
The Pro-Fibrotic Response to Lens Injury Is Signaled in a PI3K Isoform-Specific Manner



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ex vivo Post-Cataract Surgery Chick Explant Cultures and Inhibitor Treatments
2.2. Immunofluorescence
2.3. Proliferation
2.4. TUNEL Assay
2.5. Immunoblotting
2.6. Confocal Microscopy Imaging
2.7. Statistical Analysis
3. Results
3.1. PI3K Signals Transition to a Fibrotic Phenotype in Response to a Mock Cataract Surgery Wounding
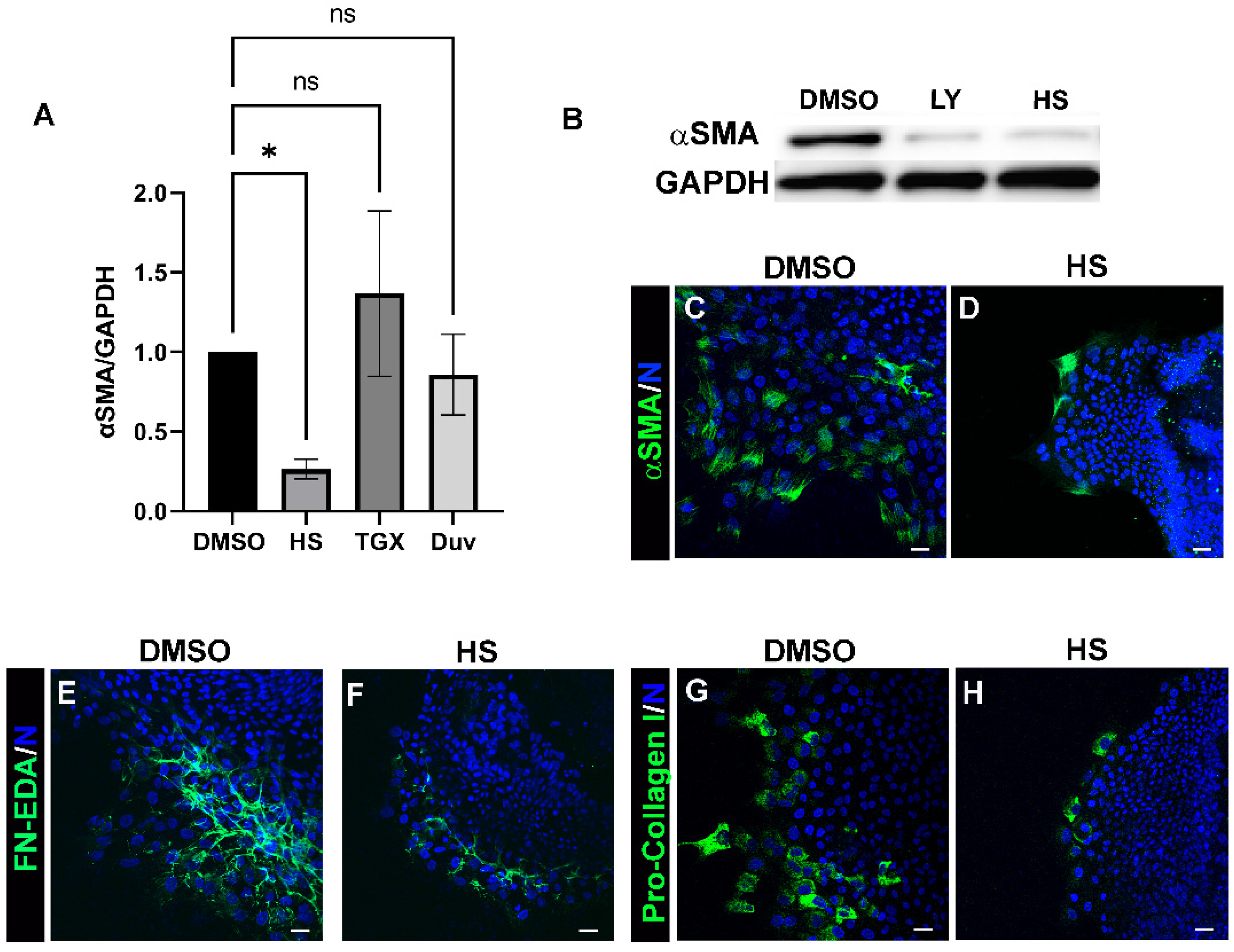
3.2. Acquisition of an αSMA+ Myofibroblast Phenotype in Response to Lens Injury Is Induced in a PI3K-Isoform Specific Manner
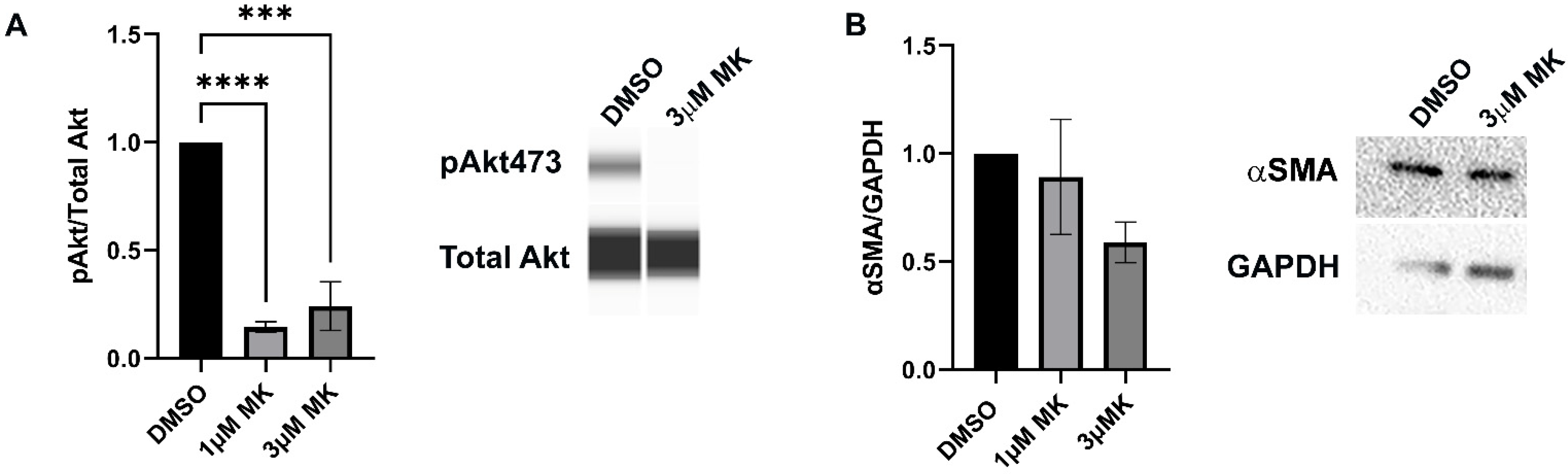
3.3. Blocking Akt Activation Is Alone Insufficient to Block Myofibroblast Emergence
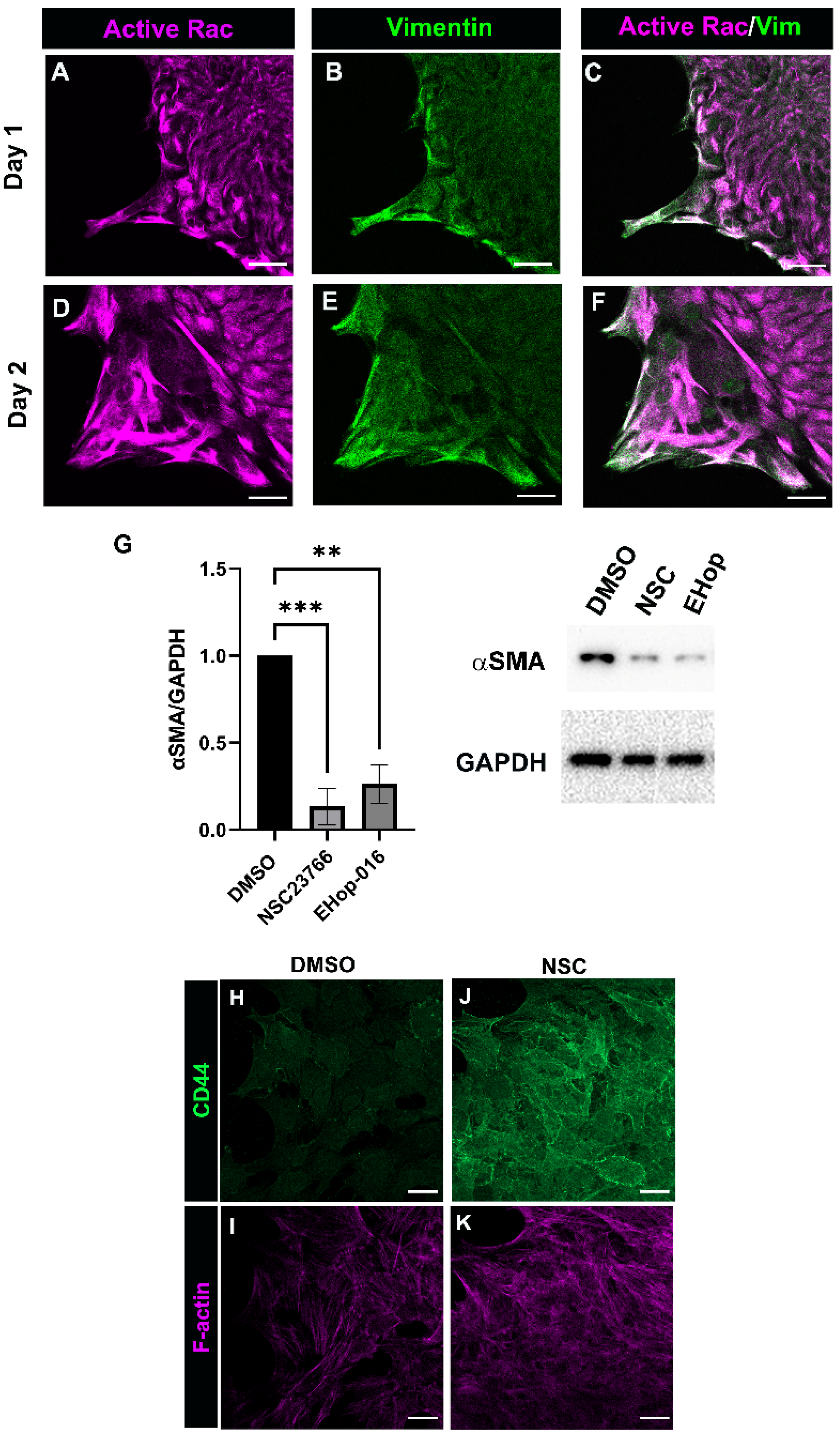
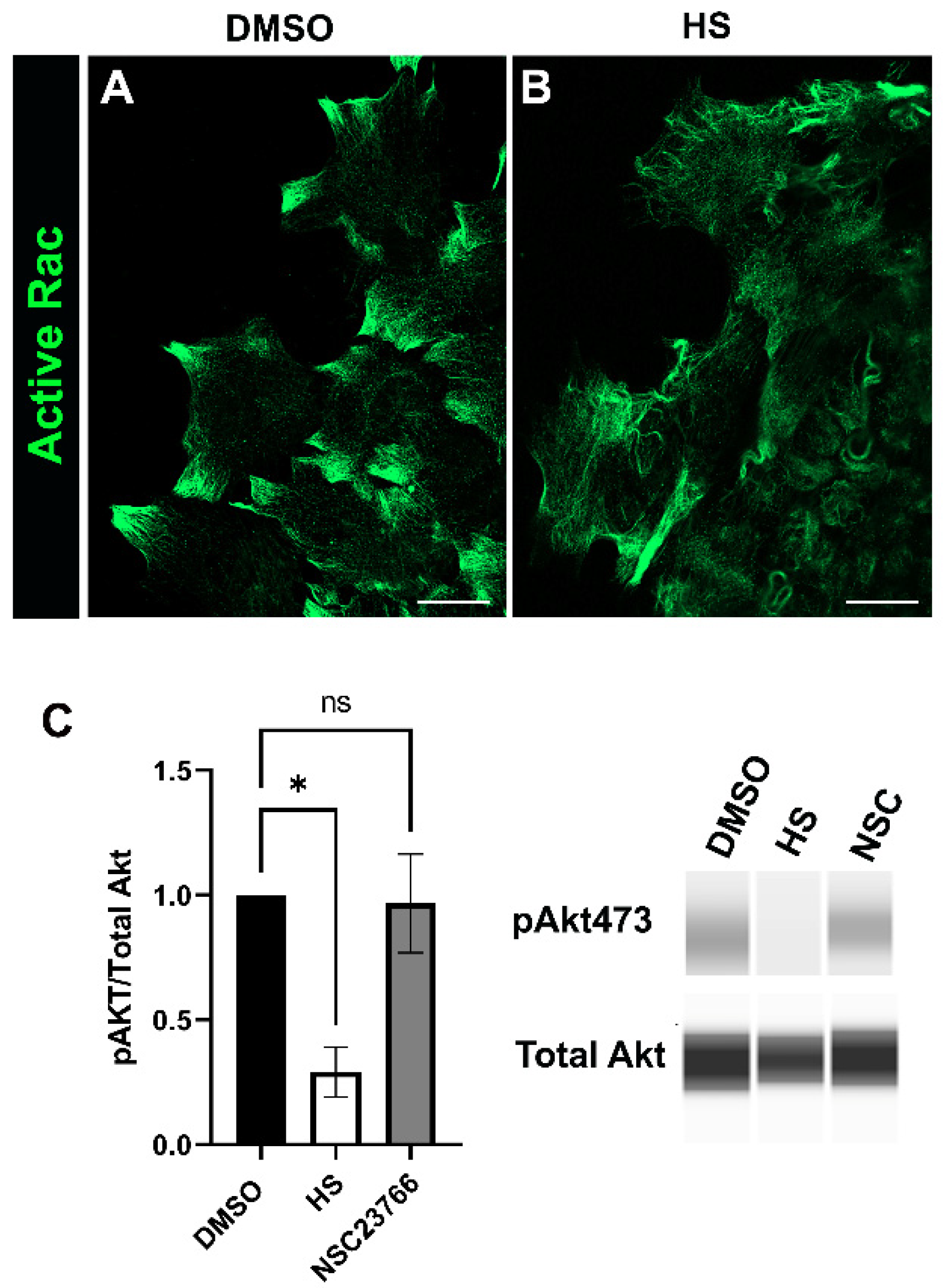
3.4. Rac Signaling Is Involved in Promoting αSMA+ Myofibroblast Differentiation in Response to Cataract Surgery Wounding
3.5. Relationship between Rac and PI3K Signaling in Response to Cataract Surgery Wounding
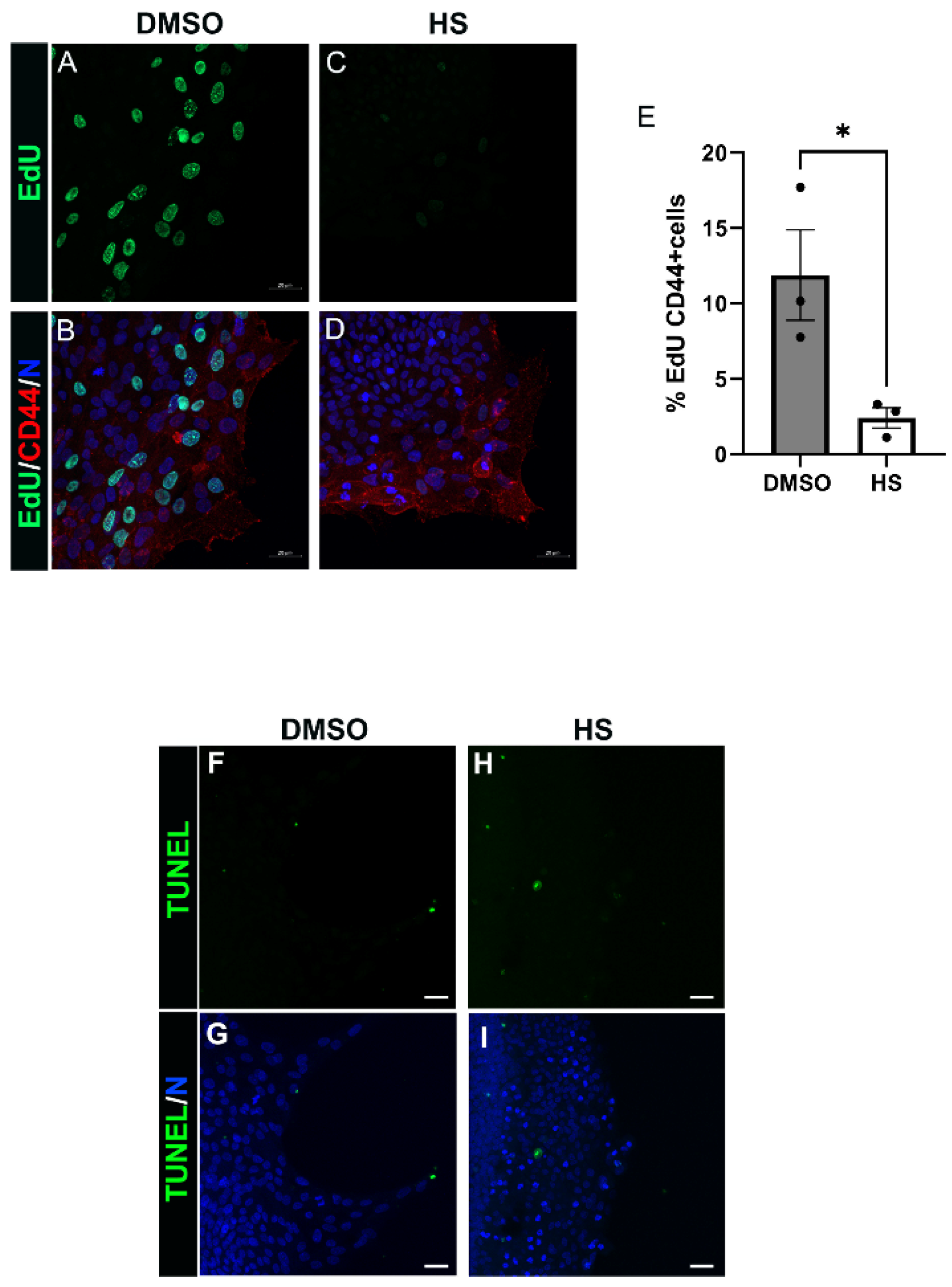
3.6. Induction of Leader Cell Proliferation by PI3K p110α Is Linked to the Emergence and Persistence of Myofibroblasts Post-Mock Cataract Surgery Wounding
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Woods, E.L.; Grigorieva, I.V.; Midgley, A.C.; Brown, C.V.M.; Lu, Y.A.; Phillips, A.O.; Bowen, T.; Meran, S.; Steadman, R. CD147 mediates the CD44s-dependent differentiation of myofibroblasts driven by transforming growth factor-β(1). J. Biol. Chem. 2021, 297, 100987. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef]
- Basta, M.D.; Paulson, H.; Walker, J.L. The local wound environment is a key determinant of the outcome of TGFβ signaling on the fibrotic response of CD44(+) leader cells in an ex vivo post-cataract-surgery model. Exp. Eye Res. 2021, 213, 108829. [Google Scholar] [CrossRef]
- Karppinen, S.M.; Heljasvaara, R.; Gullberg, D.; Tasanen, K.; Pihlajaniemi, T. Toward understanding scarless skin wound healing and pathological scarring. F1000Research 2019, 8, 787. [Google Scholar] [CrossRef]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Tawara, I.; Yoshida, T. Tenascin-C in cardiac disease: A sophisticated controller of inflammation, repair, and fibrosis. Am. J. Physiol. Cell Physiol. 2020, 319, C781–C796. [Google Scholar] [CrossRef]
- de Castro Brás, L.E.; Frangogiannis, N.G. Extracellular matrix-derived peptides in tissue remodeling and fibrosis. Matrix Biol. 2020, 91–92, 176–187. [Google Scholar] [CrossRef]
- Klingberg, F.; Chau, G.; Walraven, M.; Boo, S.; Koehler, A.; Chow, M.L.; Olsen, A.L.; Im, M.; Lodyga, M.; Wells, R.G.; et al. The fibronectin ED-A domain enhances recruitment of latent TGF-β-binding protein-1 to the fibroblast matrix. J. Cell Sci. 2018, 131, jcs201293. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Hinz, B. Myofibroblasts. Exp. Eye Res. 2016, 142, 56–70. [Google Scholar] [CrossRef]
- Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis. Biomolecules 2021, 11, 1095. [Google Scholar] [CrossRef]
- Little, K.; Llorián-Salvador, M.; Tang, M.; Du, X.; Marry, S.; Chen, M.; Xu, H. Macrophage to myofibroblast transition contributes to subretinal fibrosis secondary to neovascular age-related macular degeneration. J. Neuroinflamm. 2020, 17, 355. [Google Scholar] [CrossRef]
- Lodyga, M.; Hinz, B. TGF-β1—A truly transforming growth factor in fibrosis and immunity. Semin Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef]
- Menko, A.S.; Bleaken, B.M.; Libowitz, A.A.; Zhang, L.; Stepp, M.A.; Walker, J.L. A central role for vimentin in regulating repair function during healing of the lens epithelium. Mol. Biol. Cell 2014, 25, 776–790. [Google Scholar] [CrossRef]
- Walker, J.L.; Bleaken, B.M.; Wolff, I.M.; Menko, A.S. Establishment of a Clinically Relevant Ex Vivo Mock Cataract Surgery Model for Investigating Epithelial Wound Repair in a Native Microenvironment. J. Vis. Exp. 2015, 5, e52886. [Google Scholar] [CrossRef]
- Walraven, M.; Hinz, B. Therapeutic approaches to control tissue repair and fibrosis: Extracellular matrix as a game changer. Matrix Biol. 2018, 71–72, 205–224. [Google Scholar] [CrossRef]
- Taiyab, A.; West-Mays, J. Lens Fibrosis: Understanding the Dynamics of Cell Adhesion Signaling in Lens Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2022, 10, 886053. [Google Scholar] [CrossRef]
- Shu, D.Y.; Ng, K.; Wishart, T.F.L.; Chui, J.; Lundmark, M.; Flokis, M.; Lovicu, F.J. Contrasting roles for BMP-4 and ventromorphins (BMP agonists) in TGFβ-induced lens EMT. Exp. Eye Res. 2021, 206, 108546. [Google Scholar] [CrossRef]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Jia, S.; Roberts, T.M.; Zhao, J.J. Should individual PI3 kinase isoforms be targeted in cancer? Curr. Opin. Cell Biol. 2009, 21, 199–208. [Google Scholar] [CrossRef]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.; Okkenhaug, K.; Vanhaesebroeck, B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef]
- Du, Z.; Lin, Z.; Wang, Z.; Liu, D.; Tian, D.; Xia, L. SPOCK1 overexpression induced by platelet-derived growth factor-BB promotes hepatic stellate cell activation and liver fibrosis through the integrin α5β1/PI3K/Akt signaling pathway. Lab. Investig. 2020, 100, 1042–1056. [Google Scholar] [CrossRef]
- Kulkarni, A.A.; Thatcher, T.H.; Olsen, K.C.; Maggirwar, S.B.; Phipps, R.P.; Sime, P.J. PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: Implications for therapy of fibrosis. PLoS ONE 2011, 6, e15909. [Google Scholar] [CrossRef]
- Fang, L.; Chen, H.; Kong, R.; Que, J. Endogenous tryptophan metabolite 5-Methoxytryptophan inhibits pulmonary fibrosis by downregulating the TGF-β/SMAD3 and PI3K/AKT signaling pathway. Life Sci. 2020, 260, 118399. [Google Scholar] [CrossRef]
- Menko, A.S.; DeDreu, J.; Logan, C.M.; Paulson, H.; Levin, A.V.; Walker, J.L. Resident immune cells of the avascular lens: Mediators of the injury and fibrotic response of the lens. FASEB J. 2021, 35, e21341. [Google Scholar] [CrossRef]
- Menko, A.S.; Romisher, A.; Walker, J.L. The Pro-fibrotic Response of Mesenchymal Leader Cells to Lens Wounding Involves Hyaluronic Acid, Its Receptor RHAMM, and Vimentin. Front. Cell Dev. Biol. 2022, 10, 862423. [Google Scholar] [CrossRef]
- Walker, J.L.; Wolff, I.M.; Zhang, L.; Menko, A.S. Activation of SRC kinases signals induction of posterior capsule opacification. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2214–2223. [Google Scholar] [CrossRef][Green Version]
- Bleaken, B.M.; Menko, A.S.; Walker, J.L. Cells activated for wound repair have the potential to direct collective invasion of an epithelium. Mol. Biol. Cell 2016, 27, 451–465. [Google Scholar] [CrossRef]
- Menko, A.S.; Bleaken, B.M.; Walker, J.L. Regional-specific alterations in cell-cell junctions, cytoskeletal networks and myosin-mediated mechanical cues coordinate collectivity of movement of epithelial cells in response to injury. Exp. Cell Res. 2014, 322, 133–148. [Google Scholar] [CrossRef]
- Walker, J.L.; Bleaken, B.M.; Romisher, A.R.; Alnwibit, A.A.; Menko, A.S. In wound repair vimentin mediates the transition of mesenchymal leader cells to a myofibroblast phenotype. Mol. Biol. Cell 2018, 29, 1555–1570. [Google Scholar] [CrossRef]
- Gheyas, R.; Ortega-Alvarez, R.; Chauss, D.; Kantorow, M.; Menko, A.S. Suppression of PI3K signaling is linked to autophagy activation and the spatiotemporal induction of the lens organelle free zone. Exp. Cell Res. 2022, 412, 113043. [Google Scholar] [CrossRef]
- Matsuo, M.; Sakurai, H.; Ueno, Y.; Ohtani, O.; Saiki, I. Activation of MEK/ERK and PI3K/Akt pathways by fibronectin requires integrin alphav-mediated ADAM activity in hepatocellular carcinoma: A novel functional target for gefitinib. Cancer Sci. 2006, 97, 155–162. [Google Scholar] [CrossRef]
- Shaw, L.M.; Rabinovitz, I.; Wang, H.H.; Toker, A.; Mercurio, A.M. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell 1997, 91, 949–960. [Google Scholar] [CrossRef]
- Suwanabol, P.A.; Seedial, S.M.; Zhang, F.; Shi, X.; Si, Y.; Liu, B.; Kent, K.C. TGF-beta and Smad3 modulate PI3K/Akt signaling pathway in vascular smooth muscle cells. Am. J. Physiol Heart Circ. Physiol. 2012, 302, H2211–H2219. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Cary, L.A.; Guan, J.L. Focal adhesion kinase in integrin-mediated signaling. Front. Biosci. 1999, 4, D102–D113. [Google Scholar] [CrossRef]
- Menko, A.S.; Walker, J.L.; Stepp, M.A. Fibrosis: Shared Lessons From the Lens and Cornea. Anat. Rec. 2020, 303, 1689–1702. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Qiao, M.; Sheng, S.; Pardee, A.B. Metastasis and AKT activation. Cell Cycle 2008, 7, 2991–2996. [Google Scholar] [CrossRef]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef]
- Tapon, N.; Hall, A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol. 1997, 9, 86–92. [Google Scholar] [CrossRef]
- Hall, A.; Nobes, C.D. Rho GTPases: Molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R. Soc. Lond B Biol. Sci. 2000, 355, 965–970. [Google Scholar] [CrossRef]
- Choi, S.S.; Sicklick, J.K.; Ma, Q.; Yang, L.; Huang, J.; Qi, Y.; Chen, W.; Li, Y.X.; Goldschmidt-Clermont, P.J.; Diehl, A.M. Sustained activation of Rac1 in hepatic stellate cells promotes liver injury and fibrosis in mice. Hepatology 2006, 44, 1267–1277. [Google Scholar] [CrossRef]
- Xu, S.W.; Liu, S.; Eastwood, M.; Sonnylal, S.; Denton, C.P.; Abraham, D.J.; Leask, A. Rac inhibition reverses the phenotype of fibrotic fibroblasts. PLoS ONE 2009, 4, e7438. [Google Scholar] [CrossRef][Green Version]
- Patel, S.; Tang, J.; Overstreet, J.M.; Anorga, S.; Lian, F.; Arnouk, A.; Goldschmeding, R.; Higgins, P.J.; Samarakoon, R. Rac-GTPase promotes fibrotic TGF-beta1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways. FASEB J. 2019, 33, 9797–9810. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Wierzbicka-Patynowski, I.; Schwarzbauer, J.E. Regulatory role for SRC and phosphatidylinositol 3-kinase in initiation of fibronectin matrix assembly. J. Biol. Chem. 2002, 277, 19703–19708. [Google Scholar] [CrossRef]
- Di-Luoffo, M.; Ben-Meriem, Z.; Lefebvre, P.; Delarue, M.; Guillermet-Guibert, J. PI3K functions as a hub in mechanotransduction. Trends Biochem. Sci. 2021, 46, 878–888. [Google Scholar] [CrossRef]
- Vedham, V.; Phee, H.; Coggeshall, K.M. Vav activation and function as a rac guanine nucleotide exchange factor in macrophage colony-stimulating factor-induced macrophage chemotaxis. Mol. Cell Biol. 2005, 25, 4211–4220. [Google Scholar] [CrossRef]
- Fleming, I.N.; Gray, A.; Downes, C.P. Regulation of the Rac1-specific exchange factor Tiam1 involves both phosphoinositide 3-kinase-dependent and -independent components. Biochem. J. 2000, 351, 173–182. [Google Scholar] [CrossRef]
- Yokoyama, K.; Kimoto, K.; Itoh, Y.; Nakatsuka, K.; Matsuo, N.; Yoshioka, H.; Kubota, T. The PI3K/Akt pathway mediates the expression of type I collagen induced by TGF-beta2 in human retinal pigment epithelial cells. Graefes Arch. Clin. Exp. Ophthalmol. 2012, 250, 15–23. [Google Scholar] [CrossRef]
- Hu, X.; Xu, Q.; Wan, H.; Hu, Y.; Xing, S.; Yang, H.; Gao, Y.; He, Z. PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab. Investig. 2020, 100, 801–811. [Google Scholar] [CrossRef]
- Magaye, R.R.; Savira, F.; Hua, Y.; Xiong, X.; Huang, L.; Reid, C.; Flynn, B.L.; Kaye, D.; Liew, D.; Wang, B.H. Attenuating PI3K/Akt- mTOR pathway reduces dihydrosphingosine 1 phosphate mediated collagen synthesis and hypertrophy in primary cardiac cells. Int. J. Biochem. Cell Biol. 2021, 134, 105952. [Google Scholar] [CrossRef]
- Wang, J.; You, J.; Gong, D.; Xu, Y.; Yang, B.; Jiang, C. PDGF-BB induces conversion, proliferation, migration, and collagen synthesis of oral mucosal fibroblasts through PDGFR-beta/PI3K/AKT signaling pathway. Cancer Biomark. 2021, 30, 407–415. [Google Scholar] [CrossRef]
- Wegner, K.A.; Mueller, B.R.; Unterberger, C.J.; Avila, E.J.; Ruetten, H.; Turco, A.E.; Oakes, S.R.; Girardi, N.M.; Halberg, R.B.; Swanson, S.M.; et al. Prostate epithelial-specific expression of activated PI3K drives stromal collagen production and accumulation. J. Pathol. 2020, 250, 231–242. [Google Scholar] [CrossRef]
- Lu, Y.; Azad, N.; Wang, L.; Iyer, A.K.; Castranova, V.; Jiang, B.H.; Rojanasakul, Y. Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am. J. Respir. Cell Mol. Biol. 2010, 42, 432–441. [Google Scholar] [CrossRef]
- Conte, E.; Fruciano, M.; Fagone, E.; Gili, E.; Caraci, F.; Iemmolo, M.; Crimi, N.; Vancheri, C. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: The role of class I P110 isoforms. PLoS ONE 2011, 6, e24663. [Google Scholar] [CrossRef]
- Son, M.K.; Ryu, Y.L.; Jung, K.H.; Lee, H.; Lee, H.S.; Yan, H.H.; Park, H.J.; Ryu, J.K.; Suh, J.K.; Hong, S.; et al. HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis. Sci. Rep. 2013, 3, 3470. [Google Scholar] [CrossRef]
- Hettiarachchi, S.U.; Li, Y.H.; Roy, J.; Zhang, F.; Puchulu-Campanella, E.; Lindeman, S.D.; Srinivasarao, M.; Tsoyi, K.; Liang, X.; Ayaub, E.A.; et al. Targeted inhibition of PI3 kinase/mTOR specifically in fibrotic lung fibroblasts suppresses pulmonary fibrosis in experimental models. Sci. Transl. Med. 2020, 12, eaay3724. [Google Scholar] [CrossRef]
- Spriggs, L.L.; Hill, S.M.; Jeter, J.R., Jr. Proliferation is required for induction of terminal differentiation of Friend erythroleukemia cells. Biochem. Cell Biol. 1992, 70, 555–564. [Google Scholar] [CrossRef]
- Ruijtenberg, S.; van den Heuvel, S. Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 2016, 15, 196–212. [Google Scholar] [CrossRef]
- Sellitto, C.; Li, L.; Vaghefi, E.; Donaldson, P.J.; Lin, R.Z.; White, T.W. The Phosphoinosotide 3-Kinase Catalytic Subunit p110alpha is Required for Normal Lens Growth. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3145–3151. [Google Scholar] [CrossRef][Green Version]
- Walker, J.L.; Zhai, N.; Zhang, L.; Bleaken, B.M.; Wolff, I.; Gerhart, J.; George-Weinstein, M.; Menko, A.S. Unique precursors for the mesenchymal cells involved in injury response and fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 13730–13735. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menko, A.S.; Walker, J.L. The Pro-Fibrotic Response to Lens Injury Is Signaled in a PI3K Isoform-Specific Manner. Biomolecules 2022, 12, 1181. https://doi.org/10.3390/biom12091181
Menko AS, Walker JL. The Pro-Fibrotic Response to Lens Injury Is Signaled in a PI3K Isoform-Specific Manner. Biomolecules. 2022; 12(9):1181. https://doi.org/10.3390/biom12091181
Chicago/Turabian StyleMenko, A. Sue, and Janice L. Walker. 2022. "The Pro-Fibrotic Response to Lens Injury Is Signaled in a PI3K Isoform-Specific Manner" Biomolecules 12, no. 9: 1181. https://doi.org/10.3390/biom12091181
APA StyleMenko, A. S., & Walker, J. L. (2022). The Pro-Fibrotic Response to Lens Injury Is Signaled in a PI3K Isoform-Specific Manner. Biomolecules, 12(9), 1181. https://doi.org/10.3390/biom12091181