
Lipotoxicity as a Barrier for T Cell-Based Therapies


 , and
, and
Abstract
:1. Introduction
2. Sources of Lipids within the Tumor Microenvironment
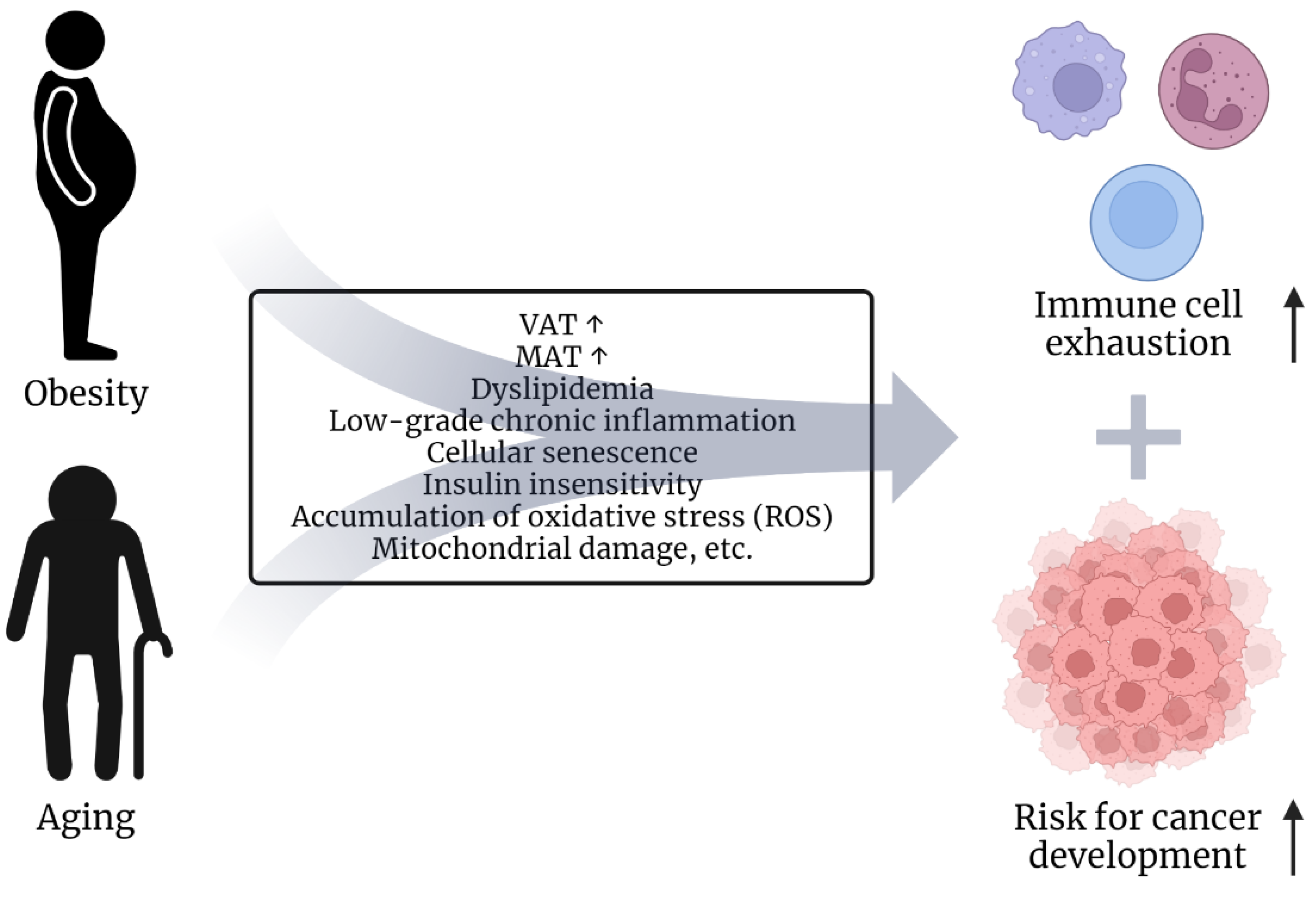
2.1. Risk Factors Obesity and Age
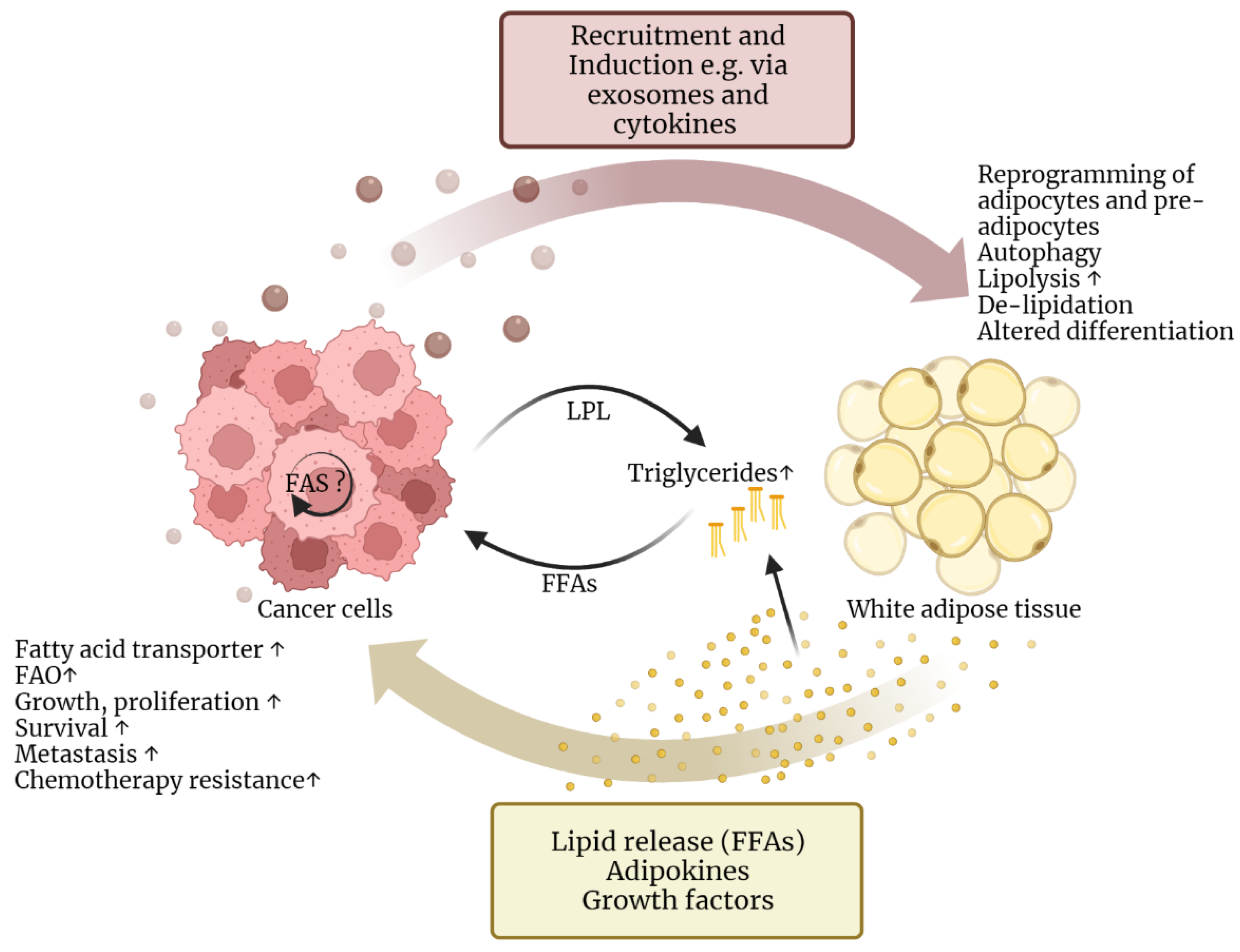
2.2. Lipids Support Cancer Cell Survival and Progression
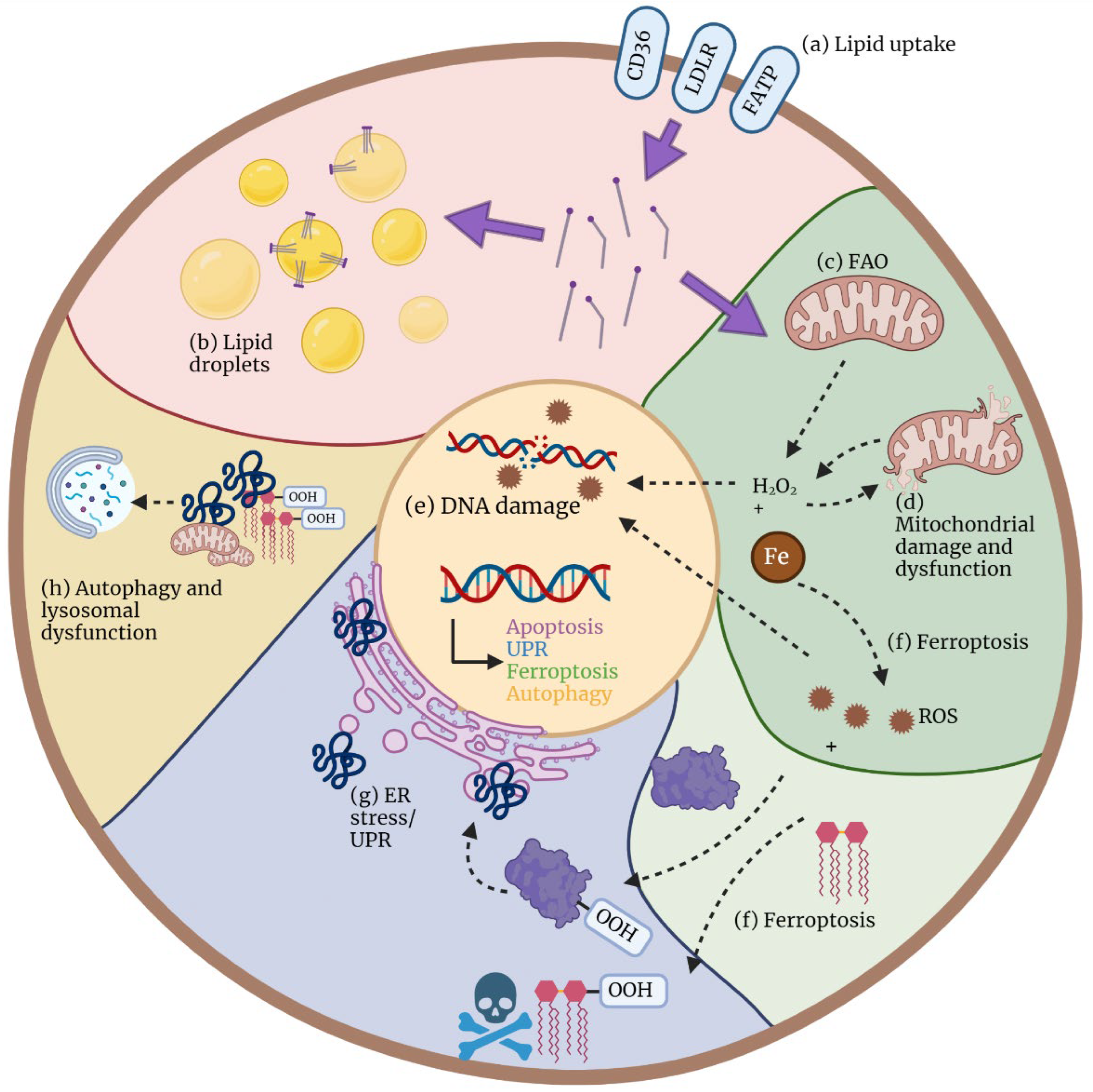
3. Mechanisms of Cellular Lipotoxicity
3.1. Oxidative Stress and Mitochondrial Dysfunction
3.2. Endoplasmatic Reticulum Stress
3.3. Ferroptosis
3.4. Autophagy and Lysosomal Dysfunction
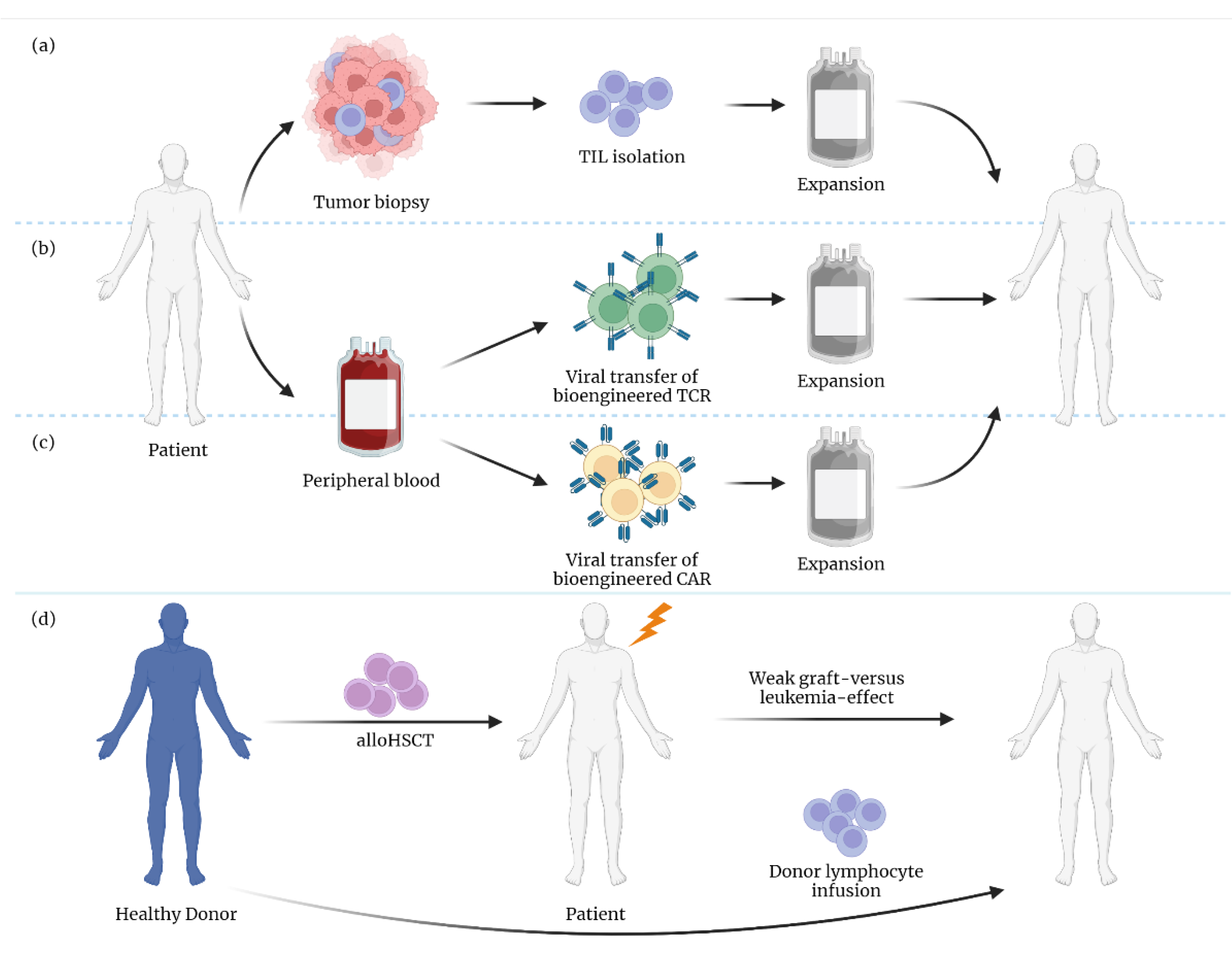
4. Implications of Lipotoxicity on T Cell-Based Therapies
4.1. Differential Effects of FAs in T Cell Subsets
4.2. The Role of CD36 Expression and Ferroptosis in T Cells
5. Conclusions and Perspectives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Tumor Entity | Target | Effect | Source | |
|---|---|---|---|---|---|
| Clinical studies | Metformin (monotherapy/combination therapy) | Various human tumor entities | Inhibition of hepatic gluconeogenesis and lipid synthesis Decreased adipose tissue fatty acid synthesis and lipolysis Decreased pancreatic insulin secretion Increased muscle glucose uptake | No beneficial effects proven | [156] |
| Statins | Various human tumor entities | Inhibition of HMG-CoA reductase | Improvement of patients’ survival in several tumor entities | [157,160] | |
| CLL | Inhibition of HMG-CoA reductase | Improvement of patients’ survival and outcome; | [64,65,158,159] | ||
| delayed need for chemotherapy | [64] | ||||
| Fibrates + immune checkpoint blockade | Non-small cell lung cancer (NSCLC) | Peroxisome proliferator-activated receptor agonist | Improved overall survival | [172] | |
| In vivo | Thioridazine + BRAF/MEK inhibitors | Human melanoma-bearing mice | Peroxisomal FAO inhibitor | Increased sensitivity to BRAF/MEK inhibitors in melanoma persister cells | [165] |
| Simvastatin | B16 tumor mouse model | Inhibition of HMG-CoA reductase | Lower CD8+ T cell exhaustion | [138] | |
| Nanoparticles carrying fenofibrate and surface anti-CD3e f(ab)2 fragment | B16 tumor mouse model | Fatty acid metabolism of TILs | Increased FAO and mitochondrial functions of TILs; improved T cell anti-tumor function | [174] | |
| Ferrostatin-1 | B16 tumor mouse model | Pretreatment of T cells with ferroptosis inhibitor before adoptive transfer | Improved T cell anti-tumor function | [144] | |
| Fenofibrate (+PD-1 blockade) | Mouse melanoma models | Fatty acid metabolism of TILs | Improved T cell anti-tumor function | [173] | |
| In vitro | Simvastatin + pentoxifylline | Triple-negative MDA-MB231 breast cancer cells | Inhibition of HMG-CoA reductase | Apoptosis, autophagy, and cell cycle arrest in cancer cells | [161] |
| Statin + venetoclax | CLL | Inhibition of HMG-CoA reductase | Enhanced the sensitivity to venetoclax | [162] | |
| Etomoxir/ranolazine + ABT-737 (Bcl-2/Bcl-xL inhibitor) | Human leukemic cells | Inhibition of FAO | Increased apoptosis sensitivity in leukemic cells | [163] | |
| Ibrutinib | CLL | B cell receptor inhibition | Reduction of LPL expression and FFA metabolism in CLL | [164] | |
| CD36-blocking antibody | Multiple melanoma | Fatty acid metabolism of TILs | Reduction of ferroptosis in T cells | [144] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: Review of the literature and challenges ahead. J. Immunother. Cancer 2021, 9, e002723. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Riley, J.L.; Levine, B.L. Engineering T cells to survive and thrive in the hostile tumor microenvironment. Curr. Opin. Biomed. Eng. 2022, 21, 100360. [Google Scholar] [CrossRef]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9, e55185. [Google Scholar] [CrossRef]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Manzo, T.; Prentice, B.M.; Anderson, K.G.; Raman, A.; Schalck, A.; Codreanu, G.S.; Lauson, C.B.N.; Tiberti, S.; Raimondi, A.; Jones, M.A.; et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J. Exp. Med. 2020, 217, e20191920. [Google Scholar] [CrossRef]
- Howie, D.; Bokum, A.T.; Necula, A.S.; Cobbold, S.P.; Waldmann, H. The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front. Immunol. 2017, 8, 1949. [Google Scholar] [CrossRef]
