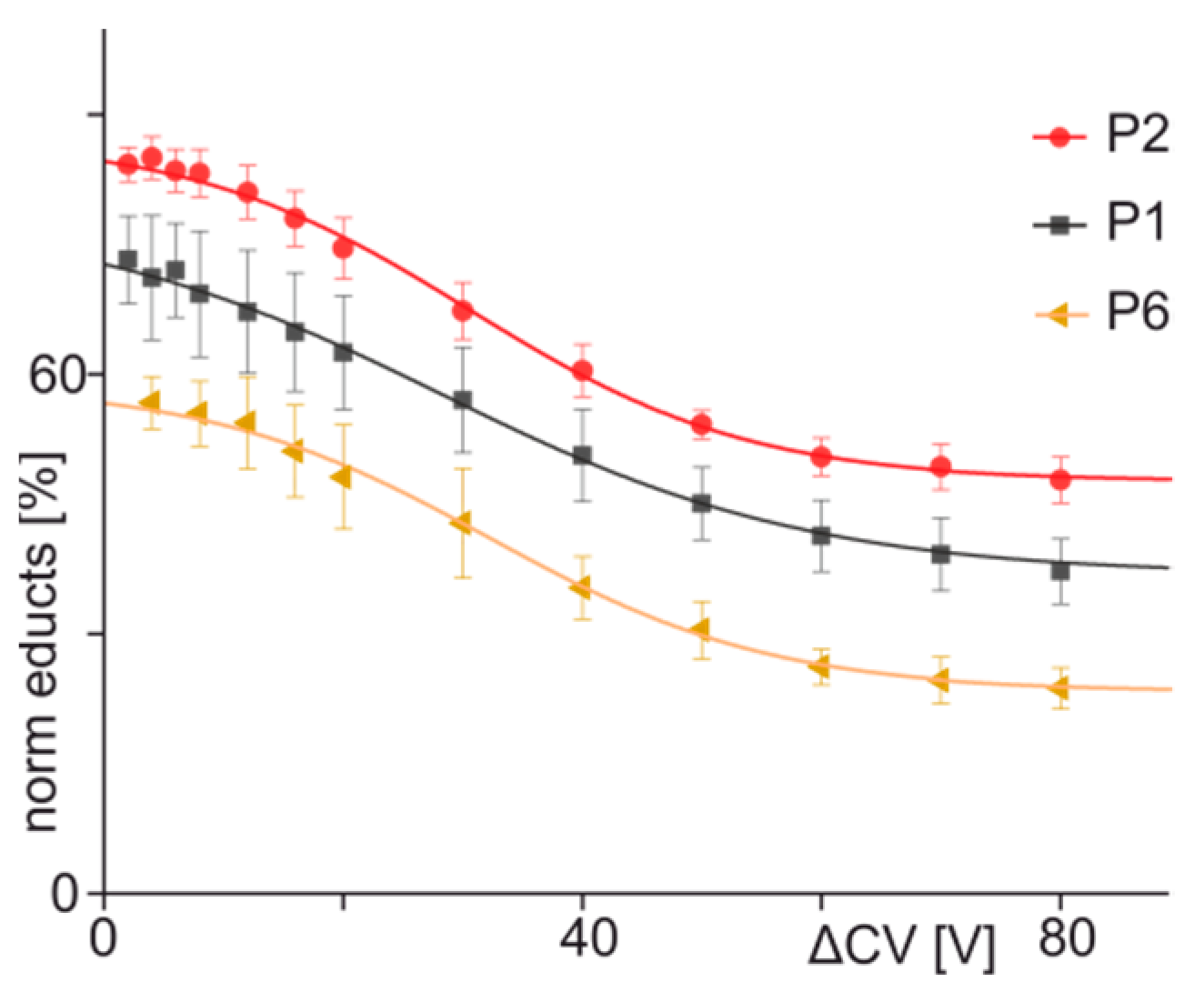
| 1 | ENREVGDWRKNIDAL | 77.36 | 36.24 | 30.0 | 15.2 | −0.67 | 0.997 |
| 2 | ENQEVGDWRKNIDAL | 87.39 | 47.63 | 30.3 | 11.7 | −0.85 | 0.998 |
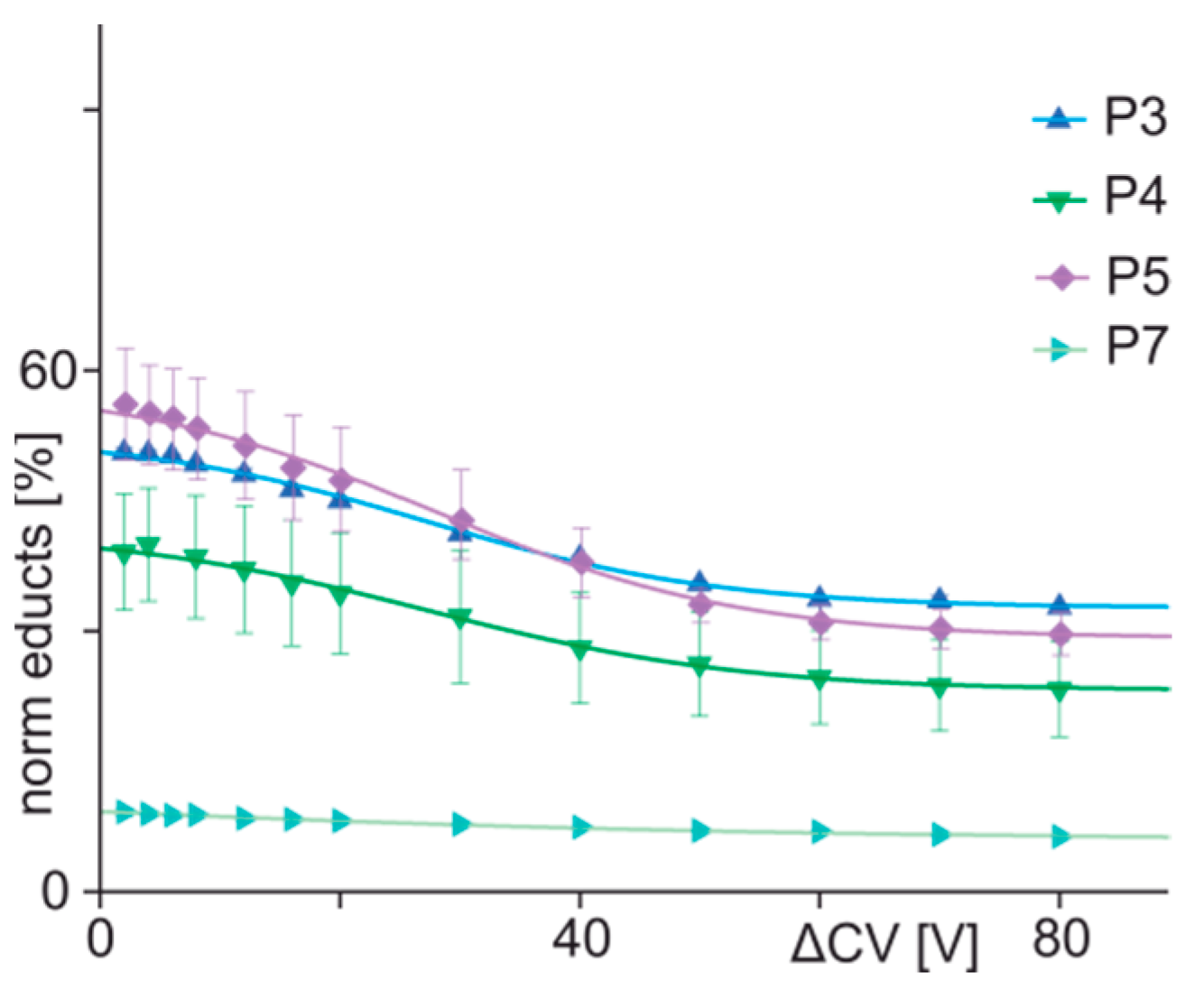
| 3 | ENREVGDWHKNIDAL | 52.55 | 32.68 | 27.1 | 12.3 | −0.40 | 0.997 |
| 4 | ENREVGDWLKNIDAL | 41.60 | 23.12 | 27.2 | 13.2 | −0.35 | 0.996 |
| 5 | ENREVGDWCKNIDAL | 58.77 | 29.41 | 27.7 | 13.0 | −0.57 | 0.996 |
| 6 | ENREVGGWRKNIDAL | 58.83 | 23.33 | 32.1 | 11.8 | −0.75 | 0.997 |
| 7 | ENREVGDWPKNIDAL | 15.37 | 5.90 | n.a. | n.a. | n.a. | n.a. |
| 8 | ENREVGDWPENIDAL | 0.00 | 0.00 | n.a. | n.a. | n.a. | n.a. |
3.3. ITEM-FOUR Analysis of Immune Complex Dissociation in the Gas Phase
3.4. Isothermal Titration Calorimetry of Immune Complex Formation in Solution
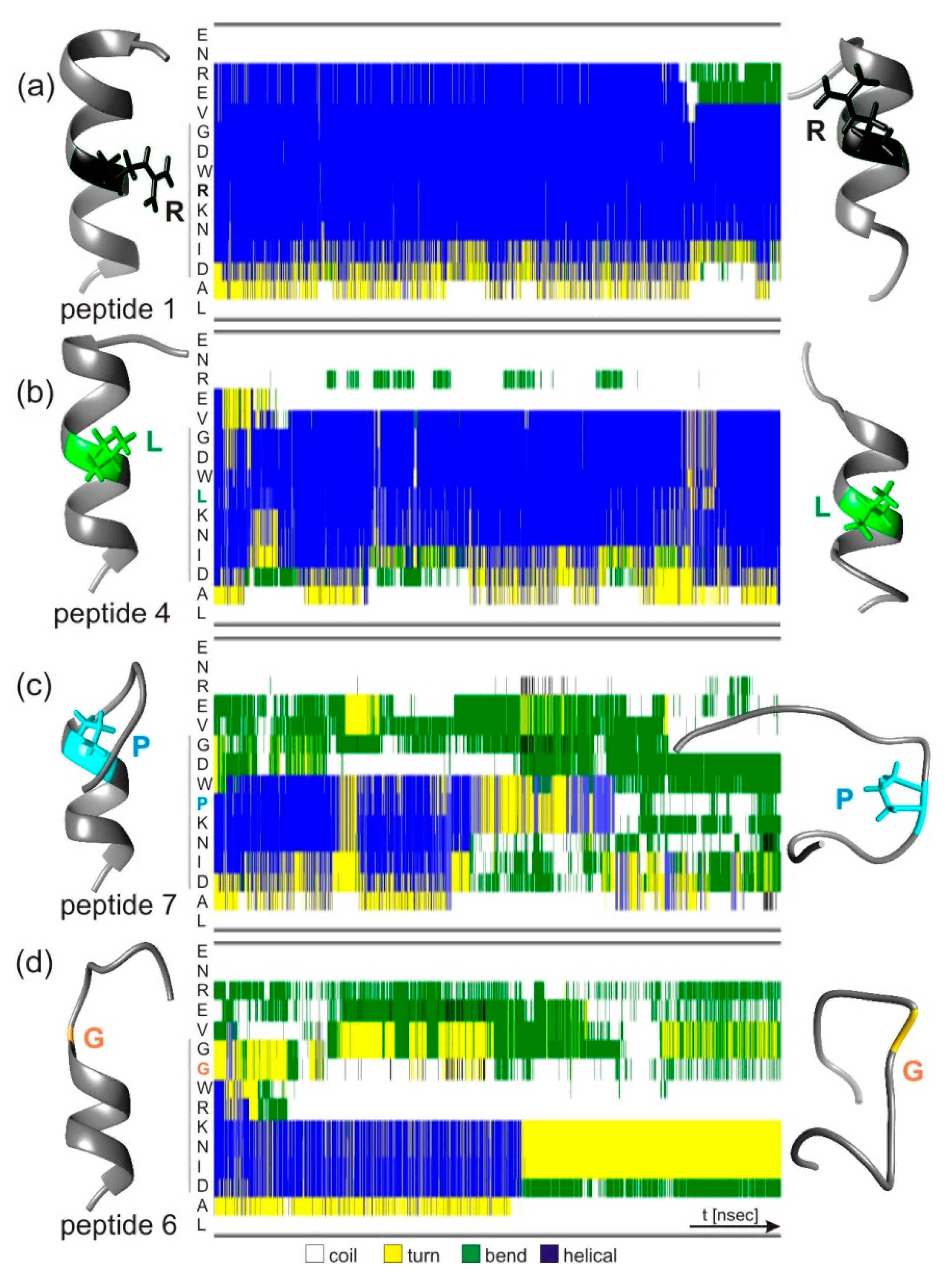
3.5. Atomistic Molecular Dynamics Simulations of hcTn I Epitope Peptide Structure Changes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Badrick, T. Evidence-based laboratory medicine. Clin. Biochem. Rev. 2013, 34, 43–46. [Google Scholar] [PubMed]
- Vogenberg, F.R.; Isaacson Barash, C.; Pursel, M. Personalized medicine: Part 1: Evolution and development into theranostics. Pharm. Ther. 2010, 35, 560–576. [Google Scholar]
- Reichlin, T.; Hochholzer, W.; Bassetti, S.; Steuer, S.; Stelzig, C.; Hartwiger, S.; Biedert, S.; Schaub, N.; Buerge, C.; Potocki, M.; et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N. Engl. J. Med. 2009, 361, 858–867. [Google Scholar] [CrossRef]
- Keller, T.; Zeller, T.; Peetz, D.; Tzikas, S.; Roth, A.; Czyz, E.; Bickel, C.; Baldus, S.; Warnholtz, A.; Frohlich, M.; et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N. Engl. J. Med. 2009, 361, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Cassedy, A.; O’Kennedy, R. The role of antibody-based troponin detection in cardiovascular disease: A critical assessment. J. Immunol. Methods 2021, 497, 113108. [Google Scholar] [CrossRef] [PubMed]
- Hyytia, H.; Heikkila, T.; Hedberg, P.; Puolakanaho, T.; Pettersson, K. Skeletal troponin I cross-reactivity in different cardiac troponin I assay versions. Clin. Biochem. 2015, 48, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Apple, F.S.; Collinson, P.O. Analytical characteristics of high-sensitivity cardiac troponin assays. Clin. Chem. 2012, 58, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bunk, D.M.; Dalluge, J.J.; Welch, M.J. Heterogeneity in Human Cardiac Troponin I Standards. Anal. Biochem. 2000, 284, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Daubert, M.A.; Jeremias, A. The utility of troponin measurement to detect myocardial infarction: Review of the current findings. Vasc. Health Risk Manag. 2010, 6, 691–699. [Google Scholar]
- Salisbury, B.A.; Pungliya, M.; Choi, J.Y.; Jiang, R.; Sun, X.J.; Stephens, J.C. SNP and haplotype variation in the human genome. Mutat. Res. 2003, 526, 53–61. [Google Scholar] [CrossRef]
- Lippi, G.; Cervellin, G. Genetic polymorphisms of human cardiac troponins as an unrecognized challenge for diagnosing myocardial injury. Int. J. Cardiol. 2014, 171, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Katrukha, I.A. Human cardiac troponin complex. Structure and functions. Biochemistry 2013, 78, 1447–1465. [Google Scholar] [CrossRef] [PubMed]
- Yefremova, Y.; Melder, F.T.; Danquah, B.D.; Opuni, K.F.M.; Koy, C.; Ehrens, A.; Frommholz, D.; Illges, H.; Koelbel, K.; Sobott, F.; et al. Apparent activation energies of protein–protein complex dissociation in the gas–phase determined by electrospray mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Danquah, B.D.; Yefremova, Y.; Opuni, K.F.M.; Rower, C.; Koy, C.; Glocker, M.O. Intact Transition Epitope Mapping—Thermodynamic Weak-force Order (ITEM—TWO). J. Proteomics 2020, 212, 103572. [Google Scholar] [CrossRef] [PubMed]
- Yefremova, Y.; Opuni, K.F.M.; Danquah, B.D.; Thiesen, H.J.; Glocker, M.O. Intact Transition Epitope Mapping (ITEM). J. Am. Soc. Mass Spectrom. 2017, 28, 1612–1622. [Google Scholar] [CrossRef] [PubMed]
- Danquah, B.D.; Opuni, K.F.M.; Roewer, C.; Koy, C.; Glocker, M.O. Mass Spectrometric Analysis of Antibody—Epitope Peptide Complex Dissociation: Theoretical Concept and Practical Procedure of Binding Strength Characterization. Molecules 2020, 25, 4776. [Google Scholar] [CrossRef]
- Danquah, B.D.; Rower, C.; Opuni, K.M.; El-Kased, R.; Frommholz, D.; Illges, H.; Koy, C.; Glocker, M.O. Intact Transition Epitope Mapping—Targeted High-Energy Rupture of Extracted Epitopes (ITEM-THREE). Mol. Cell Proteomics 2019, 18, 1543–1555. [Google Scholar] [CrossRef]
- Opuni, K.F.M.; Koy, C.; Russ, M.; Reepmeyer, M.; Danquah, B.D.; Weresow, M.; Alef, A.; Lorenz, P.; Thiesen, H.J.; Glocker, M.O. ITEM-THREE analysis of a monoclonal anti-malaria antibody reveals its assembled epitope on the pfMSP119 antigen. J. Biol. Chem. 2020, 295, 14987–14997. [Google Scholar] [CrossRef]
- Scherf, M.; Danquah, B.D.; Koy, C.; Lorenz, P.; Steinbeck, F.; Neamtu, A.; Thiesen, H.J.; Glocker, M.O. Epitope Fine Mapping by Mass Spectrometry: Investigations of Immune Complexes Consisting of Monoclonal Anti-HpTGEKP Antibody and Zinc Finger Protein Linker Phospho-Hexapeptides. ChemBioChem 2022, 23, e202200390. [Google Scholar] [CrossRef]
- Opuni, K.F.M.; Al-Majdoub, M.; Yefremova, Y.; El-Kased, R.F.; Koy, C.; Glocker, M.O. Mass spectrometric epitope mapping. Mass Spectrom. Rev. 2018, 37, 229–241. [Google Scholar] [CrossRef]
- Christenson, R.H.; Jacobs, E.; Uettwiller-Geiger, D.; Estey, M.P.; Lewandrowski, K.; Koshy, T.I.; Kupfer, K.; Li, Y.; Wesenberg, J.C. Comparison of 13 Commercially Available Cardiac Troponin Assays in a Multicenter North American Study. J. Appl. Lab. Med. 2017, 2, 134. [Google Scholar] [CrossRef]
- Jarolim, P. High sensitivity cardiac troponin assays in the clinical laboratories. Clin. Chem. Lab. Med. 2015, 53, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhu, J.; Liu, W.; Cao, F. High-sensitivity cardiac troponins I sandwich assay by immunomagnetic microparticle and quantum dots. Front. Lab. Med. 2017, 1, 107–113. [Google Scholar] [CrossRef]
- Savukoski, T.; Twarda, A.; Hellberg, S.; Ristiniemi, N.; Wittfooth, S.; Sinisalo, J.; Pettersson, K. Epitope specificity and IgG subclass distribution of autoantibodies to cardiac troponin. Clin. Chem. 2013, 59, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Wen, H.; Edwards, T.; Metzger, J.M. Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ. Res. 2007, 100, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Liu, J.; Feng, H.Z.; Hossain, M.M.; Gobara, N.; Zhang, C.; Li, Y.; Jean-Charles, P.Y.; Jin, J.P.; Huang, X.P. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2604–H2613. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhang, C.; Liu, J.; Sidky, C.; Huang, X.P. A point mutation (R192H) in the C-terminus of human cardiac troponin I causes diastolic dysfunction in transgenic mice. Arch. Biochem. Biophys. 2006, 456, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.V.; Liang, J.; Potter, J.D. Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J. Biol. Chem. 2005, 280, 30909–30915. [Google Scholar] [CrossRef]
- Lakdawala, N.K.; Funke, B.H.; Baxter, S.; Cirino, A.L.; Roberts, A.E.; Judge, D.P.; Johnson, N.; Mendelsohn, N.J.; Morel, C.; Care, M.; et al. Genetic testing for dilated cardiomyopathy in clinical practice. J. Card. Fail. 2012, 18, 296–303. [Google Scholar] [CrossRef]
- Millat, G.; Chanavat, V.; Crehalet, H.; Rousson, R. Development of a high resolution melting method for the detection of genetic variations in hypertrophic cardiomyopathy. Clin. Chim. Acta 2010, 411, 1983–1991. [Google Scholar] [CrossRef]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.V. Troponin I: Inhibitor or facilitator. Mol. Cell Biochem. 1999, 190, 9–32. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Hewapathirana, S.; Garcia-Seisdedos, D.; Kamatchinathan, S.; Kundu, D.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucl. Acids Res. 2022, 50, 543–552. [Google Scholar] [CrossRef]
- Koy, C.; Opuni, K.F.M.; Danquah, B.D.; Neamtu, A.; Glocker, M.O. Mass Spectrometric and Bio-Computational Binding Strength Analysis of Multiply Charged RNAse S Gas-Phase Complexes Obtained by Electrospray Ionization from Varying In-Solution Equilibrium Conditions. Int. J. Mol. Sci. 2021, 22, 10183. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Linnebacher, M.; Lorenz, P.; Koy, C.; Jahnke, A.; Born, N.; Steinbeck, F.; Wollbold, J.; Latzkow, T.; Thiesen, H.J.; Glocker, M.O. Clonality characterization of natural epitope-specific antibodies against the tumor-related antigen topoisomerase IIa by peptide chip and proteome analysis: A pilot study with colorectal carcinoma patient samples. Anal. Bioanal. Chem. 2012, 403, 227–238. [Google Scholar] [CrossRef]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef]
- Freire, E. Do Enthalpy and Entropy Distinguish First in Class from Best in Class? Drug Discov. Today 2008, 13, 19–20. [Google Scholar] [CrossRef]
- Imai, K.; Mitaku, S. Mechanisms of secondary structure breakers in soluble proteins. Biophysics 2005, 1, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Breuker, K.; McLafferty, F.W. Stepwise evolution of protein native structure with electrospray into the gas phase, 10(–12) to 10(2) s. Proc. Natl. Acad. Sci. USA 2008, 105, 18145–18152. [Google Scholar] [CrossRef] [PubMed]
- Yefremova, Y.; Danquah, B.D.; Opuni, K.F.; El-Kased, R.; Koy, C.; Glocker, M.O. Mass spectrometric characterization of protein structures and protein complexes in condensed and gas phase. Eur. J. Mass Spectrom. 2017, 23, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog. Mol. Biol. Transl. Sci. 2019, 166, 1–17. [Google Scholar] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Takeda, S.; Yamashita, A.; Maeda, K.; Maéda, Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 2003, 424, 35–41. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Geng, H.; Chen, F.; Ye, J.; Jiang, F. Applications of Molecular Dynamics Simulation in Structure Prediction of Peptides and Proteins. Comput. Struct. Biotechnol. J. 2019, 17, 1162–1170. [Google Scholar] [CrossRef]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic. Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- de Beer, T.A.P.; Laskowski, R.A.; Parks, S.L.; Sipos, B.; Goldmann, N.; Thornton, J.M. Amino Acid Changes in Disease-Associated Variants Differ Radically from Variants Observed in the 1000 Genomes Project Dataset. Comput. Biol. 2013, 9, 1–15. [Google Scholar] [CrossRef]
- Glocker, M.O.; Opuni, K.F.M.; Thiesen, H.J. From Free Binding Energy Calculations of SARS-CoV-2-Receptor Interactions to Cellular Immune Responses. Medicina 2022, 58, 226. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Russell, R.B. Amino Acid Properties and Consequences of Substitutions. In Bioinformatics for Geneticists; Barnes, M.R., Gray, I.C., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2003; pp. 2289–2316. [Google Scholar]
- Amos, W. Even small SNP clusters are non-randomly distributed: Is this evidence of mutational non-independence? Proc. Biol. Sci. 2010, 277, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, S.; Yamauchi, K.; Kawanabe, A.; Yamamoto, A.; Nakaza, M.; Kubota, T.; Kato, S.; Sasaki, R.; Okamura, Y.; Takahashi, M.P. Functional analysis of a double-point mutation in the KCNJ2 gene identified in a family with Andersen-Tawil syndrome. J. Neurol. Sci. 2019, 407, 116521. [Google Scholar] [CrossRef] [PubMed]
- Arthur-Farraj, P.; Mirsky, R.; Parkinson, D.B.; Jessen, K.R. A double point mutation in the DNA-binding region of Egr2 switches its function from inhibition to induction of proliferation: A potential contribution to the development of congenital hypomyelinating neuropathy. Neurobiol. Dis. 2006, 24, 159–169. [Google Scholar] [CrossRef]
- Chung, S.-H.; Kim, S.-K.; Kim, J.K.; Yang, Y.-R.; Suh, P.-G.; Chang, J.-S. A double point mutation in PCL-γ1 (Y509A/F510A) enhances Y783 phosphorylation and inositol phospholipid-hydrolyzing activity upon EGF stimulation. Exp. Mol. Med. 2010, 42, 216–222. [Google Scholar] [CrossRef]
- Scherf, M.; Koy, C.; Röwer, C.; Neamtu, A.; Glocker, M.O. Characterization of Phosphorylation-dependent Antibody Binding to Cancer-mutated Linkers of C2H2 Zinc Finger Proteins by Intact Transition Epitope Mapping—Thermodynamic Weak-Force Order Analysis. J. Am. Soc. Mass Spectrom. 2023; in press. [Google Scholar]
- Al-Majdoub, M.; Koy, C.; Lorenz, P.; Thiesen, H.J.; Glocker, M.O. Mass spectrometric and peptide chip characterization of an assembled epitope: Analysis of a polyclonal antibody model serum directed against the Sjogren/systemic lupus erythematosus autoantigen TRIM21. J. Mass Spectrom. 2013, 48, 651–659. [Google Scholar] [CrossRef]
- de Olazarra, A.S.; Cortade, D.L.; Wang, S.X. From saliva to SNP: Non-invasive, point-of-care genotyping for precision medicine applications using recombinase polymerase amplification and giant magnetoresistive nanosensors. Lab. Chip. 2022, 22, 2131–2144. [Google Scholar] [CrossRef]
- Xu, H.; Xia, A.; Wang, D.R.; Zhang, Y.; Deng, S.; Lu, W.; Luo, J.; Zhomg, Q.; Zhang, F.; Zhou, L.; et al. An ultraportable and versatile point-of-care DNA testing platform. Sci. Adv. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Dhawan, A.P. Collaborative Paradigm of Preventive, Personalized, and Precision Medicine with Point-of-Care Technologies. IEEE J. Transl. Eng. Health Med. 2016, 4, 2800908. [Google Scholar] [CrossRef]
- Glocker, M.O.; Guthke, R.; Kekow, J.; Thiesen, H.J. Rheumatoid arthritis, a complex multifactorial disease: On the way toward individualized medicine. Med. Res. Rev. 2006, 26, 63–87. [Google Scholar] [CrossRef] [PubMed]





| Peptide No. | Amino Acid Sequence (a) | [1/s] (b) | [Ø] (b,c) | [kJ/mol] (b) | [kJ/mol] (b) | [kJ/mol] (b,d) |
|---|---|---|---|---|---|---|
| 1 | ENREVGDWRKNIDAL | 1.87 × 1012 | 4.55 × 10−12 | 64.70 | −1.91 | −66.60 |
| 2 | ENQEVGDWRKNIDAL | 7.83 × 1011 | 4.41 × 10−12 | 64.78 | −1.57 | −66.33 |
| 3 | ENREVGDWHKNIDAL | 5.09 × 1012 | 4.71 × 10−12 | 64.62 | −2.21 | −66.82 |
| 4 | ENREVGDWLKNIDAL | 7.37 × 1012 | 4.77 × 10−12 | 64.59 | −2.25 | −66.82 |
| 5 | ENREVGDWCKNIDAL | 3.89 × 1012 | 4.67 × 10−12 | 64.64 | −2.04 | −66.67 |
| 6 | ENREVGGWRKNIDAL | 2.90 × 1012 | 4.62 × 10−12 | 64.67 | −1.85 | −66.50 |
| 7 | ENREVGDWPKNIDAL | n.d. | n.d. | n.d. | n.d. | n.d. |
| 8 | ENREVGDWPENIDAL | n.d. | n.d. | n.d. | n.d. | n.d. |
| Peptide No. | Amino Acid Sequence (a) | [Ø] (b) | [Ø] (b) | ∆G [kJ/mol] | ∆H [kJ/mol] | T∆S [kJ/mol] (c) |
|---|---|---|---|---|---|---|
| 1 | ENREVGDWRKNIDAL | 2.19 × 10−7 | 4.58 × 106 | −38.05 | −60.40 | −22.35 |
| 2 | ENQEVGDWRKNIDAL | 0.36 × 10−7 | 27.9 × 106 | −42.59 | −109.10 | −66.51 |
| 3 | ENREVGDWHKNIDAL | n.d. | n.d. | n.d. | n.d. | n.d. |
| 4 | ENREVGDWLKNIDAL | 2.43 × 10−7 | 4.12 × 106 | −37.77 | −52.10 | −14.33 |
| 5 | ENREVGDWCKNIDAL | n.d. | n.d. | n.d. | n.d. | n.d. |
| 6 | ENREVGGWRKNIDAL | 4.11 × 10−7 | 2.43 × 106 | −36.49 | −45.40 | −8.91 |
| 7 | ENREVGDWPKNIDAL | n.b. | n.b. | n.b. | n.b. | n.b. |
| 8 | ENREVGDWPENIDAL | n.b. | n.b. | n.b. | n.b. | n.b. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Röwer, C.; Ortmann, C.; Neamtu, A.; El-Kased, R.F.; Glocker, M.O. Intact Transition Epitope Mapping—Force Differences between Original and Unusual Residues (ITEM-FOUR). Biomolecules 2023, 13, 187. https://doi.org/10.3390/biom13010187
Röwer C, Ortmann C, Neamtu A, El-Kased RF, Glocker MO. Intact Transition Epitope Mapping—Force Differences between Original and Unusual Residues (ITEM-FOUR). Biomolecules. 2023; 13(1):187. https://doi.org/10.3390/biom13010187
Chicago/Turabian StyleRöwer, Claudia, Christian Ortmann, Andrei Neamtu, Reham F. El-Kased, and Michael O. Glocker. 2023. "Intact Transition Epitope Mapping—Force Differences between Original and Unusual Residues (ITEM-FOUR)" Biomolecules 13, no. 1: 187. https://doi.org/10.3390/biom13010187
APA StyleRöwer, C., Ortmann, C., Neamtu, A., El-Kased, R. F., & Glocker, M. O. (2023). Intact Transition Epitope Mapping—Force Differences between Original and Unusual Residues (ITEM-FOUR). Biomolecules, 13(1), 187. https://doi.org/10.3390/biom13010187