
Roles of Cullin-RING Ubiquitin Ligases in Cardiovascular Diseases


{kind=link}
{kind=link}
Abstract
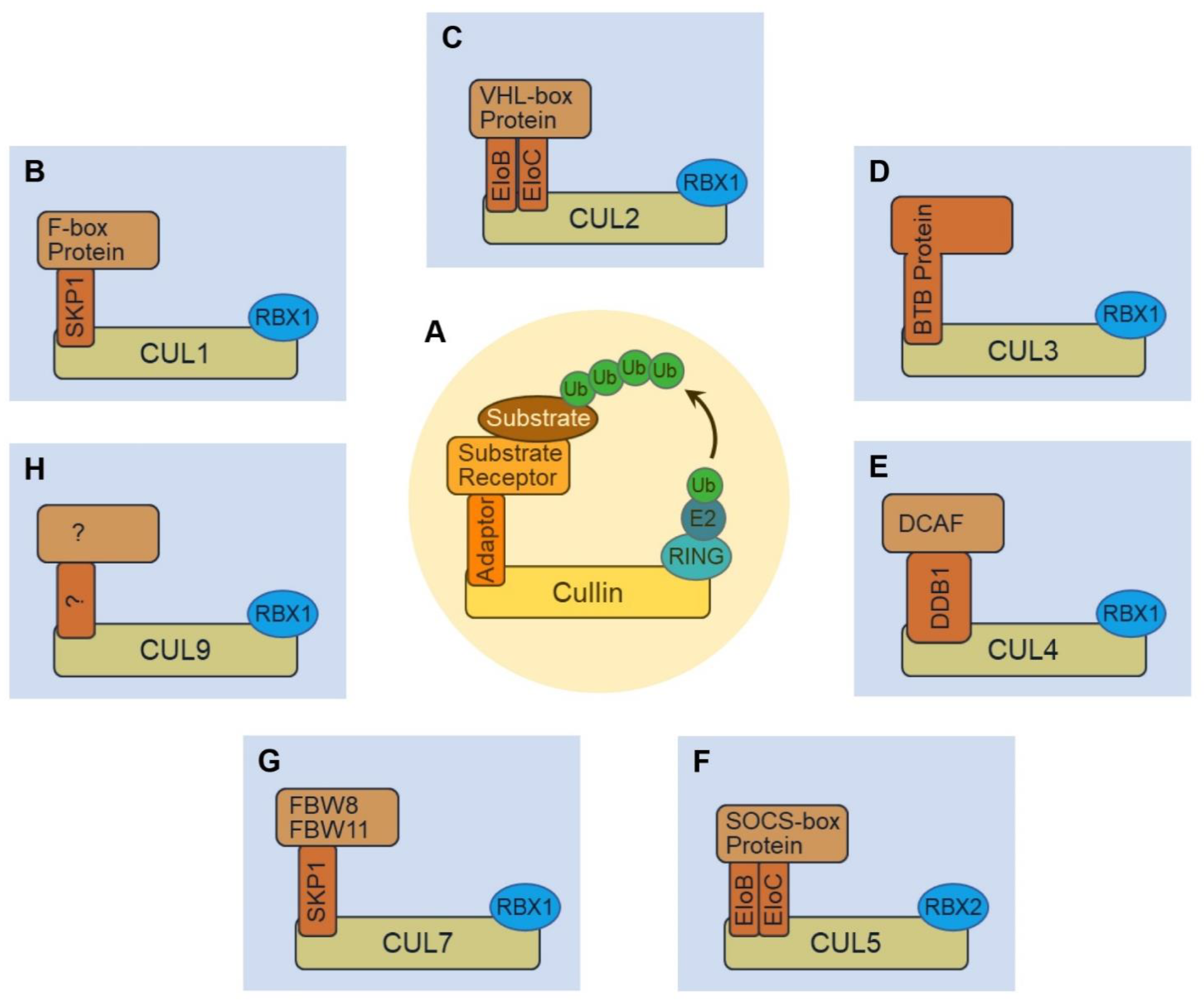
1. Introduction
2. The CUL1-RING Ligase (CRL1)
2.1. Implications for CRL1FBXO32 in Cardiac Function
2.2. CRL1FBXL2 and Ca2+ Homeostasis
2.3. A Role for CRL1FBXW7 in Oxidative Stress
2.4. CRL1SKP2 in Proliferation and Senescence
3. The CUL2-RING Ligase (CRL2)
3.1. CRL2VHL-HIF1α Regulatory Axis and Cardiovascular Diseases
3.2. CRL2VHL and PROTACs
4. The CUL3-RING Ligase (CRL3)
4.1. CUL3 Mutations and Cardiovascular Diseases
4.2. CRL3KLHL3-WNK Regulatory Axis
4.3. CRL3KEAP1-NRF2 Regulatory Axis
4.4. Other BTB Proteins Involved in Cardiovascular Diseases
5. The CUL4-RING Ligase (CRL4)
5.1. CRL4DCAF8 and CRL4DDB1-GRK5
5.2. Non-Canonical CRL4s Involved in Cardiovascular Diseases
5.3. CRL4 and PROTACs
6. The CUL5-RING Ligase (CRL5)
CRL5ASB2 in Cardiac Development
7. The CUL7-RING Ligase (CRL7) and Cardiac Signal Transduction
8. The CUL9-RING Ligase (CRL9) and Cardiovascular Disease
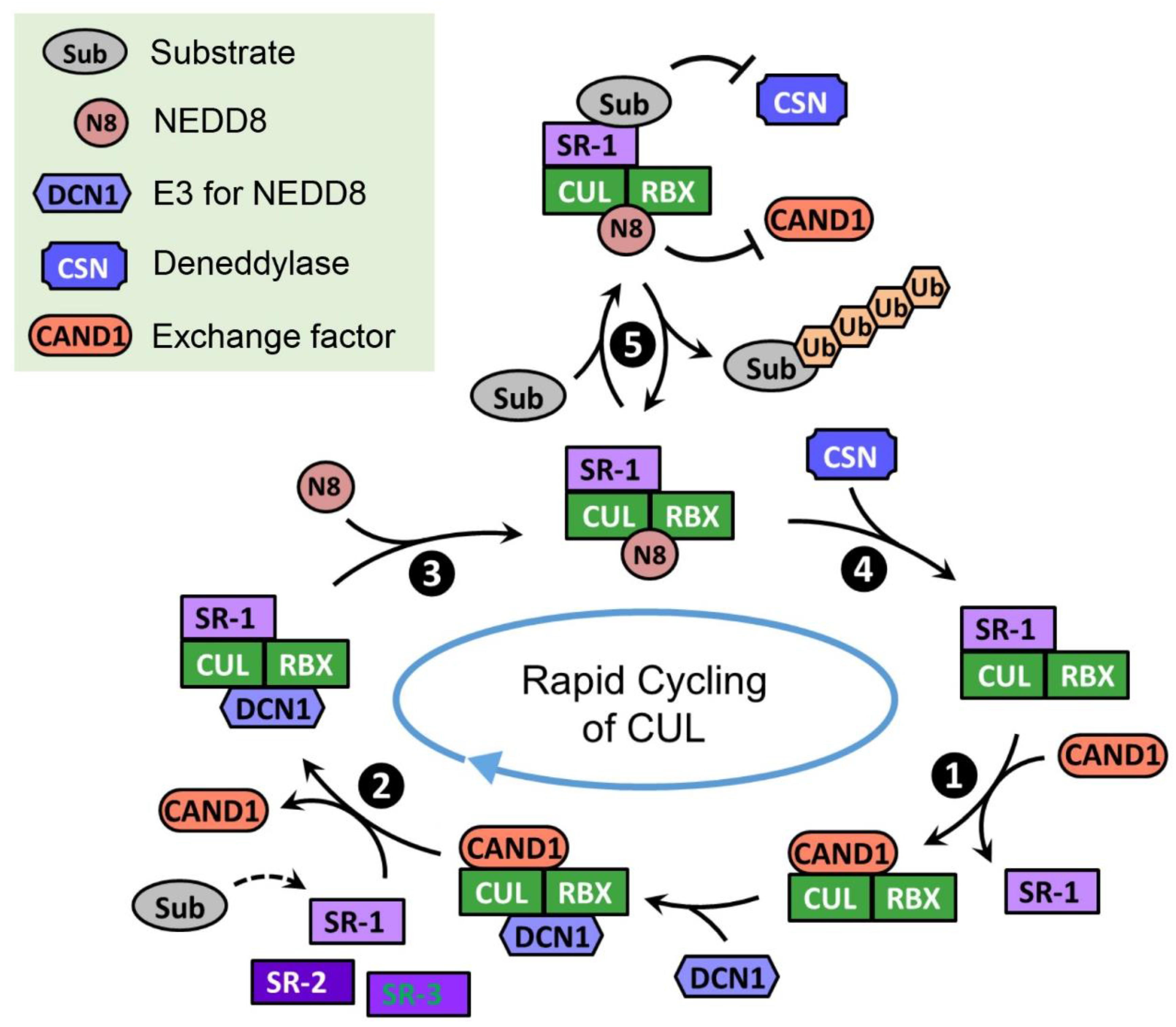
9. Regulators of CRLs
9.1. Neural Precursor Cell Expressed, Developmentally Downregulated 8 (NEDD8)
9.1.1. Neddylation Promotes the Activity of CRLs
9.1.2. Impaired Neddylation Results in Cardiomyopathy
9.2. COP9 Signalosome (CSN)
9.2.1. CSN Is Required for Maintaining the CRL Activities in Cells
9.2.2. Multifaceted Roles of CSN in Cardiac Physiology
9.3. Cullin-Associated NEDD8-Dissociated Protein 1/2 (CAND1/2)
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- GBD 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef]
- Harper, J.W.; Schulman, B.A. Cullin-RING Ubiquitin Ligase Regulatory Circuits: A Quarter Century Beyond the F-Box Hypothesis. Annu. Rev. Biochem. 2021, 90, 403–429. [Google Scholar] [CrossRef]
- Rusnac, D.V.; Zheng, N. Structural Biology of CRL Ubiquitin Ligases. Adv. Exp. Med. Biol. 2020, 1217, 9–31. [Google Scholar] [CrossRef]
- Michel, J.J.; McCarville, J.F.; Xiong, Y. A role for Saccharomyces cerevisiae Cul8 ubiquitin ligase in proper anaphase progression. J. Biol. Chem. 2003, 278, 22828–22837. [Google Scholar] [CrossRef]
- Mimura, S.; Yamaguchi, T.; Ishii, S.; Noro, E.; Katsura, T.; Obuse, C.; Kamura, T. Cul8/Rtt101 forms a variety of protein complexes that regulate DNA damage response and transcriptional silencing. J. Biol. Chem. 2010, 285, 9858–9867. [Google Scholar] [CrossRef]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Demasi, M.; Laurindo, F.R. Physiological and pathological role of the ubiquitin-proteasome system in the vascular smooth muscle cell. Cardiovasc. Res. 2012, 95, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Patterson, C. Into the heart: The emerging role of the ubiquitin-proteasome system. J. Mol. Cell. Cardiol. 2006, 41, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Pagan, J.; Seto, T.; Pagano, M.; Cittadini, A. Role of the ubiquitin proteasome system in the heart. Circ. Res. 2013, 112, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Gu, D.; Wang, S.; Kuiatse, I.; Wang, H.; He, J.; Dai, Y.; Jones, R.J.; Bjorklund, C.C.; Yang, J.; Grant, S.; et al. Inhibition of the MDM2 E3 Ligase induces apoptosis and autophagy in wild-type and mutant p53 models of multiple myeloma, and acts synergistically with ABT-737. PLoS ONE 2014, 9, e103015. [Google Scholar] [CrossRef]
- Hanna, J.; Guerra-Moreno, A.; Ang, J.; Micoogullari, Y. Protein Degradation and the Pathologic Basis of Disease. Am. J. Pathol. 2019, 189, 94–103. [Google Scholar] [CrossRef]
- Lydeard, J.R.; Schulman, B.A.; Harper, J.W. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013, 14, 1050–1061. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, P.; Inuzuka, H.; Wei, W. Roles of F-box proteins in cancer. Nat. Rev. Cancer 2014, 14, 233–247. [Google Scholar] [CrossRef]
- Peris-Moreno, D.; Cussonneau, L.; Combaret, L.; Polge, C.; Taillandier, D. Ubiquitin Ligases at the Heart of Skeletal Muscle Atrophy Control. Molecules 2021, 26, 407. [Google Scholar] [CrossRef]
