
Targeting the Ubiquitylation and ISGylation Machinery for the Treatment of COVID-19

 , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
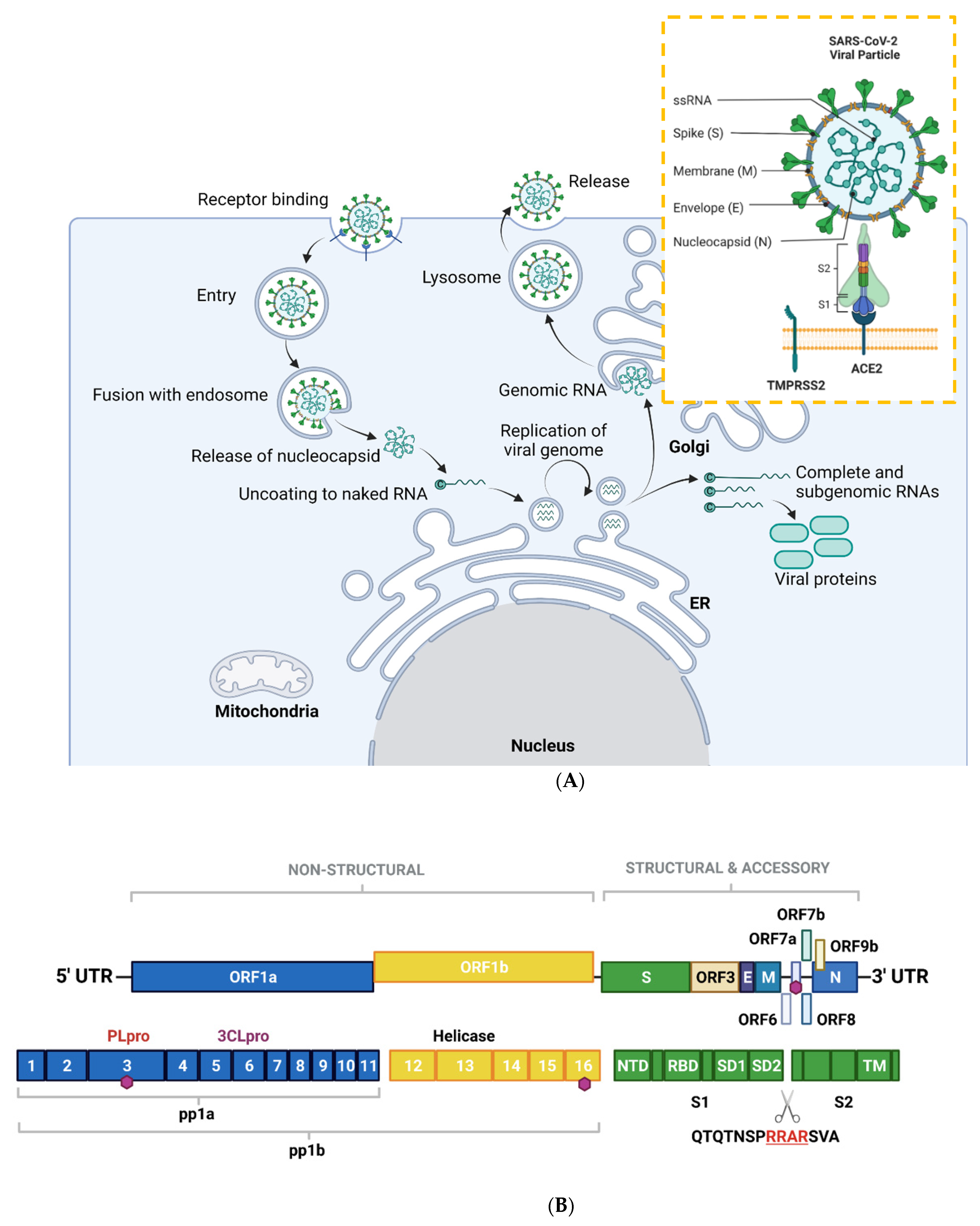
2. SARS-CoV-2-General Biology and Mechanisms of Infection
2.1. SARS-CoV-2 Viral Genome Architecture
2.2. SARS-CoV-2 Mode of Entry and Proliferation
2.3. Clinical Manifestations of COVID-19
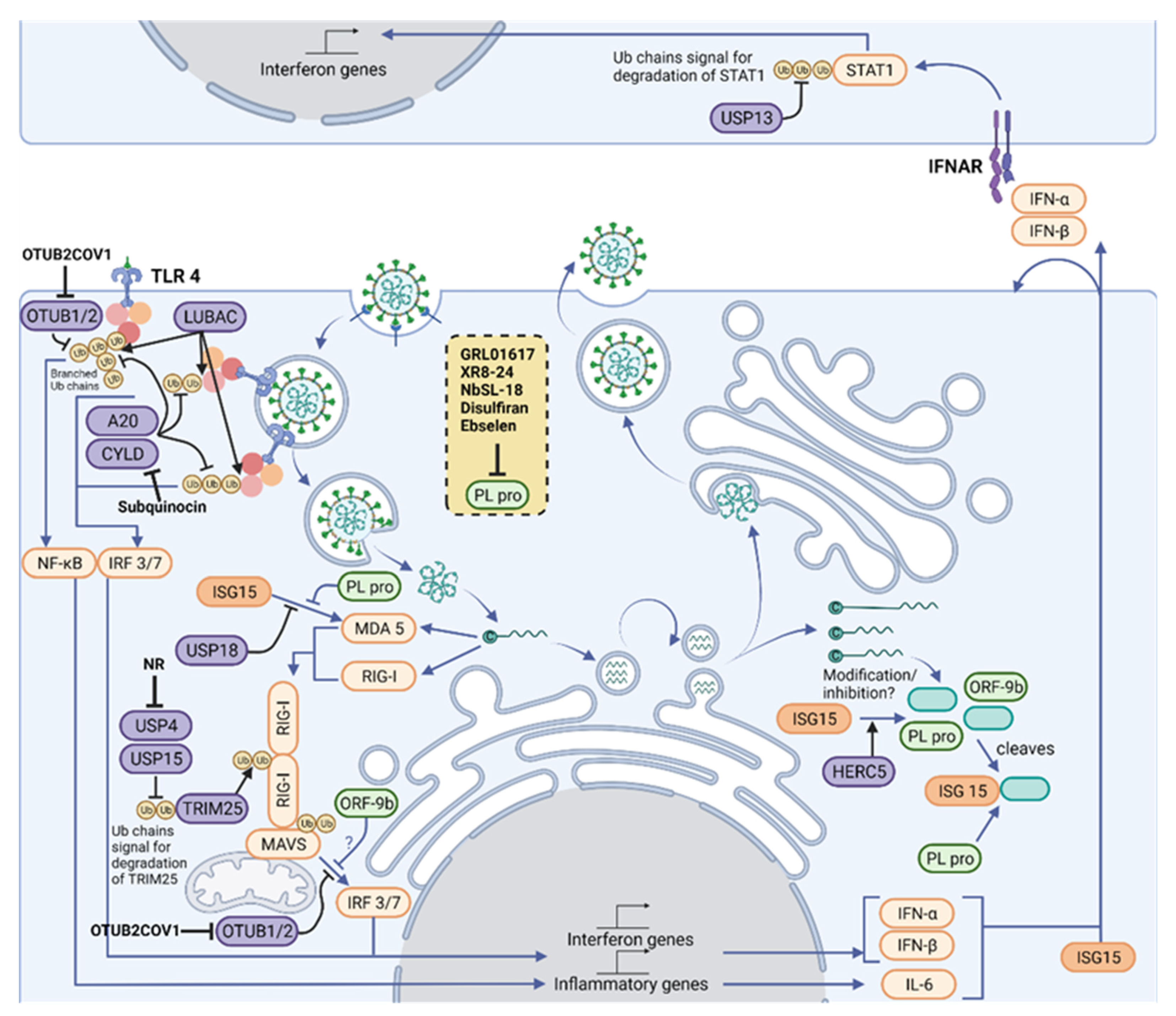
3. Ubiquitylation, ISGylation and Their Roles in Human Antiviral Responses
3.1. Ubiquitylation
3.2. Ubiquitin in Innate Immune Sensing Pathways
3.3. TLR Signaling
3.4. RLR Signaling
3.5. Ubls in Antiviral Defense: ISGylation
3.6. Viral Modulation of Ubiquitin and ISG15 Signals
4. Therapeutics-the Ub and ISG15 Machinery as a Target for COVID Treatment Regimes
4.1. Host Proteases
4.2. Viral Proteases
4.2.1. PLpro inhibitors
4.2.2. CLpro/Mpro Inhibitors
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Zhong, N.S.; Zheng, B.J.; Li, Y.M.; Poon; Xie, Z.H.; Chan, K.H.; Li, P.H.; Tan, S.Y.; Chang, Q.; Xie, J.P.; et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020 19:3 2020, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Andreata-Santos, R.; Janini, L.M.R.; Duraes-Carvalho, R. From Alpha to Omicron SARS-CoV-2 variants: What their evolutionary signatures can tell us? J. Med. Virol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Mielech, A.M.; Chen, Y.; Mesecar, A.D.; Baker, S.C. Nidovirus papain-like proteases: Multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 2014, 194, 184–190. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020 5:1 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Follis, K.E.; York, J.; Nunberg, J.H. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 2006, 350, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, S.; Liu, J.; Zhang, Z.; Wan, X.; Huang, B.; Chen, Y.; Zhang, Y. COVID-19: Immunopathogenesis and Immunotherapeutics. Signal Transduct. Target. Ther. 2020, 5, 1–8. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, S.; Haendler, B. The Ubiquitin System in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of Proteins by Ubiquitin and Ubiquitin-Like Proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clague, M.J.; Heride, C.; Urb, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtake, F.; Tsuchiya, H.; Tanaka, K.; Saeki, Y. Methods to measure ubiquitin chain length and linkage. Methods Enzymol. 2019, 618, 105–133. [Google Scholar] [CrossRef]
- Swatek, K.N.; Usher, J.L.; Kueck, A.F.; Gladkova, C.; Mevissen, T.E.T.; Pruneda, J.N.; Skern, T.; Komander, D. Insights into ubiquitin chain architecture using Ub-clipping. Nature 2019, 572, 533–537. [Google Scholar] [CrossRef]
- Heaton, S.M.; Borg, N.A.; Dixit, V.M. Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 2016, 213, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Paludan, S.R.; Mogensen, T.H. Innate immunological pathways in COVID-19 pathogenesis. Sci. Immunol. 2022, 7, eabm5505. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, J.-J.; Yoo, J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, L.; Fossali, T.; Frangipane, V.; Bozzini, S.; Morosini, M.; D’Amato, M.; Lettieri, S.; Urtis, M.A. Broncho-alveolar inflammation in COVID-19 patients: A correlation with clinical outcome. BMC BMC Pulm. Med. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, R.S.; Sanchez Sevilla Uruchurtu, A.; Siggins, M.K.; Liew, F.; Russell, C.D.; Moore, S.C.; Fairfield, C.; Carter, E.; Abrams, S.; Short, C.E.; et al. Inflammatory profiles across the spectrum of disease reveal a distinct role for GM-CSF in severe COVID-19. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Lester, S.; Li, K. Toll-Like Receptors in Antiviral Innate Immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Balka, K.R.a. Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR4. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-d. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Moynagh, P.N. The roles of Pellino E3 ubiquitin ligases in immunity. Nat. Rev. Immunol. 2014, 14, 122–131. [Google Scholar] [CrossRef]
- Enesa, K.; Ordureau, A.; Smith, H.; Barford, D.; Cheung, P.C.F.; Patterson-Kane, J.; Arthur, J.S.C.; Cohen, P. Pellino1 is required for interferon production by viral double-stranded RNA. J. Biol. Chem. 2012, 287, 34825–34835. [Google Scholar] [CrossRef] [Green Version]
- Zinngrebe, J.; Rieser, E.; Taraborrelli, L.; Peltzer, N.; Hartwig, T.; Ren, H.; Kovcs, I.; Endres, C.; Draber, P.; Darding, M.a. LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J. Exp. Med. 2016, 213, 2671–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, I.; Almazn, F.; Ziga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [Green Version]
- van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, Z.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Bigio, B.; Yang, R.; Arias, A.A.; Zhou, Q.; Han, J.E.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bortolotti, D.; Gentili, V.; Rizzo, S.; Schiuma, G.; Beltrami, S.; Strazzabosco, G.; Fernandez, M.; Caccuri, F.; Caruso, A.; Rizzo, R. TLR3 and TLR7 RNA Sensor Activation during SARS-CoV-2 Infection. Microorganisms 2021, 9, 1820. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Zhang, X. Murine Coronavirus Induces Type I Interferon in Oligodendrocytes through Recognition by RIG-I and MDA5. J. Virol. 2010, 84, 6472–6482. [Google Scholar] [CrossRef] [Green Version]
- Zalinger, Z.B.; Elliott, R.; Rose, K.M.; Weiss, S.R. MDA5 Is Critical to Host Defense during Infection with Murine Coronavirus. J. Virol. 2015, 89, 12330–12340. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Peisley, A.; Wu, B.; Xu, H.; Chen, Z.J.; Hur, S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 2014, 509, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Yan, J.; Li, Q.; Mao, A.P.; Hu, M.M.; Shu, H.B. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell Biol. 2014, 6, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Song, Y.; Li, Y.; Zhu, Q.; Tan, P.; Qin, Y.; Wang, H.Y.; Wang, R.-f. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Nat. Publ. Group 2014, 24, 400–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Crdenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The ubiquitin ligase riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [Green Version]
- Pauli, E.K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, W.; Zhang, M.; Wang, P.; Zhao, K.; Zhao, X.; Yang, S.; Gao, C. USP4 Positively Regulates RIG-I-Mediated Antiviral Response through Deubiquitination and Stabilization of RIG-I. J. Virol. 2013, 87, 4507–4515. [Google Scholar] [CrossRef] [Green Version]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef]
- Liu, G.; Lee, J.-h.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; Gent, M.V.; Riedl, W.; Davis-gardner, M.E.; Wies, E.; Chiang, C.; et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef]
- Rebendenne, A.a. SARS-CoV-2 Triggers an MDA-5-Dependent Interferon Response Which Is Unable To Control Replication in Lung Epithelial Cells. J. Virol. 2021, 95, e02415-20. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, N.G.; Chauveau, L.; Hertzog, J.; Bridgeman, A.; Fowler, G.; Moonen, J.P.; Dupont, M.; Russell, R.A.; Noerenberg, M.; Rehwinkel, J. The RNA sensor MDA5 detects SARS - CoV - 2 infection. Sci. Rep. 2021, 1–10. [Google Scholar] [CrossRef]
- Thorne, L.G.; Reuschl, A.-k.; Zuliani-alvarez, L.; Whelan, M.V.X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G.J. SARS-CoV-2 sensing by RIG-I and MDA 5 links epithelial infection to macrophage inflammation. EMBO J. 2021, 40, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.a. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Rep. 2021, 34. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Sato, S.; Sotoyama, Y.; Orba, Y.; Sawa, H.; Yamauchi, H.; Sasaki, M.; Takaoka, A. RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nat. Immunol. 2021, 22, 820–828. [Google Scholar] [CrossRef]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidre, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L.; et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021, 34, 108761. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.; Liebau, E. The Ufm1 Cascade. Cells 2014, 3, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kalveram, B.; Raasi, S.; Groettrup, M.; Schmidtke, G. FAT10, a Ubiquitin-Independent Signal for Proteasomal Degradation. Mol. Cell. Biol. 2005, 25, 3483–3491. [Google Scholar] [CrossRef] [Green Version]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Wong, J.J.; Pung, Y.F.; Sze, N.S.; Chin, K.C. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. USA 2006, 103, 10735–10740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketscher, L.; Basters, A.; Prinz, M.; Knobeloch, K.P. mHERC6 is the essential ISG15 E3 ligase in the murine system. Biochem. Biophys. Res. Commun. 2012, 417, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Okumura, A.J.; Uematsu, K.; Hatakeyama, S.; Zhang, D.E.; Kamura, T. Activation of double-stranded rna-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 2013, 288, 2839–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Zhong, G.; Zhu, L.; Liu, X.; Shan, Y.; Feng, H.; Bu, Z.; Chen, H.; Wang, C. Herc5 Attenuates Influenza A Virus by Catalyzing ISGylation of Viral NS1 Protein. J. Immunol. 2010, 184, 5777–5790. [Google Scholar] [CrossRef] [Green Version]
- Skaug, B.; Chen, Z.J. Emerging Role of ISG15 in Antiviral Immunity. Cell 2010, 143, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Munnur, D.; Teo, Q.; Eggermont, D.; Lee, H.H.Y.; Thery, F.; Ho, J.; Leur, S.W.V.; Ng, W.W.S.; Siu, L.Y.L.; Beling, A.; et al. Altered ISGylation drives aberrant macrophage-dependent immune responses during SARS-CoV-2 infection. Nat. Immunol. 2021. [Google Scholar] [CrossRef]
- Swaim, C.D.; Scott, A.F.; Canadeo, L.A.; Huibregtse, J.M. Extracellular ISG15 Signals Cytokine Secretion through the LFA-1 Integrin Receptor. Mol. Cell 2017, 68, 581–590.e5. [Google Scholar] [CrossRef] [Green Version]
- Kikkert, M. Innate Immune Evasion by Human Respiratory RNA Viruses. J. Innate. Immun. 2020, 12, 4–20. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Vere, G.; Kealy, R.; Kessler, B.M.; Pinto-Fernandez, A. Ubiquitomics: An overview and future. Biomolecules 2020, 10, 1453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bouch, R.J.; Blekhman, M.G.; He, Z. USP19 Suppresses Th17-Driven Pathogenesis in Autoimmunity. J. Immunol. 2021, 207, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Bez-Santos, Y.M.a. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Frias-Staheli, N.; Giannakopoulos, N.V.; Kikkert, M.; Taylor, S.L.; Bridgen, A.; Paragas, J.; Richt, J.A.; Rowland, R.R.; Schmaljohn, C.S.; Lenschow, D.J.; et al. Ovarian Tumor Domain-Containing Viral Proteases Evade Ubiquitin- and ISG15-Dependent Innate Immune Responses. Cell Host Microbe 2007, 2, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Swatek, K.N.; Aumayr, M.; Pruneda, J.N.; Visser, L.J.; Berryman, S.; Kueck, A.F.; Geurink, P.P.; Ovaa, H.; van Kuppeveld, F.J.M.; Tuthill, T.J.; et al. Irreversible inactivation of ISG15 by a viral leader protease enables alternative infection detection strategies. Proc. Natl. Acad. Sci. USA 2018, 115, 2371–2376. [Google Scholar] [CrossRef] [Green Version]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.C.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, 1–17. [Google Scholar] [CrossRef]
- Lindner, H.A.; Fotouhi-Ardakani, N.; Lytvyn, V.; Lachance, P.; Sulea, T.; Mnard, R. The Papain-Like Protease from the Severe Acute Respiratory Syndrome Coronavirus Is a Deubiquitinating Enzyme. J. Virol. 2005, 79, 15199–15208. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef]
- Leite, W.C.; Weiss, K.L.; Phillips, G.; Zhang, Q.; Qian, S.; Tsutakawa, S.E.; Coates, L.; O’Neill, H. Conformational Dynamics in the Interaction of SARS-CoV-2 Papain-like Protease with Human Interferon-Stimulated Gene 15 Protein. J. Phys. Chem. Lett. 2021, 12, 5608–5615. [Google Scholar] [CrossRef]
- Yan, S.; Ahmad, R.; Rosen, E.D.; Yan, S.; Kumari, M.; Xiao, H.; Jacobs, C.; Kochumon, S.; Jedrychowski, M.; Chouchani, E.; et al. IRF3 reduces adipose thermogenesis via ISG15- mediated reprogramming of glycolysis. J. Phys. Chem. Lett. 2021, 131. [Google Scholar] [CrossRef]
- Zhang, Y.; Thery, F.; Wu, N.C.; Luhmann, E.K.; Dussurget, O.; Foecke, M.; Bredow, C.; Jimnez-Fernndez, D.; Leandro, K.; Beling, A.; et al. The in vivo ISGylome links ISG15 to metabolic pathways and autophagy upon Listeria monocytogenes infection. Nat. Commun. 2019, 10, 5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxana, A.M.; Keun Il, K.; Jiann Kae, L.; Weiguo, Z.; Kumar, K.G.S.; Serge, Y.F.; Ke, S.; Dong Er, Z. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef]
- Eleanor, Q.; Hasan, T.; Poornima, K.; Robert, H.; Rajeev, J. Treatment of COVID-19: A review of current and prospective pharmacotherapies. Br. J. Hosp. Med. 2021, 82, 50. [Google Scholar] [CrossRef]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. 2021, 6, 255. [Google Scholar] [CrossRef] [PubMed]
- Daniel, B.-M.; Benjamin, E.N.-P.; Wen Chun, L.; Skyler, U.; Daisy, H.; Rasmus, M.; Tristan, X.J.; Kohei, O.; Maryline, P.; David, S.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef]
- Jérôme, H.; Nader, Y.; Laura, B.; Aurélien, C.; Jeremy, B.; Nikaïa, S.; Hélène, P.; Bruno, C.; Vincent, B.; Camille, C.-G.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Yamanaka, S.; Sato, Y.; Oikawa, D.; Goto, E.; Fukai, S.; Tokunaga, F.; Takahashi, H.; Sawasaki, T. Subquinocin, a small molecule inhibitor of CYLD and USP-family deubiquitinating enzymes, promotes NF-kappaB signaling. Biochem. Biophys. Res. Commun. 2020, 524, 1–7. [Google Scholar] [CrossRef]
- Resnick, E.; Bradley, A.; Gan, J.; Douangamath, A.; Krojer, T.; Sethi, R.; Geurink, P.P.; Aimon, A.; Amitai, G.; Bellini, D.; et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.H.; Kim, T.; Nguyen, T.; Hahn, M.J.; Yun, S.I.; Kim, K.K. A Selective Inhibitor of Ubiquitin-Specific Protease 4 Suppresses Colorectal Cancer Progression by Regulating beta-Catenin Signaling. Cell Physiol. Biochem. 2019, 53, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Daniel Jiménez, F.; Sandra, H.; Klaus Peter, K. Strategies to Target ISG15 and USP18 Toward Therapeutic Applications. Front. Chem. 2020, 7, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiaodong, H.; Vishva, M.D. Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 2016, 26, 484–498. [Google Scholar] [CrossRef]
- Zhenyi, H.; Craig, M.C. Recent Developments in PROTAC-Mediated Protein Degradation: From Bench to Clinic. ChemBioChem 2021. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, H.; Zhu, J.; Dong, Q.; Wang, J.; Fan, H.; Chen, Y.; Zhang, X.; Han, X.; Li, Q.; et al. Ubiquitin-Modified Proteome of SARS-CoV-2-Infected Host Cells Reveals Insights into Virus-Host Interaction and Pathogenesis. J. Proteome Res. 2021, 20, 2224–2239. [Google Scholar] [CrossRef] [PubMed]
- Hom-Ming, Y.; Chia-Yi, Y.; Ho-Chun, Y.; Shih-Han, K.; Ching-Len, L.; Yi-Ling, L. Ubiquitin-Specific Protease 13 Regulates IFN Signaling by Stabilizing STAT1. J. Immunol. 2013, 191, 3328–3336. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, D.; Zhong, H.; Luo, R.; Shang, M.; Liu, D.; Chen, H.; Fang, L.; Xiao, S. Ubiquitin-specific Protease 15 Negatively Regulates Virus-induced Type I Interferon Signaling via Catalytically-dependent and -independent Mechanisms. Sci. Rep. 2015, 5, 11220. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Wang, D.; Fang, L.; Zhang, H.; Luo, R.; Shang, M.; Ouyang, C.; Ouyang, H.; Chen, H.; Xiao, S. Ubiquitin-specific proteases 25 negatively regulates virus-induced type I interferon signaling. PLoS ONE 2013, 8, e80976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hylemariam Mihiretie, M.; Tebelay, D.; Tengchuan, J. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 7. [Google Scholar] [CrossRef]
- Anja, B.; Paul, P.G.; Farid El, O.; Lars, K.; Marco, S.C.; Eberhard, K.; Huib, O.; Klaus Peter, K.; Günter, F. Molecular characterization of ubiquitin-specific protease 18 reveals substrate specificity for interferon-stimulated gene 15. FEBS J. 2014, 281, 1918–1928. [Google Scholar] [CrossRef]
- Brendan, T.F.; Ian, A.D.; Jackelyn, M.; Jaron, E.L.; Holden, C.M.; David, C.; Robert Jeff, H.; Ralph, A.T.; Scott, D.P. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Mostafa, J.; Ebrahim, B.; Fathollah, G.-B. Structure-Based Screening to Discover New Inhibitors for Papain-like Proteinase of SARS-CoV-2: An in Silico Study. J. Proteome Res. 2021, 20, 1015–1026. [Google Scholar] [CrossRef]
- Shuvasish, C.; Debojyoti, M.; Anupom, B.; Purbajyoti, S.; Muhammed Khairujjaman, M. In search of drugs to alleviate suppression of the host’s innate immune responses against SARS-CoV-2 using a molecular modeling approach. Silico Pharmacol. 2021, 9. [Google Scholar] [CrossRef]
- Jung Keun, C.; Marcus, J.C.-L.; Kon Ho, L.; Dae Wook, K.; Hyung Won, R.; Heung Joo, Y.; Ki Hun, P. Geranylated flavonoids displaying SARS-CoV papain-like protease inhibition from the fruits of Paulownia tomentosa. Bioorganic Med. Chem. 2013, 21, 3051–3057. [Google Scholar] [CrossRef]
- Rajesh, G.; Ayon, C.; Ashis, B.; Snehasis, C. Identification of polyphenols from Broussonetia papyrifera as SARS CoV-2 main protease inhibitors using in silico docking and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2021, 39, 6747–6760. [Google Scholar] [CrossRef]
- Lingyu, L.; Liyan, M.; Yue, H.; Xiaoxue, L.; Meng, Y.; Hai, S.; Zhongmei, Z. Natural biflavones are potent inhibitors against SARS-CoV-2 papain-like protease. Phytochemistry 2021, 193, 112984. [Google Scholar] [CrossRef]
- Chung Yi, W.; Jia Tsrong, J.; Shiou Hwa, M.; Chin Jung, K.; Hsueh Fen, J.; Yih Shyun, E.C.; Hsien Hua, H.; Hsuan Cheng, H.; Douglass, W.; Ashraf, B.; et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [Google Scholar] [CrossRef] [Green Version]
- Zinuo, C.; Qinghua, C.; Laura, C.; Pin, Z.; Hyun, L.; Zhaoyu, C.; Yanyan, W.; Xiaoyun, L.; Lijun, R.; Ruikun, D. Ginkgolic acid and anacardic acid are specific covalent inhibitors of SARS-CoV-2 cysteine proteases. Cell Biosci. 2021, 11. [Google Scholar] [CrossRef]
- Naveen, N.; Deepak, T.N. Vitamin B12 may inhibit RNA-dependent-RNA polymerase activity of nsp12 from the SARS-CoV-2 virus. IUBMB life 2020, 72, 2112–2120. [Google Scholar] [CrossRef]
- Sheng Teng, H.; Yeh, C.; Wei Chao, C.; Hsiao Fan, C.; Hsiang Chun, L.; Yu Chun, L.; Wei Jan, W.; Yu Chuan, W.; Chia Shin, Y.; Shao Chun, W.; et al. Scutellaria barbata d. Don inhibits the main proteases (mpro and tmprss2) of severe acute respiratory syndrome coronavirus 2 (sars-cov-2) infection. Viruses 2021, 13, 826. [Google Scholar] [CrossRef]
- Lee, A.A.; Sven, M.L.; Virginia Dee, C.; Stephen, P.M.; Raja Sekhar, N.; Isobel, C.; Anthony, H.; Fraser, C.; Rachel, T.; Rukmini, M.; et al. Biochemical characterization of protease activity of Nsp3 from SARS-CoV-2 and its inhibition by nanobodies. PLoS ONE 2021, 16, e0253364. [Google Scholar] [CrossRef]
- Zhengnan, S.; Kiira, R.; Laura, C.; Deyu, K.; Hyun, L.; Youngjin, K.; Yangfeng, L.; Saad, A.; Fei, H.; Oleksii, D.; et al. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Narayanan, A.; Toner, S.A.; Jose, J. Structure-based inhibitor design and repurposing clinical drugs to target SARS-CoV-2 proteases. Biochem. Soc. Trans. 2022. [Google Scholar] [CrossRef]
- Dafydd, R.O.; Charlotte, M.N.A.; Annaliesa, S.A.; Lisa, A.; Melissa, A.; Simon, B.; Britton, B.; Rhonda, D.C.; Anthony, C.; Karen, J.C.; et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Robert, L.H.; Robert, S.K.; Mary, A.B.; Jay, F.D.; Rose, A.F.; Ketan, S.G.; Mingying, H.; Robert, J.H.; Kirk, K.; Lilian, Y.L.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Linlin, Z.; Daizong, L.; Xinyuanyuan, S.; Ute, C.; Christian, D.; Lucie, S.; Stephan, B.; Katharina, R.; Rolf, H. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Britton, B.; Rhys, M.J.; Brandon, J.A.; Dan, A.; Lisa, A.; Malina, A.B.; Nathan, B.; Joseph, B.; Emily, C.; Heather, E.; et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Temitope, I.A.; Misbaudeen, A.-H.; Emmanuel, M.O.; Qudus, K.O.; Ibrahim, D.B.; Ibrahim, O.A.; Olamide, T.O.; Ajayi, A.F.; Oladipo, E.K. Molecular docking assessment of clinically approved antiviral drugs against Mpro, spike glycoprotein and angiotensin converting enzyme-2 revealed probable anti-SARS-CoV-2 potential. Trop. J. Nat. Prod. Res. 2021, 5, 778–791. [Google Scholar] [CrossRef]
- Chunlong, M.; Michael Dominic, S.; Brett, H.; Julia Alma, T.; Yanmei, H.; Tommy, S.; Xiujun, Z.; Bart, T.; Michael Thomas, M.; Yu, C.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Dale, L.B.; Valerie, D.H.; Jared, B.; Donald, F.S.; John, D.M.; Michael, J.O.; Robert, W.S. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and beta-D-N4-hydroxycytidine. Antivir. Chem. Chemother. 2004, 15, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Bin, C.; Yeming, W.; Danning, W.; Wen, L.; Jingli, W.; Guohui, F.; Lianguo, R.; Bin, S.; Yanping, C.; Ming, W.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Peter, W.H.; Marion, M.; Jennifer, L.B.; Louise, L.; Natalie, S.; Jonathan, E.; Adrian, P.; Jason, R.; Einas, E.; Benjamin, P.; et al. Lopinavir–ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2020, 396, 1345–1352. [Google Scholar] [CrossRef]
- Terracciano, R.; Preiano, M.; Fregola, A.; Pelaia, C.; Montalcini, T.; Savino, R. Mapping the SARS-CoV-2-Host Protein-Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics. Int. J. Mol. Sci. 2021, 22, 532. [Google Scholar] [CrossRef]
- Schiller, H.B.; van Breugel, M.; Nawijn, M.C. SARS-CoV-2-specific hotspots in virus-host interaction networks. Nat. Immunol. 2021, 22, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [Green Version]
- Cannalire, R.; Barreca, M.L.; Manfroni, G.; Cecchetti, V. A Journey around the Medicinal Chemistry of Hepatitis C Virus Inhibitors Targeting NS4B: From Target to Preclinical Drug Candidates. J. Med. Chem. 2016, 59, 16–41. [Google Scholar] [CrossRef]
- Arimoto, K.I.; Lochte, S.; Stoner, S.A.; Burkart, C.; Zhang, Y.; Miyauchi, S.; Wilmes, S.; Fan, J.B.; Heinisch, J.J.; Li, Z.; et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat. Struct. Mol. Biol. 2017, 24, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, S.; Berset, C.; La Rue, J.; Friedman, K.; Nakayama, K.; Nakayama, K.; Zhang, D.E.; Lanker, S. The ISG15 isopeptidase UBP43 is regulated by proteolysis via the SCFSkp2 ubiquitin ligase. J. Biol. Chem. 2004, 279, 46424–46430. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.L.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef]
- Pinto-Fernandez, A.; Kessler, B.M. DUBbing Cancer: Deubiquitylating Enzymes Involved in Epigenetics, DNA Damage and the Cell Cycle As Therapeutic Targets. Front. Genet. 2016, 7, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, M.E.; Schot, R.; Buta, S.; Oudesluijs, G.; Tinschert, S.; Speer, S.D.; Li, Z.; van Unen, L.; Heijsman, D.; Goldmann, T.; et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 2016, 213, 1163–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, C.; Martin-Fernandez, M.; Ailal, F.; Qiu, X.; Taft, J.; Altman, J.; Rosain, J.; Buta, S.; Bousfiha, A.; Casanova, J.L.; et al. Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Conroy, M.; Naidoo, J. Immune-related adverse events and the balancing act of immunotherapy. Nat. Commun. 2022, 13, 392. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vere, G.; Alam, M.R.; Farrar, S.; Kealy, R.; Kessler, B.M.; O’Brien, D.P.; Pinto-Fernández, A. Targeting the Ubiquitylation and ISGylation Machinery for the Treatment of COVID-19. Biomolecules 2022, 12, 300. https://doi.org/10.3390/biom12020300
Vere G, Alam MR, Farrar S, Kealy R, Kessler BM, O’Brien DP, Pinto-Fernández A. Targeting the Ubiquitylation and ISGylation Machinery for the Treatment of COVID-19. Biomolecules. 2022; 12(2):300. https://doi.org/10.3390/biom12020300
Chicago/Turabian StyleVere, George, Md Rashadul Alam, Sam Farrar, Rachel Kealy, Benedikt M. Kessler, Darragh P. O’Brien, and Adán Pinto-Fernández. 2022. "Targeting the Ubiquitylation and ISGylation Machinery for the Treatment of COVID-19" Biomolecules 12, no. 2: 300. https://doi.org/10.3390/biom12020300
APA StyleVere, G., Alam, M. R., Farrar, S., Kealy, R., Kessler, B. M., O’Brien, D. P., & Pinto-Fernández, A. (2022). Targeting the Ubiquitylation and ISGylation Machinery for the Treatment of COVID-19. Biomolecules, 12(2), 300. https://doi.org/10.3390/biom12020300