- De Jong, A.J.; Kloppenburg, M.; Toes, R.E.; Ioan-Facsinay, A. Fatty acids, lipid mediators, and T-cell function. Front. Immunol. 2014, 5, 483. [Google Scholar] [CrossRef]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef]
- Kleinfeld, A.M.; Okada, C. Free fatty acid release from human breast cancer tissue inhibits cytotoxic T-lymphocyte-mediated killing. J. Lipid. Res. 2005, 46, 1983–1990. [Google Scholar] [PubMed]
- Goodwin, P.J.; Stambolic, V. Impact of the obesity epidemic on cancer. Annu. Rev. Med. 2015, 66, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; García-Cañaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e26. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Lichtman, M.A. Obesity and the risk for a hematological malignancy: Leukemia, lymphoma, or myeloma. Oncologist 2010, 15, 1083–1101. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Body mass index and risk of non-Hodgkin’s and Hodgkin’s lymphoma: A meta-analysis of prospective studies. Eur. J. Cancer 2011, 47, 2422–2430. [Google Scholar] [CrossRef]
- Castillo, J.J.; Reagan, J.L.; Ingham, R.R.; Furman, M.; Dalia, S.; Merhi, B.; Nemr, S.; Zarrabi, A.; Mitri, J. Obesity but not overweight increases the incidence and mortality of leukemia in adults: A meta-analysis of prospective cohort studies. Leuk. Res. 2012, 36, 868–875. [Google Scholar] [CrossRef]
- Genkinger, J.M.; Kitahara, C.M.; Bernstein, L.; de Gonzalez, A.B.; Brotzman, M.; Elena, J.W.; Giles, G.G.; Hartge, P.; Singh, P.N.; Stolzenberg-Solomon, R.Z.; et al. Central adiposity, obesity during early adulthood, and pancreatic cancer mortality in a pooled analysis of cohort studies. Ann. Oncol. 2015, 26, 2257–2266. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, K.; Giovannucci, E.L.; Ma, J.; Colditz, G.A.; Fuchs, C.S.; Willett, W.C.; Stampfer, M.J.; Nimptsch, K.; Ogino, S.; et al. Early life body fatness and risk of colorectal cancer in u.s. Women and men-results from two large cohort studies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 690–697. [Google Scholar] [CrossRef]
- Teras, L.R.; Kitahara, C.M.; Birmann, B.; Hartge, P.A.; Wang, S.S.; Robien, K.; Patel, A.V.; Adami, H.-O.; Weiderpass, E.; Giles, G.; et al. Body size and multiple myeloma mortality: A pooled analysis of 20 prospective studies. Br. J. Haematol. 2014, 166, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Ataey, A.; Jafarvand, E.; Adham, D.; Moradi-Asl, E. The Relationship Between Obesity, Overweight, and the Human Development Index in World Health Organization Eastern Mediterranean Region Countries. J. Prev. Med. Public Health 2020, 53, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Inoue, S.; Isoda, F.; Waseda, M.; Ishihara, M.; Yamakawa, T.; Sugiyama, A.; Takamura, Y.; Okuda, K. Impaired immunity in obesity: Suppressed but reversible lymphocyte responsiveness. Int. J. Obes. Relat. Metab. Disord. 1993, 17, 631–636. [Google Scholar] [PubMed]
- Tanaka, S.I.; Isoda, F.; Ishihara, Y.; Kimura, M.; Yamakawa, T. T lymphopaenia in relation to body mass index and TNF-alpha in human obesity: Adequate weight reduction can be corrective. Clin. Endocrinol. 2001, 54, 347–354. [Google Scholar] [CrossRef]
- Boynton, A.; Neuhouser, M.L.; Wener, M.H.; Wood, B.; Sorensen, B.; Chen-Levy, Z.; Kirk, A.E.; Yasui, Y.; Lacroix, K.; McTiernan, A.; et al. Associations between healthy eating patterns and immune function or inflammation in overweight or obese postmenopausal women. Am. J. Clin. Nutr. 2007, 86, 1445–1455. [Google Scholar]
- Misumi, I.; Starmer, J.; Uchimura, T.; Beck, M.A.; Magnuson, T.; Whitmire, J.K. Obesity Expands a Distinct Population of T Cells in Adipose Tissue and Increases Vulnerability to Infection. Cell Rep. 2019, 27, 514–524.e5. [Google Scholar] [CrossRef]
- Wijngaarden, L.H.; Taselaar, A.E.; Nuijten, F.; van der Harst, E.; Klaassen, R.A.; Kuijper, T.M.; Jongbloed, F.; Ambagtsheer, G.; Klepper, M.; Ijzermans, J.N.M.; et al. T and B Cell Composition and Cytokine Producing Capacity Before and After Bariatric Surgery. Front. Immunol. 2022, 13, 888278. [Google Scholar] [CrossRef]
- Yang, H.; Youm, Y.-H.; Vandanmagsar, B.; Rood, J.; Kumar, K.G.; Butler, A.; Dixit, V.D. Obesity accelerates thymic aging. Blood 2009, 114, 3803–3812. [Google Scholar] [CrossRef]
- Porsche, C.E.; Delproposto, J.B.; Geletka, L.; O’Rourke, R.; Lumeng, C.N. Obesity results in adipose tissue T cell exhaustion. JCI Insight 2021, 6, e139793. [Google Scholar] [CrossRef]
- Sheridan, P.A.; Paich, H.A.; Handy, J.; Karlsson, E.A.; Hudgens, M.G.; Sammon, A.B.; Holland, L.A.; Weir, S.; Noah, T.L.; Beck, M.A. Obesity is associated with impaired immune response to influenza vaccination in humans. Int. J. Obes. 2012, 36, 1072–1077. [Google Scholar] [CrossRef]
- Kado, T.; Nawaz, A.; Takikawa, A.; Usui, I.; Tobe, K. Linkage of CD8(+) T cell exhaustion with high-fat diet-induced tumourigenesis. Sci. Rep. 2019, 9, 12284. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Blomberg, B.B.; Paganelli, R. Aging, Obesity, and Inflammatory Age-Related Diseases. Front. Immunol. 2017, 8, 1745. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Palmer, A.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, A.T.; Sepe, A.; O Johnson, K.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 2015, 4, e12997. [Google Scholar] [CrossRef]
- Gustafson, B.; Nerstedt, A.; Smith, U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat. Commun. 2019, 10, 2757. [Google Scholar] [CrossRef] [PubMed]
- Mitterberger, M.C.; Lechner, S.; Mattesich, M.; Zwerschke, W. Adipogenic differentiation is impaired in replicative senescent human subcutaneous adipose-derived stromal/progenitor cells. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.S.; Shuman, W.P.; Bradbury, V.L.; Cain, K.C.; Fellingham, G.W.; Beard, J.C.; Kahn, S.E.; Stratton, J.R.; Cerqueira, M.D.; Abrass, I.B. Body fat distribution in healthy young and older men. J. Gerontol. 1990, 45, M181–M185. [Google Scholar] [CrossRef]
- Shimokata, H.; Tobin, J.D.; Muller, D.C.; Elahi, D.; Coon, P.J.; Andres, R. Studies in the distribution of body fat: I. Effects of age, sex, and obesity. J. Gerontol. 1989, 44, M66–M73. [Google Scholar] [CrossRef]
- Justesen, J.; Stenderup, K.; Ebbesen, E.; Mosekilde, L.; Steiniche, T.; Kassem, M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2001, 2, 165–171. [Google Scholar] [CrossRef]
- Liu, H.H.; Li, J.J. Aging and dyslipidemia: A review of potential mechanisms. Ageing Res. Rev. 2015, 19, 43–52. [Google Scholar] [CrossRef]
- Bays, H. Central obesity as a clinical marker of adiposopathy; increased visceral adiposity as a surrogate marker for global fat dysfunction. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 345–351. [Google Scholar] [CrossRef]
- Yang, Y.-H.K.; Ogando, C.R.; See, C.W.; Chang, T.-Y.; Barabino, G.A. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem. Cell Res. Ther. 2018, 9, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, L.; Brennan, T.A.; Russell, E.; Kim, J.-H.; Chen, Q.; Johnson, F.B.; Pignolo, R.J. Aging alters bone-fat reciprocity by shifting in vivo mesenchymal precursor cell fate towards an adipogenic lineage. Bone 2016, 85, 29–36. [Google Scholar] [CrossRef]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.-M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e6. [Google Scholar] [CrossRef] [PubMed]
- Kohlgruber, A.C.; LaMarche, N.M.; Lynch, L. Adipose tissue at the nexus of systemic and cellular immunometabolism. Semin. Immunol. 2016, 28, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Li, J.; Sun, S.; Yuan, J.; Sun, S. Cancer-associated adipocytes as immunomodulators in cancer. Biomark. Res. 2021, 9, 2. [Google Scholar] [CrossRef]
- Tam, B.T.; Morais, J.A.; Santosa, S. Obesity and ageing: Two sides of the same coin. Obes. Rev. 2020, 21, e12991. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Cerne, D.; Melkič, E.; Trošt, Z.; Sok, M.; Marc, J. Lipoprotein lipase activity and gene expression in lung cancer and in adjacent noncancer lung tissue. Exp. Lung Res. 2007, 33, 217–225. [Google Scholar] [CrossRef]
- Cao, D.; Song, X.; Che, L.; Li, X.; Pilo, M.G.; Vidili, G.; Porcu, A.; Solinas, A.; Cigliano, A.; Pes, G.M.; et al. Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells. Liver Int. 2017, 37, 80–89. [Google Scholar] [CrossRef]
- Dong, W.; Gong, H.; Zhang, G.; Vuletic, S.; Albers, J.; Zhang, J.; Liang, H.; Sui, Y.; Zheng, J. Lipoprotein lipase and phospholipid transfer protein overexpression in human glioma cells and their effect on cell growth, apoptosis, and migration. Acta Biochim. Biophys. Sin. 2017, 49, 62–73. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, J.; Yeo, J.H.; Kwon, D.; Min, B.S.; Cha, Y.J.; Koo, J.S.; Jeong, J.; Lee, J.; Choi, J. Interaction between CD36 and FABP4 modulates adipocyte-induced fatty acid import and metabolism in breast cancer. NPJ Breast Cancer 2021, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, I.M.; Napier, D.L.; Weiss, H.L.; Evers, B.M.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Fernandez, L.P.; Gomez de Cedron, M.; Ramirez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Orellana, M.; Brooks, J.; Madabushi, S.S.; Vishwasrao, P.; Parra, L.E.; Sanchez, J.; Salhotra, A.; Stein, A.; Chen, C.-C.; et al. Exosomes-driven lipolysis and bone marrow niche remodeling supports leukemia expansion. Haematologica 2020, 106, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, Q.; Cang, H.; Wang, Z.; Hu, X.; Pan, R.; Yang, Y.; Chen, Y. Acute Myeloid Leukemia Cells Educate Mesenchymal Stromal Cells toward an Adipogenic Differentiation Propensity with Leukemia Promotion Capabilities. Adv. Sci. 2022, 9, 2105811. [Google Scholar] [CrossRef]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef]
- Salunkhe, S.; Mishra, S.V.; Ghorai, A.; Hole, A.; Chandrani, P.; Dutt, A.; Chilakapati, M.; Dutt, S. Metabolic rewiring in drug resistant cells exhibit higher OXPHOS and fatty acids as preferred major source to cellular energetics. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148300. [Google Scholar] [CrossRef]
- Bottcher, M.; Panagiotidis, K.; Bruns, H.; Stumpf, M.; Völkl, S.; Geyh, S.; Dietel, B.; Schroeder, T.; Mackensen, A.; Mougiakakos, D. Bone marrow stroma cells promote induction of a chemoresistant and prognostic unfavorable S100A8/A9high AML cell subset. Blood Adv. 2022. [Google Scholar] [CrossRef] [PubMed]
- Thurgood, L.A.; Best, O.G.; Rowland, A.; Lower, K.M.; Brooks, D.A.; Kuss, B.J. Lipid uptake in chronic lymphocytic leukemia. Exp. Hematol. 2022, 106, 58–67. [Google Scholar] [CrossRef] [PubMed]
- McCaw, L.; Shi, Y.; Wang, G.; Li, Y.-J.; Spaner, D.E. Low Density Lipoproteins Amplify Cytokine-signaling in Chronic Lymphocytic Leukemia Cells. EBioMedicine 2017, 15, 24–35. [Google Scholar] [PubMed]
- Chow, S.; Buckstein, R.; Spaner, D.E. A link between hypercholesterolemia and chronic lymphocytic leukemia. Leuk. Lymphoma 2016, 57, 797–802. [Google Scholar] [CrossRef]
- Mozessohn, L.; Earle, C.; Spaner, D.; Cheng, S.Y.; Kumar, M.; Buckstein, R. The Association of Dyslipidemia With Chronic Lymphocytic Leukemia: A Population-Based Study. J. Natl. Cancer Inst. 2017, 109, djw226. [Google Scholar] [CrossRef]
- Masoodi, M.; Lee, E.; Eiden, M.; Bahlo, A.; Shi, Y.; Ceddia, R.B.; Baccei, C.; Prasit, P.; Spaner, D.E. A role for oleoylethanolamide in chronic lymphocytic leukemia. Leukemia 2014, 28, 1381–1387. [Google Scholar] [CrossRef]
- Spaner, D.E.; Lee, E.; Shi, Y.; Wen, F.; Li, Y.; Tung, S.; McCaw, L.; Wong, K.; Gary-Gouy, H.; Dalloul, A.; et al. PPAR-alpha is a therapeutic target for chronic lymphocytic leukemia. Leukemia 2013, 27, 1090–1099. [Google Scholar] [CrossRef]
- Prieto, D.; Seija, N.; Uriepero, A.; Souto-Padrón, T.; Oliver, A.; Irigoin, V.; Guillermo, C.; Navarrete, M.A.; Landoni, A.I.; Dighiero, G.; et al. LPL protein in Chronic Lymphocytic Leukaemia have different origins in Mutated and Unmutated patients. Advances for a new prognostic marker in CLL. Br. J. Haematol. 2018, 182, 521–525. [Google Scholar] [CrossRef]
- Bilous, N.; Abramenko, I.; Chumak, A.; Dyagil, I.; Martina, Z. Analysis of LPL gene expression in patients with chronic lymphocytic leukemia. Exp. Oncol. 2019, 41, 39–45. [Google Scholar] [CrossRef]
- Kaderi, M.A.; Kanduri, M.; Buhl, A.M.; Sevov, M.; Cahill, N.; Gunnarsson, R.; Jansson, M.; Smedby, K.E.; Hjalgrim, H.; Jurlander, J.; et al. LPL is the strongest prognostic factor in a comparative analysis of RNA-based markers in early chronic lymphocytic leukemia. Haematologica 2011, 96, 1153–1160. [Google Scholar]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.R.; Butler, L.M.; Hoy, A.J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 2021, 9, 2. [Google Scholar] [PubMed]
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 11818–11823. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Hussain, A.R.; Ahmed, M.; Bu, R.; Ahmed, S.O.; Ajarim, D.; Al-Dayel, F.; Bavi, P.; Al-Kuraya, K.S. Inhibition of fatty acid synthase suppresses c-Met receptor kinase and induces apoptosis in diffuse large B-cell lymphoma. Mol. Cancer Ther. 2010, 9, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Simeone, P.; Tacconi, S.; Longo, S.; Lanuti, P.; Bravaccini, S.; Pirini, F.; Ravaioli, S.; Dini, L.; Giudetti, A. Expanding Roles of De Novo Lipogenesis in Breast Cancer. Int. J. Environ. Res. Public Health 2021, 18, 3575. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Hua, W.; Huang, H.-Z.; Tan, L.-T.; Wan, J.-M.; Gui, H.-B.; Zhao, L.; Ruan, X.-Z.; Chen, X.-M.; Du, X.-G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef]
- Penzo, D.; Tagliapietra, C.; Colonna, R.; Petronilli, V.; Bernardi, P. Effects of fatty acids on mitochondria: Implications for cell death. Biochim. Biophys. Acta 2002, 1555, 160–165. [Google Scholar] [CrossRef]
- Kumar, B.; Kowluru, A.; Kowluru, R.A. Lipotoxicity augments glucotoxicity-induced mitochondrial damage in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2985–2992. [Google Scholar] [CrossRef]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Silvestri, S.; Orlando, P.; Mazibuko-Mbeje, S.E.; Johnson, R.; Marcheggiani, F.; Cirilli, I.; Muller, C.J.; Louw, J.; Chellan, N.; et al. Palmitate-induced toxicity is associated with impaired mitochondrial respiration and accelerated oxidative stress in cultured cardiomyocytes: The critical role of coenzyme Q9/10. Toxicol. In Vitro 2020, 68, 104948. [Google Scholar] [CrossRef] [PubMed]
- Itami, N.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Palmitic acid induces ceramide accumulation, mitochondrial protein hyperacetylation, and mitochondrial dysfunction in porcine oocytes. Biol. Reprod. 2018, 98, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Nam, S.M.; Kim, J.-H.; Das, R.; Choi, S.-K.; Nguyen, T.T.; Quan, X.; Chung, C.H.; Lee, E.Y.; Lee, I.-K.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schroder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.; Gorman, A.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Nivala, A.M.; Reese, L.; Frye, M.; Gentile, C.L.; Pagliassotti, M.J. Fatty acid-mediated endoplasmic reticulum stress in vivo: Differential response to the infusion of Soybean and Lard Oil in rats. Metabolism 2013, 62, 753–760. [Google Scholar] [CrossRef]
- Pardo, V.; González-Rodríguez, Á.; Muntané, J.; Kozma, S.C.; Valverde, Á.M. Role of hepatocyte S6K1 in palmitic acid-induced endoplasmic reticulum stress, lipotoxicity, insulin resistance and in oleic acid-induced protection. Food Chem. Toxicol. 2015, 80, 298–309. [Google Scholar] [CrossRef]
- Volmer, R.; Ron, D. Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol. 2015, 33, 67–73. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: Implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [PubMed]
- Schroder, M.; Clark, R.; Kaufman, R.J. IRE1- and HAC1-independent transcriptional regulation in the unfolded protein response of yeast. Mol. Microbiol. 2003, 49, 591–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haywood, J.; Yammani, R.R. Free fatty acid palmitate activates unfolded protein response pathway and promotes apoptosis in meniscus cells. Osteoarthr. Cartil. 2016, 24, 942–945. [Google Scholar] [CrossRef]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef]
- Rennert, C.; Heil, T.; Schicht, G.; Stilkerich, A.; Seidemann, L.; Kegel-Hübner, V.; Seehofer, D.; Damm, G. Prolonged Lipid Accumulation in Cultured Primary Human Hepatocytes Rather Leads to ER Stress than Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 7097. [Google Scholar] [CrossRef]
- Guo, W.; Wong, S.; Xie, W.; Lei, T.; Luo, Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E576–E586. [Google Scholar] [CrossRef]
- Anusornvongchai, T.; Nangaku, M.; Jao, T.-M.; Wu, C.-H.; Ishimoto, Y.; Maekawa, H.; Tanaka, T.; Shimizu, A.; Yamamoto, M.; Suzuki, N.; et al. Palmitate deranges erythropoietin production via transcription factor ATF4 activation of unfolded protein response. Kidney Int. 2018, 94, 536–550. [Google Scholar] [CrossRef]
- Cao, J.; Dai, D.L.; Yao, L.; Yu, H.H.; Ning, B.; Zhang, Q.; Chen, J.; Cheng, W.H.; Shen, W.; Yang, Z.X. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell. Biochem. 2012, 364, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Claycombe, K.; Schommer, J.; Collins, D.; Ghribi, O. Palmitate-induced Endoplasmic Reticulum stress and subsequent C/EBPalpha Homologous Protein activation attenuates leptin and Insulin-like growth factor 1 expression in the brain. Cell. Signal. 2016, 28, 1789–1805. [Google Scholar] [PubMed]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar]
- Doll, S.; Proneth, B.; Tyurina, Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, M.I.J.; Aichler, M.; Walch, M.A.A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Beatty, A.; Singh, T.; Tyurina, Y.Y.; Tyurin, V.A.; Samovich, S.; Nicolas, E.; Maslar, K.; Zhou, Y.; Cai, K.Q.; Tan, Y.; et al. Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat. Commun. 2021, 12, 2244. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [PubMed]
- Yin, J.; Wang, Y.; Gu, L.; Fan, N.; Ma, Y.; Peng, Y. Palmitate induces endoplasmic reticulum stress and autophagy in mature adipocytes: Implications for apoptosis and inflammation. Int. J. Mol. Med. 2015, 35, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Lee, S.M.; Lee, Y.J.; Li, L.J.; Lee, S.J.; Lee, J.H.; Kim, Y.; Jun, H.S.; Lee, K.W.; Kang, Y. Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology 2009, 150, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Sun, L.Q.; Wang, B.A.; Zou, X.M.; Mu, Y.M.; Lu, J.M. Palmitate induces autophagy in pancreatic beta-cells via endoplasmic reticulum stress and its downstream JNK pathway. Int. J. Mol. Med. 2013, 32, 1401–1406. [Google Scholar] [CrossRef]
- Komiya, K.; Uchida, T.; Ueno, T.; Koike, M.; Abe, H.; Hirose, T.; Kawamori, R.; Uchiyama, Y.; Kominami, E.; Fujitani, Y.; et al. Free fatty acids stimulate autophagy in pancreatic beta-cells via JNK pathway. Biochem. Biophys. Res. Commun. 2010, 401, 561–567. [Google Scholar] [CrossRef]
- Russo, S.B.; Baicu, C.F.; Van Laer, A.; Geng, T.; Kasiganesan, H.; Zile, M.R.; Cowart, L.A. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J. Clin. Investig. 2012, 122, 3919–3930. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Kimura, T.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.-Y.; Matsui, I.; Matsusaka, T.; et al. High-Fat Diet-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney. J. Am. Soc. Nephrol. 2017, 28, 1534–1551. [Google Scholar] [CrossRef]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.-C.; Kim, K.H.; Kim, G.H.; Kim, S.-W.; Kim, H.L.; Lee, M.-K.; et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef]
- Chen, P.; Liu, H.; Xiang, H.; Zhou, J.; Zeng, Z.; Chen, R.; Zhao, S.; Xiao, J.; Shu, Z.; Chen, S.; et al. Palmitic acid-induced autophagy increases reactive oxygen species via the Ca2+/PKCalpha/NOX4 pathway and impairs endothelial function in human umbilical vein endothelial cells. Exp. Ther. Med. 2019, 17, 2425–2432. [Google Scholar] [PubMed]
- Rezaei, M.; Handali, S.; Pourahmad, J. Mitochondria as Balancers of Reduction/Oxidation for Intracellular Environment. In Mitochondrial Metabolism; Pourahmad, J., Rezaei, M., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 1–17. [Google Scholar]
- Guerrero-Ros, I.; Clement, C.C.; Reynolds, C.A.; Patel, B.; Santambrogio, L.; Cuervo, A.M.; Macian, F. The negative effect of lipid challenge on autophagy inhibits T cell responses. Autophagy 2020, 16, 223–238. [Google Scholar] [CrossRef]
- Mir, S.U.; George, N.M.; Zahoor, L.; Harms, R.; Guinn, Z.; Sarvetnick, N.E. Inhibition of autophagic turnover in beta-cells by fatty acids and glucose leads to apoptotic cell death. J. Biol. Chem. 2015, 290, 6071–6085. [Google Scholar] [CrossRef]
- Ortiz-Rodriguez, A.; Acaz-Fonseca, E.; Boya, P.; Arevalo, M.A.; Garcia-Segura, L.M. Lipotoxic Effects of Palmitic Acid on Astrocytes Are Associated with Autophagy Impairment. Mol. Neurobiol. 2019, 56, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [PubMed] [Green Version]
- Cury-Boaventura, M.F.; Gorjão, R.; de Lima, T.M.; Newsholme, P.; Curi, R. Comparative toxicity of oleic and linoleic acid on human lymphocytes. Life Sci. 2006, 78, 1448–1456. [Google Scholar] [PubMed]
- Lima, T.M.; Lima, T.; Kanunfre, C.; Pompéia, C.; Verlengia, R.; Curi, R. Ranking the toxicity of fatty acids on Jurkat and Raji cells by flow cytometric analysis. Toxicol. In Vitro 2002, 16, 741–747. [Google Scholar] [CrossRef]
- Takahashi, H.K.; Cambiaghi, T.D.; Luchessi, A.D.; Hirabara, S.M.; Vinolo, M.A.R.; Newsholme, P.; Curi, R. Activation of survival and apoptotic signaling pathways in lymphocytes exposed to palmitic acid. J. Cell. Physiol. 2012, 227, 339–350. [Google Scholar] [CrossRef]
- Zurier, R.; Rossetti, R.; Seiler, C.; Laposata, M. Human peripheral blood T lymphocyte proliferation after activation of the T cell receptor: Effects of unsaturated fatty acids. Prostaglandins Leukot. Essent. Fat. Acids 1999, 60, 371–375. [Google Scholar] [CrossRef]
- O’Sullivan, D.; van der Windt, G.J.; Huang, S.C.-C.; Curtis, J.D.; Chang, C.-H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef]
- Howie, D.; Cobbold, S.P.; Adams, E.; Ten Bokum, A.; Necula, A.S.; Zhang, W.; Huang, H.; Roberts, D.J.; Thomas, B.; Hester, S.S.; et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight 2017, 2, e89160. [Google Scholar] [CrossRef] [PubMed]
- Samartsev, V.N.; Kozhina, O.V. Oxidative stress as regulatory factor for fatty-acid-induced uncoupling involving liver mitochondrial ADP/ATP and aspartate/glutamate antiporters of old rats. Biochemistry 2008, 73, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Patane, G.; Anello, M.; Piro, S.; Vigneri, R.; Purrello, F.; Rabuazzo, A.M. Role of ATP production and uncoupling protein-2 in the insulin secretory defect induced by chronic exposure to high glucose or free fatty acids and effects of peroxisome proliferator-activated receptor-gamma inhibition. Diabetes 2002, 51, 2749–2756. [Google Scholar] [CrossRef]
- Ma, S.; Yang, D.; Li, D.; Tan, Y.; Tang, B.; Yang, Y. Inhibition of uncoupling protein 2 with genipin exacerbates palmitate-induced hepatic steatosis. Lipids Health Dis. 2012, 11, 154. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, L.; Srivastava, R.K.; Kos, F.; Shrikant, P.A. Uncoupling protein 2 regulates metabolic reprogramming and fate of antigen-stimulated CD8+ T cells. Cancer Immunol. Immunother. 2016, 65, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumar, M.; Cheng, K.; Gangaplara, A.; Patel, Y.; Vodnala, S.K.; Islam, R.; Eidizadeh, A.; Subramaniam, C.; Lee, P.; Kishton, R.; et al. Abstract 1527: Integrated computational and experimental analysis identifies the mitochondrial uncoupling protein 2 (Ucp2) as a key regulator of T cell anti-tumor function. Cancer Res. 2021, 81, 1527. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; DeOliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef]
- Wu, X.-N.; Su, D.; Mei, Y.-D.; Xu, M.-Q.; Zhang, H.; Wang, Z.-Y.; Li, L.-L.; Peng, L.; Jiang, J.-Y.; Yang, J.-Y.; et al. Identified lung adenocarcinoma metabolic phenotypes and their association with tumor immune microenvironment. Cancer Immunol. Immunother. 2021, 70, 2835–2850. [Google Scholar] [PubMed]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 2021, 54, 1561–1577.e7. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.; Camara, S.; Shakiba, M.; Scott, A.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Staveley-O’Carroll, K.; Schell, T.D.; Jimenez, M.; Mylin, L.M.; Tevethia, M.J.; Schoenberger, S.P.; Tevethia, S.S. In vivo ligation of CD40 enhances priming against the endogenous tumor antigen and promotes CD8+ T cell effector function in SV40 T antigen transgenic mice. J. Immunol. 2003, 171, 697–707. [Google Scholar] [CrossRef]
- Xiao, L.; Ma, X.; Ye, L.; Su, P.; Xiong, W.; Bi, E.; Wang, Q.; Xian, M.; Yang, M.; Qian, J.; et al. IL-9/STAT3/fatty acid oxidation-mediated lipid peroxidation contributes to Tc9 cell longevity and enhanced antitumor activity. J. Clin. Investig. 2022, 132, e153247. [Google Scholar] [CrossRef]
- Matsushita, M.; Kopf, M.; Bornkamm, G.W.; Schneider, C.; Conrad, M.; Freigang, S.B. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef]
- Xu, C.; Sun, S.; Johnson, T.; Qi, R.; Zhang, S.; Zhang, J.; Yang, K. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021, 35, 109235. [Google Scholar] [CrossRef]
- Anderson, A.S.; Martin, R.M.; Renehan, A.G.; Cade, J.; Copson, E.R.; Cross, A.J.; Grimmett, C.; Keaver, L.; King, A.; Riboli, E.; et al. Cancer survivorship, excess body fatness and weight-loss intervention-where are we in 2020? Br. J. Cancer 2021, 124, 1057–1065. [Google Scholar] [CrossRef]
- Pancreatic Nutritional Program for Weight Loss in Overweight/Obese Patients with Stage I–III Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT02432950 (accessed on 8 July 2022).
- A Study of Ketogenic Diet in Newly Diagnosed Overweight or Obese Endometrial Cancer Patients. Available online: https://ClinicalTrials.gov/show/NCT03285152 (accessed on 8 July 2022).
- A Study of the Body’s Response to Exercise and a Plant-Based Diet in Overweight Postmenopausal Women with Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT04298086 (accessed on 8 July 2022).
- Breast Cancer Weight Loss Study (BWEL Study). Available online: https://ClinicalTrials.gov/show/NCT02750826 (accessed on 8 July 2022).
- Skuli, S.J.; Alomari, S.; Gaitsch, H.; Bakayoko, A.; Skuli, N.; Tyler, B.M. Metformin and Cancer, an Ambiguanidous Relationship. Pharmaceuticals 2022, 15, 626. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.-W.; He, X.-R.; Jin, W.-L.; He, X.-Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [PubMed]
- Chae, Y.K.; Trinh, L.; Jain, P.; Wang, X.; Rozovski, U.; Wierda, W.G.; Keating, M.J.; Estrov, Z. Statin and aspirin use is associated with improved outcome of FCR therapy in relapsed/refractory chronic lymphocytic leukemia. Blood 2014, 123, 1424–1426. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.R.; Magura, L.A.; Warren, H.A.C.; Harrison, J.D.; Diehl, L.F.; Weinberg, J.B. Statin use and need for therapy in chronic lymphocytic leukemia. Leuk. Lymphoma 2010, 51, 2295–2298. [Google Scholar] [CrossRef]
- Tilija Pun, N.; Jeong, C.H. Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance. Pharmaceuticals 2021, 14, 470. [Google Scholar] [CrossRef]
- Castellanos-Esparza, Y.C.; Wu, S.; Huang, L.; Buquet, C.; Shen, R.; Sanchez-Gonzalez, B.; Latorre, E.A.G.; Boyer, O.; Varin, R.; Jimenez-Zamudio, L.A.; et al. Synergistic promoting effects of pentoxifylline and simvastatin on the apoptosis of triple-negative MDA-MB-231 breast cancer cells. Int. J. Oncol. 2018, 52, 1246–1254. [Google Scholar] [CrossRef]
- Lee, J.S.; Roberts, A.; Juarez, D.; Vo, T.-T.; Bhatt, S.; Herzog, L.-O.; Mallya, S.; Bellin, R.J.; Agarwal, S.K.; Salem, A.H.; et al. Statins enhance efficacy of venetoclax in blood cancers. Sci. Transl. Med. 2018, 10, eaaq1240. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Ferrajoli, A.; Burger, J.; Thompson, P.; Jain, N.; Wierda, W.; et al. Ibrutinib inhibits free fatty acid metabolism in chronic lymphocytic leukemia. Leuk. Lymphoma 2018, 59, 2686–2691. [Google Scholar] [CrossRef]
- Shen, S.; Faouzi, S.; Souquere, S.; Roy, S.; Routier, E.; Libenciuc, C.; André, F.; Pierron, G.; Scoazec, J.-Y.; Robert, C. Melanoma Persister Cells Are Tolerant to BRAF/MEK Inhibitors via ACOX1-Mediated Fatty Acid Oxidation. Cell Rep. 2020, 33, 108421. [Google Scholar] [CrossRef]
- Enciu, A.M.; Radu, E.; Popescu, I.D.; Hinescu, M.E.; Ceafalan, L.C. Targeting CD36 as Biomarker for Metastasis Prognostic: How Far from Translation into Clinical Practice? Biomed. Res. Int. 2018, 2018, 7801202. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Han, H.; Liu, L.; Duan, Y.; Yang, X.; Ma, C.; Zhu, Y.; Han, J.; Li, X.; Chen, Y. CD36 plays a critical role in proliferation, migration and tamoxifen-inhibited growth of ER-positive breast cancer cells. Oncogenesis 2018, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Wilkins, O.; Bang, S.; Ung, M.; Li, J.; An, J.; Del Genio, C.; Canfield, K.; DiRenzo, J.; Wells, W.; et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep. 2019, 29, 3405–3420.e5. [Google Scholar] [CrossRef] [PubMed]
- Landberg, N.; Von Palffy, S.; Askmyr, M.; Lilljebjörn, H.; Sandén, C.; Rissler, M.; Mustjoki, S.; Hjorth-Hansen, H.; Richter, J.; Ågerstam, H.; et al. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica 2018, 103, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. A drug to block fat intake and combat cancer spread. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
- Horton, B.L.; Spranger, S. CD36-the Achilles’ heel of Treg cells. Nat. Immunol. 2020, 21, 251–253. [Google Scholar] [CrossRef]
- Stokes, W.A.; Behera, M.; Jiang, R.; Gutman, D.A.; Huang, Z.; Burns, A.; Sebastian, N.T.; Sukhatme, V.; Lowe, M.C.; Ramalingam, S.S.; et al. Impact of concomitant fibrates on immunotherapy outcomes for advanced non-small cell lung cancer. Cancer Med. 2022. [Google Scholar] [CrossRef]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef]
- Kim, D.; Wu, Y.; Li, Q.; Oh, Y.-K. Nanoparticle-Mediated Lipid Metabolic Reprogramming of T Cells in Tumor Microenvironments for Immunometabolic Therapy. Nanomicro Lett. 2021, 13, 31. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böttcher-Loschinski, R.; Rial Saborido, J.; Böttcher, M.; Kahlfuss, S.; Mougiakakos, D. Lipotoxicity as a Barrier for T Cell-Based Therapies. Biomolecules 2022, 12, 1182. https://doi.org/10.3390/biom12091182
Böttcher-Loschinski R, Rial Saborido J, Böttcher M, Kahlfuss S, Mougiakakos D. Lipotoxicity as a Barrier for T Cell-Based Therapies. Biomolecules. 2022; 12(9):1182. https://doi.org/10.3390/biom12091182
Chicago/Turabian StyleBöttcher-Loschinski, Romy, Judit Rial Saborido, Martin Böttcher, Sascha Kahlfuss, and Dimitrios Mougiakakos. 2022. "Lipotoxicity as a Barrier for T Cell-Based Therapies" Biomolecules 12, no. 9: 1182. https://doi.org/10.3390/biom12091182
APA StyleBöttcher-Loschinski, R., Rial Saborido, J., Böttcher, M., Kahlfuss, S., & Mougiakakos, D. (2022). Lipotoxicity as a Barrier for T Cell-Based Therapies. Biomolecules, 12(9), 1182. https://doi.org/10.3390/biom12091182