- Schisler, J.C.; Willis, M.S.; Patterson, C. You spin me round: MaFBx/Atrogin-1 feeds forward on FOXO transcription factors (like a record). Cell Cycle 2008, 7, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.R.; Kim, H.J.; Lecker, S.H. Ubiquitin-protein ligases in muscle wasting. Int. J. Biochem. Cell Biol. 2005, 37, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Lagirand-Cantaloube, J.; Offner, N.; Csibi, A.; Leibovitch, M.P.; Batonnet-Pichon, S.; Tintignac, L.A.; Segura, C.T.; Leibovitch, S.A. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008, 27, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Tintignac, L.A.; Lagirand, J.; Batonnet, S.; Sirri, V.; Leibovitch, M.P.; Leibovitch, S.A. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem. 2005, 280, 2847–2856. [Google Scholar] [CrossRef]
- Xie, P.; Guo, S.; Fan, Y.; Zhang, H.; Gu, D.; Li, H. Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation. J. Biol. Chem. 2009, 284, 5488–5496. [Google Scholar] [CrossRef]
- Li, H.H.; Willis, M.S.; Lockyer, P.; Miller, N.; McDonough, H.; Glass, D.J.; Patterson, C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J. Clin. Investig. 2007, 117, 3211–3223. [Google Scholar] [CrossRef]
- Li, H.H.; Kedar, V.; Zhang, C.; McDonough, H.; Arya, R.; Wang, D.Z.; Patterson, C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J. Clin. Investig. 2004, 114, 1058–1071. [Google Scholar] [CrossRef]
- Zaglia, T.; Milan, G.; Ruhs, A.; Franzoso, M.; Bertaggia, E.; Pianca, N.; Carpi, A.; Carullo, P.; Pesce, P.; Sacerdoti, D.; et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Investig. 2014, 124, 2410–2424. [Google Scholar] [CrossRef]
- Mota, R.; Parry, T.L.; Yates, C.C.; Qiang, Z.; Eaton, S.C.; Mwiza, J.M.; Tulasi, D.; Schisler, J.C.; Patterson, C.; Zaglia, T.; et al. Increasing Cardiomyocyte Atrogin-1 Reduces Aging-Associated Fibrosis and Regulates Remodeling In Vivo. Am. J. Pathol. 2018, 188, 1676–1692. [Google Scholar] [CrossRef] [PubMed]
- Al-Yacoub, N.; Shaheen, R.; Awad, S.M.; Kunhi, M.; Dzimiri, N.; Nguyen, H.C.; Xiong, Y.; Al-Buraiki, J.; Al-Habeeb, W.; Alkuraya, F.S.; et al. FBXO32, encoding a member of the SCF complex, is mutated in dilated cardiomyopathy. Genome Biol. 2016, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Al-Hassnan, Z.N.; Shinwari, Z.M.; Wakil, S.M.; Tulbah, S.; Mohammed, S.; Rahbeeni, Z.; Alghamdi, M.; Rababh, M.; Colak, D.; Kaya, N.; et al. A substitution mutation in cardiac ubiquitin ligase, FBXO32, is associated with an autosomal recessive form of dilated cardiomyopathy. BMC Med. Genet. 2016, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Al-Yacoub, N.; Colak, D.; Mahmoud, S.A.; Hammonds, M.; Muhammed, K.; Al-Harazi, O.; Assiri, A.M.; Al-Buraiki, J.; Al-Habeeb, W.; Poizat, C. Mutation in FBXO32 causes dilated cardiomyopathy through up-regulation of ER-stress mediated apoptosis. Commun. Biol. 2021, 4, 884. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Sun, M.; Zhou, H.; Ajoolabady, A.; Zhou, Y.; Tao, J.; Sowers, J.R.; Zhang, Y. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci. Adv. 2020, 6, 943–953. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Lee, J.E.; Sweredoski, M.J.; Graham, R.L.; Kolawa, N.J.; Smith, G.T.; Hess, S.; Deshaies, R.J. The steady-state repertoire of human SCF ubiquitin ligase complexes does not require ongoing Nedd8 conjugation. Mol. Cell. Proteom. MCP 2011, 10, M110006460. [Google Scholar] [CrossRef]
- Kuchay, S.; Duan, S.; Schenkein, E.; Peschiaroli, A.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. FBXL2- and PTPL1-mediated degradation of p110-free p85beta regulatory subunit controls the PI(3)K signalling cascade. Nat. Cell Biol. 2013, 15, 472–480. [Google Scholar] [CrossRef]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca(2+)-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef]
- Wang, X.; Bathina, M.; Lynch, J.; Koss, B.; Calabrese, C.; Frase, S.; Schuetz, J.D.; Rehg, J.E.; Opferman, J.T. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013, 27, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.C.; Yeh, C.H. Inhibition of miR-302 Suppresses Hypoxia-Reoxygenation-Induced H9c2 Cardiomyocyte Death by Regulating Mcl-1 Expression. Oxid. Med. Cell. Longev. 2017, 2017, 7968905. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, N.; Zhang, Y.; Jia, P.; Guo, Y.; Tian, Y.; You, S.; Wu, S.; Sun, Y. E3 ligase Fbw7 participates in oxidative stressinduced myocardial cell injury via interacting with Mcl1. Mol. Med. Rep. 2019, 20, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Guo, N.; Zhao, S.; Chen, Z.; Zhang, W.; Yan, F.; Liao, H.; Chi, K. FBXW7 promotes pathological cardiac hypertrophy by targeting EZH2-SIX1 signaling. Exp. Cell Res. 2020, 393, 112059. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Su, X.; Shen, Y.; Jin, Y.; Luo, T.; Kim, I.M.; Weintraub, N.L.; Tang, Y. MiR322 mediates cardioprotection against ischemia/reperfusion injury via FBXW7/notch pathway. J. Mol. Cell. Cardiol. 2019, 133, 67–74. [Google Scholar] [CrossRef]
- Wang, L.; Qin, D.; Shi, H.; Zhang, Y.; Li, H.; Han, Q. MiR-195-5p Promotes Cardiomyocyte Hypertrophy by Targeting MFN2 and FBXW7. Biomed. Res. Int. 2019, 2019, 1580982. [Google Scholar] [CrossRef]
- Zou, J.F.; Wu, X.N.; Shi, R.H.; Sun, Y.Q.; Qin, F.J.; Yang, Y.M. Inhibition of microRNA-184 reduces H2O2-mediated cardiomyocyte injury via targeting FBXO28. Eur. Rev. Med. Pharm. Sci. 2020, 24, 11251–11258. [Google Scholar] [CrossRef]
- Wang, H.H.; Lee, Y.N.; Su, C.H.; Shu, K.T.; Liu, W.T.; Hsieh, C.L.; Yeh, H.I.; Wu, Y.J. S-Phase Kinase-associated Protein-2 Rejuvenates Senescent Endothelial Progenitor Cells and Induces Angiogenesis In Vivo. Sci. Rep. 2020, 10, 6646. [Google Scholar] [CrossRef]
- Asmamaw, M.D.; Liu, Y.; Zheng, Y.C.; Shi, X.J.; Liu, H.M. Skp2 in the ubiquitin-proteasome system: A comprehensive review. Med. Res. Rev. 2020, 40, 1920–1949. [Google Scholar] [CrossRef]
- Wu, Y.J.; Sala-Newby, G.B.; Shu, K.T.; Yeh, H.I.; Nakayama, K.I.; Nakayama, K.; Newby, A.C.; Bond, M. S-phase kinase-associated protein-2 (Skp2) promotes vascular smooth muscle cell proliferation and neointima formation in vivo. J. Vasc. Surg. 2009, 50, 1135–1142. [Google Scholar] [CrossRef]
- Cai, W.; Yang, H. The structure and regulation of Cullin 2 based E3 ubiquitin ligases and their biological functions. Cell Div. 2016, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Pause, A.; Lee, S.; Worrell, R.A.; Chen, D.Y.; Burgess, W.H.; Linehan, W.M.; Klausner, R.D. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 2156–2161. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Conrad, M.N.; Yan, Q.; Conaway, R.C.; Conaway, J.W. The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev. 1999, 13, 2928–2933. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Epstein, A.C.R.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and Mammalian Homologs Define a Family of Dioxygenases that Regulate HIF by Prolyl Hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Li, H.; Ko, H.P.; Whitlock, J.P. Induction of phosphoglycerate kinase 1 gene expression by hypoxia. Roles of Arnt and HIF1alpha. J. Biol. Chem. 1996, 271, 21262–21267. [Google Scholar] [CrossRef]
- Ema, M.; Hirota, K.; Mimura, J.; Abe, H.; Yodoi, J.; Sogawa, K.; Poellinger, L.; Fujii-Kuriyama, Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: Their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999, 18, 1905–1914. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Galli, G.; Wang, Y.; Fan, Q.; Wang, Z.; Wang, X.; Xiao, W. Novel Therapeutic Targets for Hypoxia-Related Cardiovascular Diseases: The Role of HIF-1. Front. Physiol. 2020, 11, 774. [Google Scholar] [CrossRef] [PubMed]
- Farnaby, W.; Koegl, M.; McConnell, D.B.; Ciulli, A. Transforming targeted cancer therapy with PROTACs: A forward-looking perspective. Curr. Opin. Pharm. 2021, 57, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Verma, R.; Mohl, D.; Deshaies, R.J. Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol. Cell 2020, 77, 446–460. [Google Scholar] [CrossRef]
- Wu, T.; Yoon, H.; Xiong, Y.; Dixon-Clarke, S.E.; Nowak, R.P.; Fischer, E.S. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat. Struct. Mol. Biol. 2020, 27, 605–614. [Google Scholar] [CrossRef]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I.; Roth, A.G.; Tae, H.S.; Gareiss, P.C.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew. Chem. Int. Ed. Engl. 2012, 51, 11463–11467. [Google Scholar] [CrossRef]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77.e63. [Google Scholar] [CrossRef] [PubMed]
- Cromm, P.M.; Samarasinghe, K.T.G.; Hines, J.; Crews, C.M. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. [Google Scholar] [CrossRef] [PubMed]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Zhang, X.; Thummuri, D.; Liu, X.; Hu, W.; Zhang, P.; Khan, S.; Yuan, Y.; Zhou, D.; Zheng, G. Discovery of PROTAC BCL-XL degraders as potent anticancer agents with low on-target platelet toxicity. Eur. J. Med. Chem. 2020, 192, 112186. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.V.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.; McGonagle, G.A.; Eden, J.; Kiritharan, G.; Touzet, M.; Lewell, X.; Emery, J.; Eidam, H.; Harling, J.D.; Anderson, N.A. Targeting IRAK4 for Degradation with PROTACs. ACS Med. Chem. Lett. 2019, 10, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 830. [Google Scholar] [CrossRef]
- Imaide, S.; Riching, K.M.; Makukhin, N.; Vetma, V.; Whitworth, C.; Hughes, S.J.; Trainor, N.; Mahan, S.D.; Murphy, N.; Cowan, A.D.; et al. Trivalent PROTACs enhance protein degradation via combined avidity and cooperativity. Nat. Chem. Biol. 2021, 17, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Cooper, C.D.O.; Krojer, T.; Murray, J.W.; Pike, A.C.W.; Chaikuad, A.; Keates, T.; Thangaratnarajah, C.; Hojzan, V.; Marsden, B.D.; et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J. Biol. Chem. 2013, 288, 7803–7814. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.X.; Prive, G.G. Crystal structure of KLHL3 in complex with Cullin3. PLoS ONE 2013, 8, e60445. [Google Scholar] [CrossRef]
- Dubiel, W.; Dubiel, D.; Wolf, D.A.; Naumann, M. Cullin 3-Based Ubiquitin Ligases as Master Regulators of Mammalian Cell Differentiation. Trends Biochem. Sci. 2018, 43, 95–107. [Google Scholar] [CrossRef]
- Wu, J.; McCormick, J.A.; Sigmund, C.D. Cullin-3: Renal and Vascular Mechanisms Regulating Blood Pressure. Curr. Hypertens. Rep. 2020, 22, 61. [Google Scholar] [CrossRef]
- Sakaue, T.; Maekawa, M.; Nakayama, H.; Higashiyama, S. Prospect of divergent roles for the CUL3 system in vascular endothelial cell function and angiogenesis. J. Biochem. 2017, 162, 237–245. [Google Scholar] [CrossRef][Green Version]
- Papizan, J.B.; Vidal, A.H.; Bezprozvannaya, S.; Bassel-Duby, R.; Olson, E.N. Cullin-3-RING ubiquitin ligase activity is required for striated muscle function in mice. J. Biol. Chem. 2018, 293, 8802–8811. [Google Scholar] [CrossRef] [PubMed]
- Agbor, L.N.; Nair, A.R.; Wu, J.; Lu, K.T.; Davis, D.R.; Keen, H.L.; Quelle, F.W.; McCormick, J.A.; Singer, J.D.; Sigmund, C.D. Conditional deletion of smooth muscle Cullin-3 causes severe progressive hypertension. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, T.; Fujisaki, A.; Nakayama, H.; Maekawa, M.; Hiyoshi, H.; Kubota, E.; Joh, T.; Izutani, H.; Higashiyama, S. Neddylated Cullin 3 is required for vascular endothelial-cadherin-mediated endothelial barrier function. Cancer Sci. 2017, 108, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef]
- Glover, M.; Ware, J.S.; Henry, A.; Wolley, M.; Walsh, R.; Wain, L.V.; Xu, S.; Van’t Hoff, W.G.; Tobin, M.D.; Hall, I.P.; et al. Detection of mutations in KLHL3 and CUL3 in families with FHHt (familial hyperkalaemic hypertension or Gordon’s syndrome). Clin. Sci. 2014, 126, 721–726. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Siew, K.; Zhang, J.; Johnson, C.; Wood, N.; Cleary, S.E.; Al Maskari, R.S.; Ferryman, J.T.; Hardege, I.; Yasmin; et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol. Med. 2015, 7, 1285–1306. [Google Scholar] [CrossRef]
- Abdel Khalek, W.; Rafael, C.; Loisel-Ferreira, I.; Kouranti, I.; Clauser, E.; Hadchouel, J.; Jeunemaitre, X. Severe Arterial Hypertension from Cullin 3 Mutations Is Caused by Both Renal and Vascular Effects. J. Am. Soc. Nephrol. 2019, 30, 811–823. [Google Scholar] [CrossRef]
- Agbor, L.N.; Ibeawuchi, S.C.; Hu, C.; Wu, J.; Davis, D.R.; Keen, H.L.; Quelle, F.W.; Sigmund, C.D. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight 2016, 1, e91015. [Google Scholar] [CrossRef]
- Louis-Dit-Picard, H.; Barc, J.; Trujillano, D.; Miserey-Lenkei, S.; Bouatia-Naji, N.; Pylypenko, O.; Beaurain, G.; Bonnefond, A.; Sand, O.; Simian, C.; et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012, 44, 456–460. [Google Scholar] [CrossRef]
- Hureaux, M.; Mazurkiewicz, S.; Boccio, V.; Vargas-Poussou, R.; Jeunemaitre, X. The variety of genetic defects explains the phenotypic heterogeneity of Familial Hyperkalemic Hypertension. Kidney Int. Rep. 2021, 6, 2639–2652. [Google Scholar] [CrossRef]
- Louis-Dit-Picard, H.; Kouranti, I.; Rafael, C.; Loisel-Ferreira, I.; Chavez-Canales, M.; Abdel-Khalek, W.; Argaiz, E.R.; Baron, S.; Vacle, S.; Migeon, T.; et al. Mutation affecting the conserved acidic WNK1 motif causes inherited hyperkalemic hyperchloremic acidosis. J. Clin. Investig. 2020, 130, 6379–6394. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.A.; Mutig, K.; Nelson, J.H.; Saritas, T.; Hoorn, E.J.; Yang, C.L.; Rogers, S.; Curry, J.; Delpire, E.; Bachmann, S.; et al. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 2011, 14, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Zhang, J.; Puthumana, J.; Stone, K.L.; Lifton, R.P. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc. Natl. Acad. Sci. USA 2013, 110, 7838–7843. [Google Scholar] [CrossRef]
- Ishizawa, K.; Wang, Q.; Li, J.; Yamazaki, O.; Tamura, Y.; Fujigaki, Y.; Uchida, S.; Lifton, R.P.; Shibata, S. Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc. Natl. Acad. Sci. USA 2019, 116, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Schumacher, F.R.; Mehellou, Y.; Johnson, C.; Knebel, A.; Macartney, T.J.; Wood, N.T.; Alessi, D.R.; Kurz, T. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: Disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem. J. 2013, 451, 111–122. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Sorrell, F.J.; Alessi, D.R.; Bullock, A.N.; Kurz, T. Structural and biochemical characterization of the KLHL3-WNK kinase interaction important in blood pressure regulation. Biochem. J. 2014, 460, 237–246. [Google Scholar] [CrossRef]
- Wu, G.; Peng, J.B. Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett. 2013, 587, 1717–1722. [Google Scholar] [CrossRef]
- Wakabayashi, M.; Mori, T.; Isobe, K.; Sohara, E.; Susa, K.; Araki, Y.; Chiga, M.; Kikuchi, E.; Nomura, N.; Mori, Y.; et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013, 3, 858–868. [Google Scholar] [CrossRef]
- Zhang, C.; Meermeier, N.P.; Terker, A.S.; Blankenstein, K.I.; Singer, J.D.; Hadchouel, J.; Ellison, D.H.; Yang, C.L. Degradation by Cullin 3 and effect on WNK kinases suggest a role of KLHL2 in the pathogenesis of Familial Hyperkalemic Hypertension. Biochem. Biophys. Res. Commun. 2016, 469, 44–48. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, P.; Serbulea, V.; Leitinger, N.; Eckers, A.; Haendeler, J. Nuclear Factor (Erythroid-Derived 2)-Like 2 and Thioredoxin-1 in Atherosclerosis and Ischemia/Reperfusion Injury in the Heart. Antioxid. Redox Signal. 2017, 26, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef]
- Li, J.; Zhang, C.; Xing, Y.; Janicki, J.S.; Yamamoto, M.; Wang, X.L.; Tang, D.Q.; Cui, T. Up-regulation of p27(kip1) contributes to Nrf2-mediated protection against angiotensin II-induced cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 315–324. [Google Scholar] [CrossRef]
- Strom, J.; Xu, B.; Tian, X.; Chen, Q.M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016, 30, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Cominacini, L.; Mozzini, C.; Garbin, U.; Pasini, A.; Stranieri, C.; Solani, E.; Vallerio, P.; Tinelli, I.A.; Fratta Pasini, A. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free Radic. Biol. Med. 2015, 88, 233–242. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zimmerman, M.C.; Zucker, I.H. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H928–H939. [Google Scholar] [CrossRef]
- Gao, L.; Zimmerman, M.C.; Biswal, S.; Zucker, I.H. Selective Nrf2 Gene Deletion in the Rostral Ventrolateral Medulla Evokes Hypertension and Sympathoexcitation in Mice. Hypertension 2017, 69, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Mukohda, M.; Fang, S.; Wu, J.; Agbor, L.N.; Nair, A.R.; Ibeawuchi, S.C.; Hu, C.; Liu, X.; Lu, K.T.; Guo, D.F.; et al. RhoBTB1 protects against hypertension and arterial stiffness by restraining phosphodiesterase 5 activity. J. Clin. Investig. 2019, 129, 2318–2332. [Google Scholar] [CrossRef] [PubMed]
- Rybalkin, S.D.; Yan, C.; Bornfeldt, K.E.; Beavo, J.A. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ. Res. 2003, 93, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Sewduth, R.N.; Pandolfi, S.; Steklov, M.; Sheryazdanova, A.; Zhao, P.; Criem, N.; Baietti, M.F.; Lechat, B.; Quarck, R.; Impens, F.; et al. The Noonan Syndrome Gene Lztr1 Controls Cardiovascular Function by Regulating Vesicular Trafficking. Circ. Res. 2020, 126, 1379–1393. [Google Scholar] [CrossRef]
- Schaefer, A.; Reinhard, N.R.; Hordijk, P.L. Toward understanding RhoGTPase specificity: Structure, function and local activation. Small GTPases 2014, 5, 6. [Google Scholar] [CrossRef]
- Kovacevic, I.; Sakaue, T.; Majolee, J.; Pronk, M.C.; Maekawa, M.; Geerts, D.; Fernandez-Borja, M.; Higashiyama, S.; Hordijk, P.L. The Cullin-3-Rbx1-KCTD10 complex controls endothelial barrier function via K63 ubiquitination of RhoB. J. Cell Biol. 2018, 217, 1015–1032. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef]
- Ibeawuchi, S.R.; Agbor, L.N.; Quelle, F.W.; Sigmund, C.D. Hypertension-causing Mutations in Cullin3 Protein Impair RhoA Protein Ubiquitination and Augment the Association with Substrate Adaptors. J. Biol. Chem. 2015, 290, 19208–19217. [Google Scholar] [CrossRef]
- Hedberg-Oldfors, C.; Abramsson, A.; Osborn, D.P.S.; Danielsson, O.; Fazlinezhad, A.; Nilipour, Y.; Hubbert, L.; Nennesmo, I.; Visuttijai, K.; Bharj, J.; et al. Cardiomyopathy with lethal arrhythmias associated with inactivation of KLHL24. Hum. Mol. Genet. 2019, 28, 1919–1929. [Google Scholar] [CrossRef]
- Sang, Y.; Yan, F.; Ren, X. The role and mechanism of CRL4 E3 ubiquitin ligase in cancer and its potential therapy implications. Oncotarget 2015, 6, 42590–42602. [Google Scholar] [CrossRef]
- Hannah, J.; Zhou, P. Distinct and overlapping functions of the cullin E3 ligase scaffolding proteins CUL4A and CUL4B. Gene 2015, 573, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 2007, 26, 775–780. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; McCall, C.M.; Hu, J.; Zeng, Y.; Xiong, Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006, 20, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Zhang, N.; Zhang, Y.; Qian, H.; Wu, B.; Sun, Y. Cul4a as a New Interaction Protein of PARP1 Inhibits Oxidative Stress-Induced H9c2 Cell Apoptosis. Oxid. Med. Cell. Longev. 2019, 2019, 4273261. [Google Scholar] [CrossRef]
- Li, P.; Song, Y.; Zan, W.; Qin, L.; Han, S.; Jiang, B.; Dou, H.; Shao, C.; Gong, Y. Lack of CUL4B in Adipocytes Promotes PPARgamma-Mediated Adipose Tissue Expansion and Insulin Sensitivity. Diabetes 2017, 66, 300–313. [Google Scholar] [CrossRef]
- Nowak, M.; Suenkel, B.; Porras, P.; Migotti, R.; Schmidt, F.; Kny, M.; Zhu, X.; Wanker, E.E.; Dittmar, G.; Fielitz, J.; et al. DCAF8, a novel MuRF1 interaction partner, promotes muscle atrophy. J. Cell Sci. 2019, 132, jcs233395. [Google Scholar] [CrossRef]
- Penela, P. Chapter Three—Ubiquitination and Protein Turnover of G-Protein-Coupled Receptor Kinases in GPCR Signaling and Cellular Regulation. Prog. Mol. Biol. Transl. Sci. 2016, 141, 85–140. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, Y.; Yang, T.; Gao, Q.; Yuan, M.; Ma, L. Targeted ubiquitination and degradation of G-protein-coupled receptor kinase 5 by the DDB1-CUL4 ubiquitin ligase complex. PLoS ONE 2012, 7, e43997. [Google Scholar] [CrossRef]
- Zha, Z.; Han, X.; Smith, M.D.; Liu, Y.; Giguere, P.M.; Kopanja, D.; Raychaudhuri, P.; Siderovski, D.P.; Guan, K.L.; Lei, Q.Y.; et al. A Non-Canonical Function of Gbeta as a Subunit of E3 Ligase in Targeting GRK2 Ubiquitylation. Mol. Cell 2015, 58, 794–803. [Google Scholar] [CrossRef]
- Sjogren, B.; Swaney, S.; Neubig, R.R. FBXO44-Mediated Degradation of RGS2 Protein Uniquely Depends on a Cullin 4B/DDB1 Complex. PLoS ONE 2015, 10, e0123581. [Google Scholar] [CrossRef]
- Heximer, S.P.; Knutsen, R.H.; Sun, X.; Kaltenbronn, K.M.; Rhee, M.H.; Peng, N.; Oliveira-dos-Santos, A.; Penninger, J.M.; Muslin, A.J.; Steinberg, T.H.; et al. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J. Clin. Investig. 2003, 111, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Bodenstein, J.; Sunahara, R.K.; Neubig, R.R. N-terminal residues control proteasomal degradation of RGS2, RGS4, and RGS5 in human embryonic kidney 293 cells. Mol. Pharm. 2007, 71, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kamide, K.; Kokubo, Y.; Takiuchi, S.; Tanaka, C.; Banno, M.; Miwa, Y.; Yoshii, M.; Horio, T.; Okayama, A.; et al. Genetic variations of regulator of G-protein signaling 2 in hypertensive patients and in the general population. J. Hypertens. 2005, 23, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.T.N.; Sjogren, B.; Neubig, R.R. Human Missense Mutations in Regulator of G Protein Signaling 2 Affect the Protein Function Through Multiple Mechanisms. Mol. Pharm. 2017, 92, 451–458. [Google Scholar] [CrossRef] [PubMed]
- McNabb, H.J.; Gonzalez, S.; Muli, C.S.; Sjogren, B. N-Terminal Targeting of Regulator of G Protein Signaling Protein 2 for F-Box Only Protein 44-Mediated Proteasomal Degradation. Mol. Pharm. 2020, 98, 677–685. [Google Scholar] [CrossRef]
- Chopra, R.; Sadok, A.; Collins, I. A critical evaluation of the approaches to targeted protein degradation for drug discovery. Drug Discov. Today Technol. 2019, 31, 5–13. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Lu, G.; Ito, T.; Pagarigan, B.; Lu, C.C.; Miller, K.; Fang, W.; Wang, N.Y.; Nguyen, D.; Houston, J.; et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 2016, 535, 252–257. [Google Scholar] [CrossRef]
- Petzold, G.; Fischer, E.S.; Thoma, N.H. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 2016, 532, 127–130. [Google Scholar] [CrossRef]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Slabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thoma, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362. [Google Scholar] [CrossRef]
- Hansen, J.D.; Condroski, K.; Correa, M.; Muller, G.; Man, H.W.; Ruchelman, A.; Zhang, W.; Vocanson, F.; Crea, T.; Liu, W.; et al. Protein Degradation via CRL4(CRBN) Ubiquitin Ligase: Discovery and Structure-Activity Relationships of Novel Glutarimide Analogs That Promote Degradation of Aiolos and/or GSPT1. J. Med. Chem. 2018, 61, 492–503. [Google Scholar] [CrossRef]
- Kronke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Ponthier, C.M.; Sack, R.; Seebacher, J.; Stadler, M.B.; Donovan, K.A.; Fischer, E.S. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4(CRBN) ubiquitin ligase. Nat. Commun. 2017, 8, 15398. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, 6336. [Google Scholar] [CrossRef]
- Das, A.; Dasgupta, S.; Gong, Y.; Shah, U.A.; Fradley, M.G.; Cheng, R.K.; Roy, B.; Guha, A. Cardiotoxicity as an adverse effect of immunomodulatory drugs and proteasome inhibitors in multiple myeloma: A network meta-analysis of randomized clinical trials. Hematol. Oncol. 2021, 1–10. [Google Scholar] [CrossRef]
- Zhao, Y.; Xiong, X.; Sun, Y. Cullin-RING Ligase 5: Functional characterization and its role in human cancers. Semin. Cancer Biol. 2020, 67, 61–79. [Google Scholar] [CrossRef]
- Kamura, T.; Maenaka, K.; Kotoshiba, S.; Matsumoto, M.; Kohda, D.; Conaway, R.C.; Conaway, J.W.; Nakayama, K.I. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004, 18, 3055–3065. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Mear, L.; Zhong, W.; Digre, A.; Katona, B.; Sjostedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Metais, A.; Lamsoul, I.; Melet, A.; Uttenweiler-Joseph, S.; Poincloux, R.; Stefanovic, S.; Valiere, A.; de Peredo, A.G.; Stella, A.; Burlet-Schiltz, O.; et al. Asb2 alpha-Filamin A Axis Is Essential for Actin Cytoskeleton Remodeling during Heart Development. Circ. Res. 2018, 122, e34–e48. [Google Scholar] [CrossRef]
- Heuze, M.L.; Lamsoul, I.; Baldassarre, M.; Lad, Y.; Leveque, S.; Razinia, Z.; Moog-Lutz, C.; Calderwood, D.A.; Lutz, P.G. ASB2 targets filamins A and B to proteasomal degradation. Blood 2008, 112, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Min, K.D.; Asakura, M.; Shirai, M.; Yamazaki, S.; Ito, S.; Fu, H.Y.; Asanuma, H.; Asano, Y.; Minamino, T.; Takashima, S.; et al. ASB2 is a novel E3 ligase of SMAD9 required for cardiogenesis. Sci. Rep. 2021, 11, 23056. [Google Scholar] [CrossRef]
- Shi, L.; Du, D.; Peng, Y.; Liu, J.; Long, J. The functional analysis of Cullin 7 E3 ubiquitin ligases in cancer. Oncogenesis 2020, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, Y.; Wu, L.; Ma, X.; Liu, Q.; Huang, F.; Zhang, X.; Zhang, Y.; Zhang, J.; Luo, H.; et al. CUL7 E3 Ubiquitin Ligase Mediates the Degradation of Activation-Induced Cytidine Deaminase and Regulates the Ig Class Switch Recombination in B Lymphocytes. J. Immunol. 2019, 203, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, T.; Kuwabara, H.; Arai, T.; Xiao, Y.; Decaprio, J.A. Disruption of the Fbxw8 gene results in pre- and postnatal growth retardation in mice. Mol. Cell. Biol. 2008, 28, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.C.; Dolios, G.; Wang, R.; Pan, Z.Q. CUL7: A DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc. Natl. Acad. Sci. USA 2002, 99, 16601–16606. [Google Scholar] [CrossRef]
- Scheufele, F.; Wolf, B.; Kruse, M.; Hartmann, T.; Lempart, J.; Muhlich, S.; Pfeiffer, A.F.H.; Field, L.J.; Charron, M.J.; Pan, Z.Q.; et al. Evidence for a regulatory role of Cullin-RING E3 ubiquitin ligase 7 in insulin signaling. Cell. Signal. 2014, 26, 233–239. [Google Scholar] [CrossRef]
- Xu, X.; Keshwani, M.; Meyer, K.; Sarikas, A.; Taylor, S.; Pan, Z.Q. Identification of the degradation determinants of insulin receptor substrate 1 for signaling cullin-RING E3 ubiquitin ligase 7-mediated ubiquitination. J. Biol. Chem. 2012, 287, 40758–40766. [Google Scholar] [CrossRef]
- Xu, X.; Sarikas, A.; Dias-Santagata, D.C.; Dolios, G.; Lafontant, P.J.; Tsai, S.C.; Zhu, W.; Nakajima, H.; Nakajima, H.O.; Field, L.J.; et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol. Cell 2008, 30, 403–414. [Google Scholar] [CrossRef]
- Zou, J.; Ma, W.; Li, J.; Littlejohn, R.; Zhou, H.; Kim, I.M.; Fulton, D.J.R.; Chen, W.; Weintraub, N.L.; Zhou, J.; et al. Neddylation mediates ventricular chamber maturation through repression of Hippo signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E4101–E4110. [Google Scholar] [CrossRef]
- Anger, M.; Scheufele, F.; Ramanujam, D.; Meyer, K.; Nakajima, H.; Field, L.J.; Engelhardt, S.; Sarikas, A. Genetic ablation of Cullin-RING E3 ubiquitin ligase 7 restrains pressure overload-induced myocardial fibrosis. PLoS ONE 2020, 15, e0244096. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pei, X.H.; Yan, J.; Yan, F.; Cappell, K.M.; Whitehurst, A.W.; Xiong, Y. CUL9 mediates the functions of the 3M complex and ubiquitylates survivin to maintain genome integrity. Mol. Cell 2014, 54, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Ortolano, N.A.; Romero-Morales, A.I.; Rasmussen, M.L.; Bodnya, C.; Kline, L.A.; Joshi, P.; Connelly, J.P.; Rose, K.L.; Pruett-Miller, S.M.; Gama, V. A proteomics approach for the identification of cullin-9 (CUL9) related signaling pathways in induced pluripotent stem cell models. PLoS ONE 2021, 16, e0248000. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xiong, Y. Cytoplasmic E3 ubiquitin ligase CUL9 controls cell proliferation, senescence, apoptosis and genome integrity through p53. Oncogene 2017, 36, 5212–5218. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Djurovic, S.; Thompson, W.K.; Schork, A.J.; Kendler, K.S.; O’Donovan, M.C.; Rujescu, D.; Werge, T.; van de Bunt, M.; