
Dimeric Benzodiazepines as Peptide Mimetics to Overcome p53-Dependent Drug Resistance of Tumors

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemistry
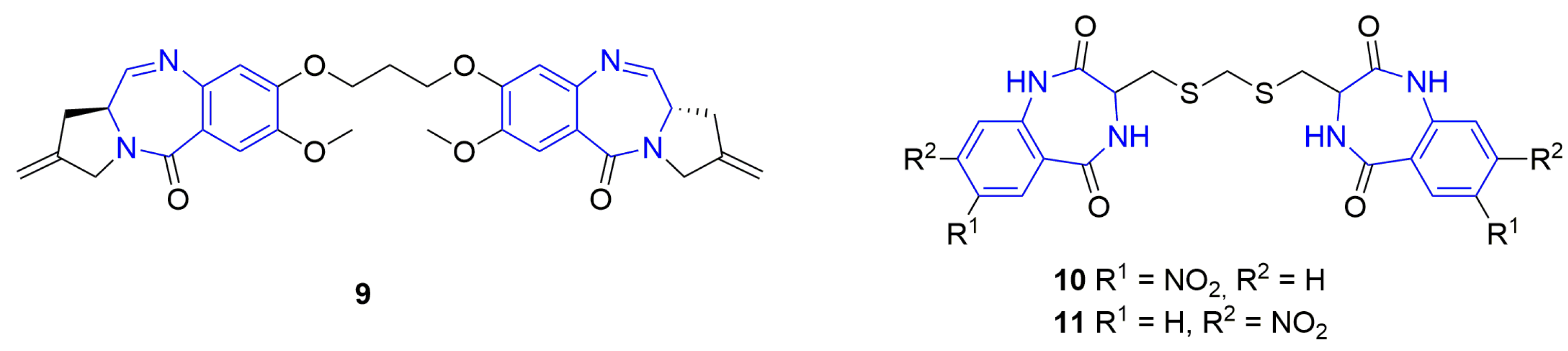
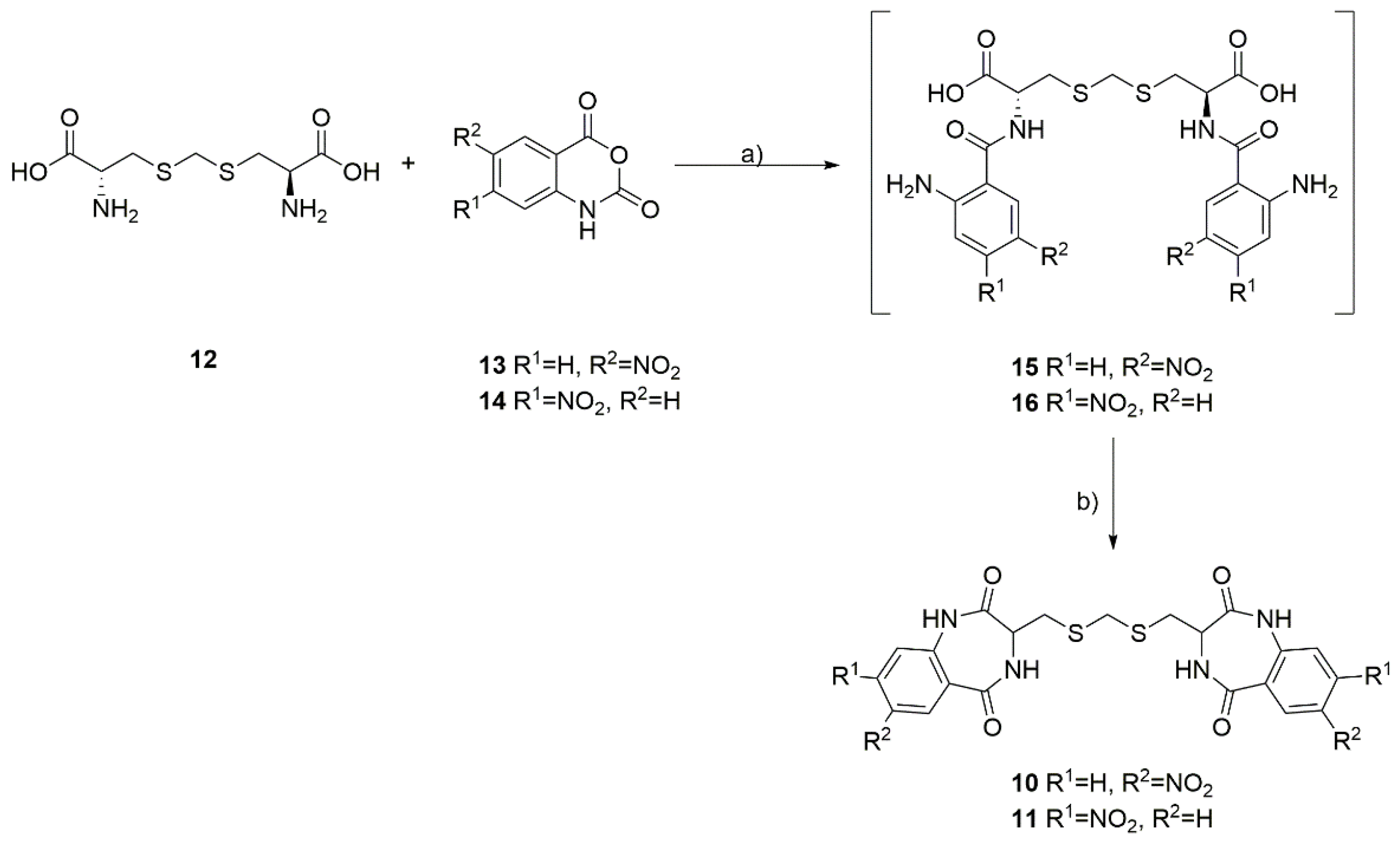
General Procedure for the Synthesis of Dimeric Benzodiazepines 10 and 11
2.2. Biology
2.2.1. Cell Lines and Culture Conditions
2.2.2. Cell Viability Assay
2.2.3. Cell Proliferation and Death Assay
2.2.4. Statistical Analysis
3. Results and Discussion
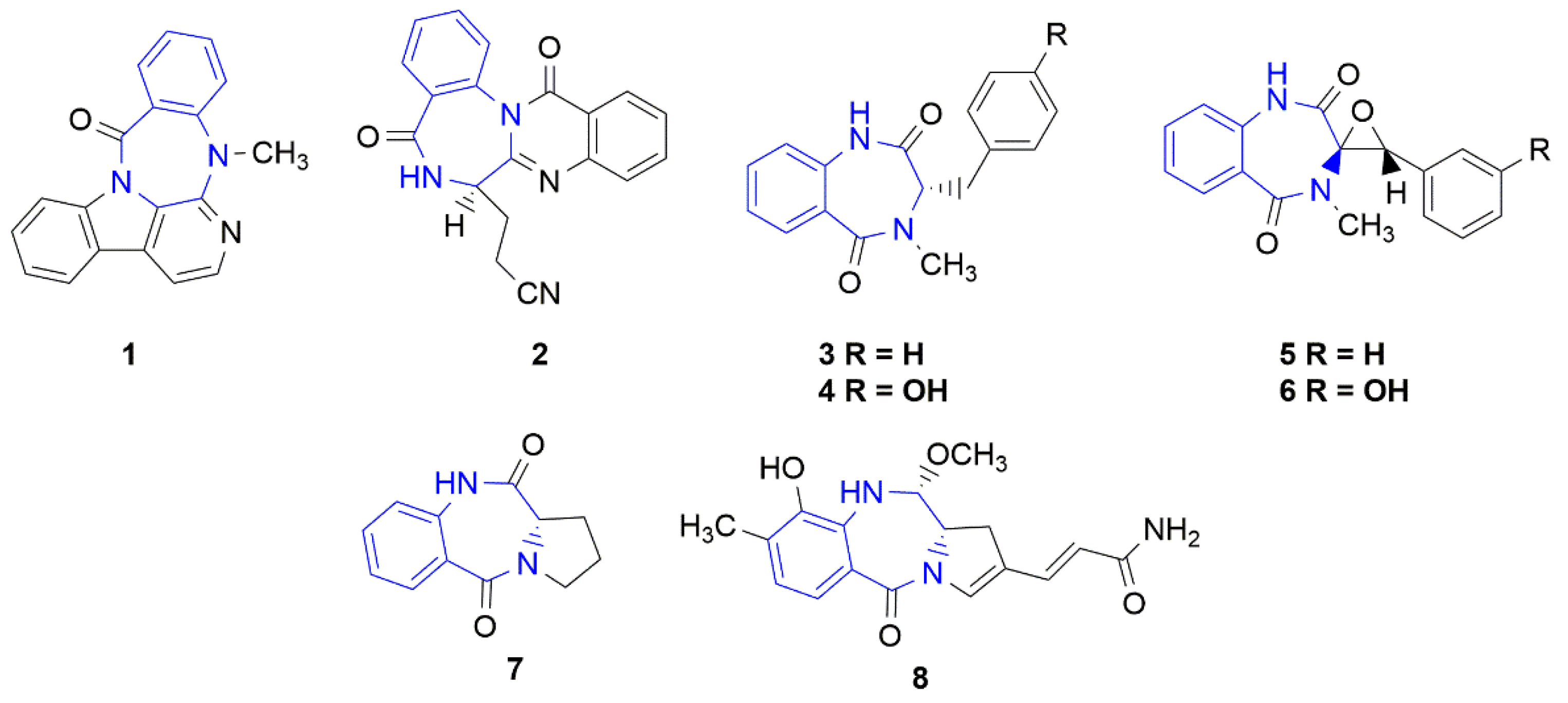
3.1. Chemistry
3.2. Biology
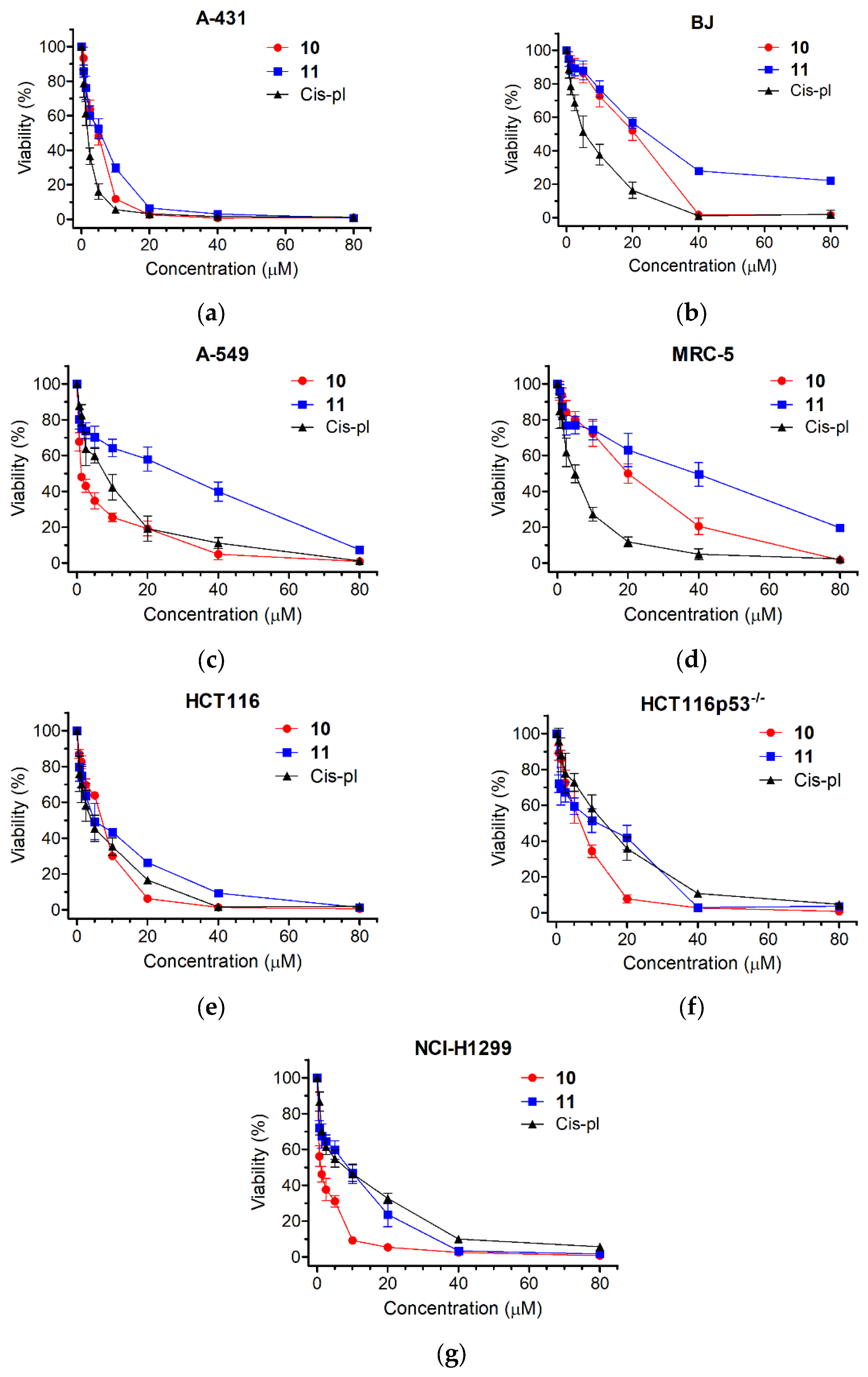
3.2.1. Cancer Cells Are Hypersensitive to 10 and 11
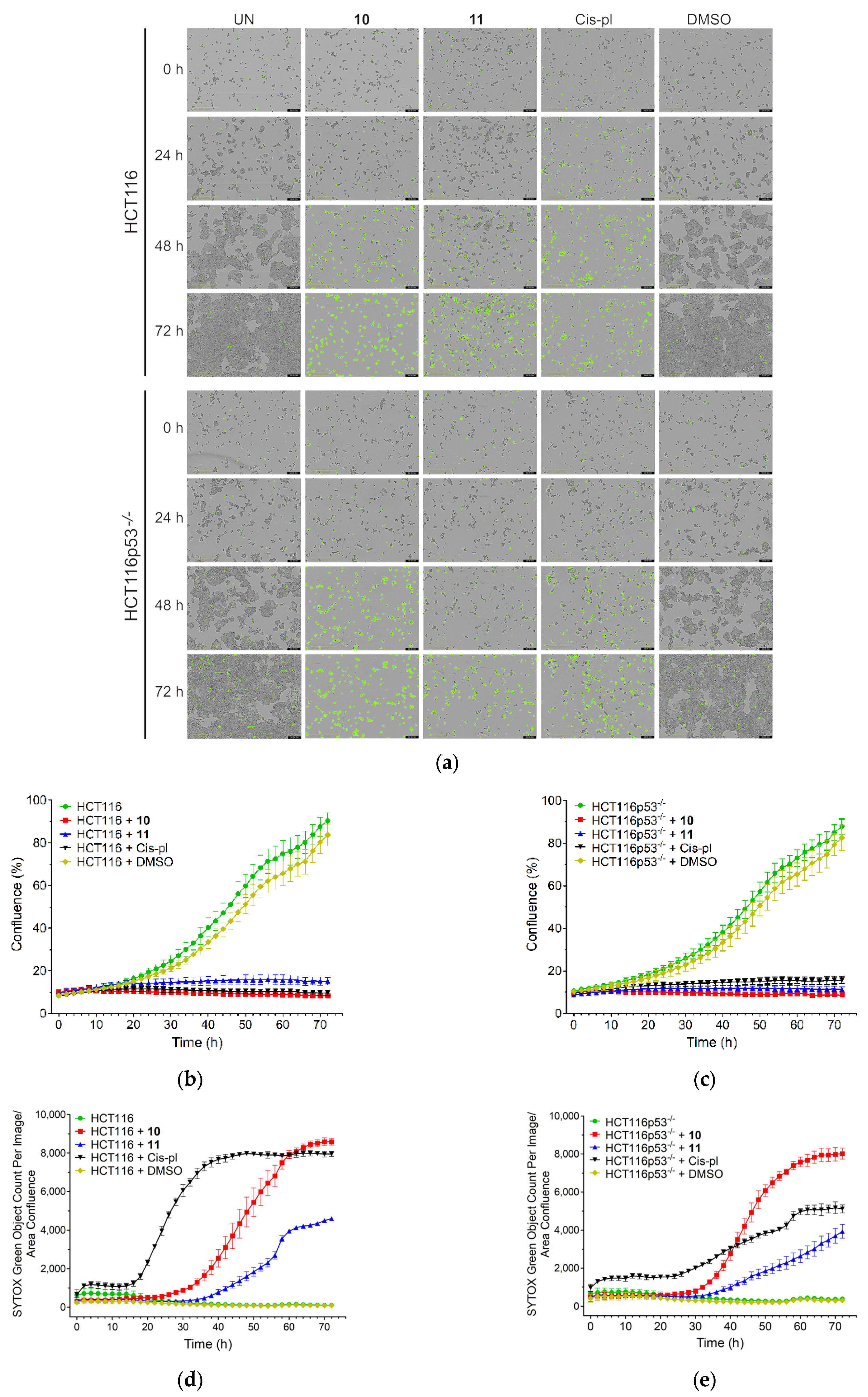
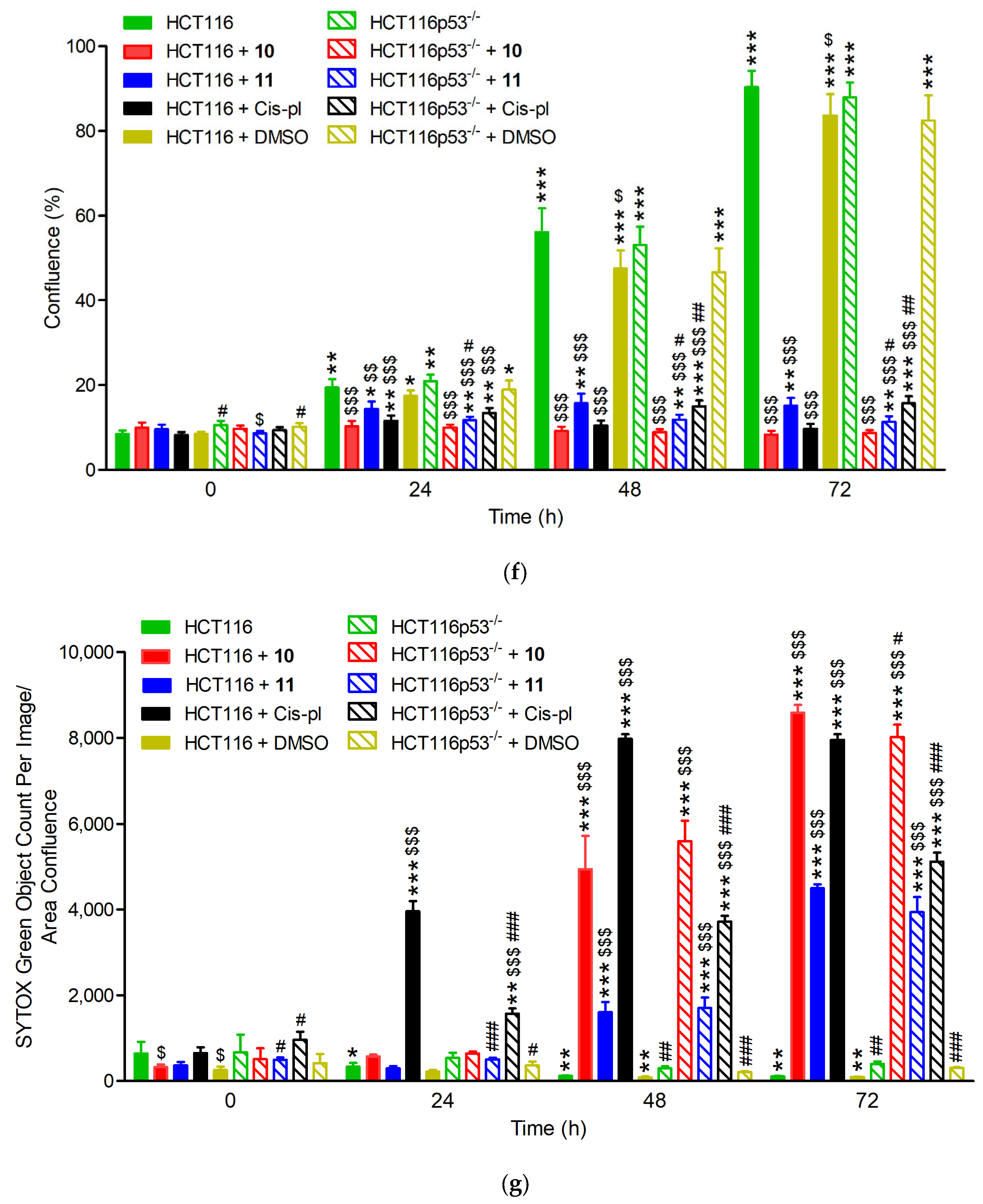
3.2.2. Treatment with 10 and 11 Inhibits Proliferation and Induces Massive Cell Death Independent of p53 Background
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellantuono, C.; Reggi, V.; Tognoni, G.; Garattini, S. Benzodiazepines: Clinical pharmacology and therapeutic use. Drugs 1980, 19, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.K.; Kaushik, S.O.; Chugh, J.; Bansal, S.; Shah, A.; Bariwal, J. Recent development in [1,4]benzodiazepines as potent anticancer agents: A review. Mini-Rev. Med. Chem. 2014, 14, 229–256. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs. 2011, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.; Rathnam, R.P.; Chowdhry, B.Z. 1,4-Benzodiazepin-2-ones in medicinal chemistry. Future Med. Chem. 2010, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, S. A Mini Review on Synthetic Approaches and Biological Activities of Benzodiazepines. Mini-Rev. Org. Chem. 2017, 14, 453–468. [Google Scholar] [CrossRef]
- Wang, Q.Z.; Liang, J.Y.; Feng, X. Evodiagenine and dievodiamine, two new indole alkaloids from Evodia rutaecarpa. Chin. Chem. Lett. 2010, 21, 596–599. [Google Scholar] [CrossRef]
- Wang, Q.Z.; Feng, X.; Wang, M.; Chen, Y.; Shan, Y.; Yin, M.; Guan, F.; Zhao, X.-Z.; Zhao, Y. Preparation Method and Activity of a New Indolequinazoline Alkaloid. China Patent CN103288827B, 11 September 2013. [Google Scholar]
- Li, J.; Zhong, Y.-s.; Yuan, J.; Zhu, X.; Lu, Y.-j.; Lin, Y.-c.; Liu, L. A New Terminal Cyano Group-containing Benzodiazepine Alkaloid from the Mangrove Endophytic Fungus Penicillium sp. Nat. Prod. Commun. 2015, 10, 1549–1551. [Google Scholar] [CrossRef]
- Toghueo, R.M.K.; Boyom, F.F. Endophytic Penicillium species and their agricultural, biotechnological, and pharmaceutical applications. 3 Biotech 2020, 10, 107. [Google Scholar] [CrossRef]
- Bracken, A.; Pocker, A.; Raistrick, H. Studies in the biochemistry of microorganisms. 93. Cyclopenin, a nitrogen-containing metabolic product of Penicillium cyclopium Westling. Biochem. J. 1954, 57, 587–595. [Google Scholar] [CrossRef]
- Birkinshaw, J.H.; Luckner, M.; Mohammed, Y.S.; Mothes, K.; Stickings, C.E. Studies in the Biochemistry of Micro-Organisms. 114. Viridicatol and Cyclopenol, Metabolites of Penicillium Viridicatum Westling and Penicillium Cyclopium Westling. Biochem. J. 1963, 89, 196–202. [Google Scholar] [CrossRef]
- Zhou, X.; Fang, P.; Tang, J.; Wu, Z.; Li, X.; Li, S.; Wang, Y.; Liu, G.; He, Z.; Gou, D.; et al. A novel cyclic dipeptide from deep marine-derived fungus Aspergillus sp. SCSIOW2. Nat. Prod. Res. 2016, 30, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Schwelle, N.; Lerbs, W.; Luckner, M. Enzymatic synthesis of cyclopeptine intermediates in Penicillium cyclopium. Phytochemistry 1985, 24, 1935–1939. [Google Scholar] [CrossRef]
- Sakaine, G.; Ture, A.; Pedroni, J.; Smits, G. Isolation, chemistry, and biology of pyrrolo[1,4]benzodiazepine natural products. Med. Res. Rev. 2022, 42, 5–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Sheng, W.; Sun, J.; Qin, G. Chemical constituents of Isatis indigotica. Planta Med. 1997, 63, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, S.; Küçüksolak, M.; Uze, A.; Bedir, E. Benzodiazepine Derivatives from Marine-Derived Streptomyces cacaoi 14CM034. Rec. Nat. Prod. 2021, 15, 602–607. [Google Scholar] [CrossRef]
- Nakatani, S.; Yamamoto, Y.; Hayashi, M.; Komiyama, K.; Ishibashi, M. Cycloanthranilylproline-derived constituents from a myxomycete Fuligo candida. Chem. Pharm. Bull. 2004, 52, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Grunberg, E.; Prince, H.N.; Titsworth, E.; Beskid, G.; Tendler, M.D. Chemotherapeutic properities of anthramycin. Chemotherapy 1966, 11, 249–260. [Google Scholar] [CrossRef]
- Antonow, D.; Thurston, D.E. Synthesis of DNA-interactive pyrrolo[2,1-c][1,4]benzodiazepines (PBDs). Chem. Rev. 2011, 111, 2815–2864. [Google Scholar] [CrossRef]
- Hartley, J.A.; Spanswick, V.J.; Brooks, N.; Clingen, P.H.; McHugh, P.J.; Hochhauser, D.; Pedley, R.B.; Kelland, L.R.; Alley, M.C.; Schultz, R.; et al. SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumor activity: Part 1: Cellular pharmacology, in vitro and initial in vivo antitumor activity. Cancer Res. 2004, 64, 6693–6699. [Google Scholar] [CrossRef]
- Alley, M.C.; Hollingshead, M.G.; Pacula-Cox, C.M.; Waud, W.R.; Hartley, J.A.; Howard, P.W.; Gregson, S.J.; Thurston, D.E.; Sausville, E.A. SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumor activity: Part 2: Efficacy evaluations. Cancer Res. 2004, 64, 6700–6706. [Google Scholar] [CrossRef]
- Mantaj, J.; Jackson, P.J.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew. Chem. Int. Ed. 2017, 56, 462–488. [Google Scholar] [CrossRef] [PubMed]
- Mieczkowski, A.; Trzybiński, D.; Wilczek, M.; Psurski, M.; Bagiński, M.; Bieszczad, B.; Mroczkowska, M.; Woźniak, K. (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—Synthesis and Crystallographic Studies. Molbank 2017, 2017, M964. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Psurski, M.; Bagiński, M.; Bieszczad, B.; Mroczkowska, M.; Wilczek, M.; Czajkowska, J.; Trzybiński, D.; Woźniak, K.; Wietrzyk, J. Novel (S)-1,3,4,12a-tetrahydropyrazino[2,1-c][1,4]benzodiazepine-6,12(2H,11H)-dione derivatives: Selective inhibition of MV-4-11 biphenotypic B myelomonocytic leukemia cells’ growth is accompanied by reactive oxygen species overproduction and apoptosis. Bioorg. Med. Chem. Lett. 2018, 28, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Bieszczad, B.; Garbicz, D.; Trzybiński, D.; Mielecki, D.; Woźniak, K.; Grzesiuk, E.; Mieczkowski, A. Unsymmetrically Substituted Dibenzo[b,f][1,5]-diazocine-6,12(5H,11H)dione-A Convenient Scaffold for Bioactive Molecule Design. Molecules 2020, 25, 906. [Google Scholar] [CrossRef] [PubMed]
- Bieszczad, B.; Garbicz, D.; Trzybiński, D.; Dudek, M.K.; Woźniak, K.; Grzesiuk, E.; Mieczkowski, A. Unsymmetrically-Substituted 5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione Scaffold-A Useful Tool for Bioactive Molecules Design. Molecules 2020, 25, 2855. [Google Scholar] [CrossRef] [PubMed]
- Bieszczad, B.; Siwek, A.; Wilczek, M.; Trzybiński, D.; Woźniak, K.; Satała, G.; Bojarski, A.J.; Mieczkowski, A. Synthesis, crystal structure and biological activity of novel analogues of tricyclic drugs. Bioorg. Med. Chem. Lett. 2020, 30, 127493. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Frączyk, T.; Psurski, M.; Wińska, P.; Siedlecki, P.; Dziełak, M.; Trzybiński, D.; Wilczek, M.; Bagiński, M.; Bieszczad, B.; et al. Design and in Vitro Characterization of Tricyclic Benzodiazepine Derivatives as Potent and Selective Antileukemic Agents. Chem. Biodivers. 2021, 18, e2000733. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Speina, E.; Trzybiński, D.; Winiewska-Szajewska, M.; Wińska, P.; Borsuk, E.M.; Podsiadła-Białoskórska, M.; Przygodzki, T.; Drabikowski, K.; Stańczyk, L.; et al. Diketopiperazine-Based, Flexible Tadalafil Analogues: Synthesis, Crystal Structures and Biological Activity Profile. Molecules 2021, 26, 794. [Google Scholar] [CrossRef]
- Bieszczad, B.; Garbicz, D.; Świtalska, M.; Dudek, M.K.; Warszycki, D.; Wietrzyk, J.; Grzesiuk, E.; Mieczkowski, A. Improved HDAC Inhibition, Stronger Cytotoxic Effect and Higher Selectivity against Leukemias and Lymphomas of Novel, Tricyclic Vorinostat Analogues. Pharmaceuticals 2021, 14, 851. [Google Scholar] [CrossRef]
- Bojarska, J.; Mieczkowski, A.; Ziora, Z.M.; Skwarczyński, M.; Toth, I.; Shalash, A.O.; Parang, K.; El-Mowafi, S.A.; Mohammed, E.H.M.; Elnagdy, S.; et al. Cyclic Dipeptides: The Biological and Structural Landscape with Special Focus on the Anti-Cancer Proline-Based Scaffold. Biomolecules 2021, 11, 1515. [Google Scholar] [CrossRef]
- Pietsch, E.C.; Sykes, S.M.; McMahon, S.B.; Murphy, M.E. The p53 family and programmed cell death. Oncogene 2008, 27, 6507–6521. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Andrade-Vieira, R.; Bay, S.N.; Brown, D.G.; Calaf, G.M.; Castellino, R.C.; Cohen-Solal, K.A.; et al. Mechanisms of environmental chemicals that enable the cancer hallmark of evasion of growth suppression. Carcinogenesis 2015, 36 (Suppl. S1), S2–S18. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, M.; Yokosawa, T.; Noguchi, T.; Shimada, T.; Yamada, M.; Sekiguchi, Y.; Hirata, Y.; Matsuzawa, A. Pro-apoptotic functions of TRAF2 in p53-mediated apoptosis induced by cisplatin. J. Toxicol. Sci. 2020, 45, 219–226. [Google Scholar] [CrossRef]
- Kigawa, J.; Sato, S.; Shimada, M.; Takahashi, M.; Itamochi, H.; Kanamori, Y.; Terakawa, N. p53 gene status and chemosensitivity in ovarian cancer. Hum. Cell 2001, 14, 165–171. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | 10 | 11 | Cisplatin |
|---|---|---|---|
| IC50 (µM) a | |||
| A-431 (epidermoid cancer) BJ (normal skin) SI index: IC50 BJ/IC50 A-431 | 3.47 ± 0.32 (**) b | 3.97 ± 0.87 (**) b | 1.56 ± 0.29 |
| 16.72 ± 2.77 (**) b | 23.1 ± 2.34 (***) b | 5.06 ± 0.94 | |
| 4.88 ± 1.08 (**) c (ns) b | 5.89 ± 0.58 (**) c (**) b | 3.26 ± 0.41 (**) c | |
| A549 (lung cancer) MRC-5 (normal lung) SI index: IC50 MRC-5/IC50 A549 | 1.6 ± 0.21 (**) b | 16.29 ± 4.65 (*) b | 5.95 ± 1.28 |
| 16.86 ± 2.96 (**) b | 28.7 ± 7.6 (**) b | 4.17 ± 0.88 | |
| 10.54 ± 1.54 (***) c (***) b | 1.86 ± 0.79 (ns) c (ns) b | 0.74 ± 0.3 (ns) c | |
| NCI-H1299 (lung cancer) MRC-5 (normal lung) SI index: IC50 MRC-5/IC50 NCI-H1299 | 1.07 ± 0.25 (***) b | 4.91 ± 1.45 (ns) b | 5.7 ± 0.88 |
| 16.86 ± 2.96 (**) b | 28.7 ± 7.6 (**) b | 4.17 ± 0.88 | |
| 16.08 ± 3.43 (***) c (**) b | 6.42 ± 2.95 (**) c (*) b | 0.75 ± 0.22 (ns) c | |
| HCT116 (colon cancer) HCT116p53−/− (colon cancer) SI index: IC50 HCT116p53−/−/IC50 HCT116 | 5.31 ± 0.23 (*) b | 4.9 ± 0.53 (ns) b | 3.51 ± 1.16 |
| 5.56 ± 0.74 (*) b | 6.07 ± 0.9 (*) b | 10.77 ± 2.86 | |
| 1.05 ± 0.19 (ns) c (*) b | 1.26 ± 0.3 (ns) c (*) b | 3.18 ± 0.79 (*) c | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Speina, E.; Wilczek, M.; Mieczkowski, A. Dimeric Benzodiazepines as Peptide Mimetics to Overcome p53-Dependent Drug Resistance of Tumors. Biomolecules 2023, 13, 291. https://doi.org/10.3390/biom13020291
Speina E, Wilczek M, Mieczkowski A. Dimeric Benzodiazepines as Peptide Mimetics to Overcome p53-Dependent Drug Resistance of Tumors. Biomolecules. 2023; 13(2):291. https://doi.org/10.3390/biom13020291
Chicago/Turabian StyleSpeina, Elżbieta, Marcin Wilczek, and Adam Mieczkowski. 2023. "Dimeric Benzodiazepines as Peptide Mimetics to Overcome p53-Dependent Drug Resistance of Tumors" Biomolecules 13, no. 2: 291. https://doi.org/10.3390/biom13020291
APA StyleSpeina, E., Wilczek, M., & Mieczkowski, A. (2023). Dimeric Benzodiazepines as Peptide Mimetics to Overcome p53-Dependent Drug Resistance of Tumors. Biomolecules, 13(2), 291. https://doi.org/10.3390/biom13020291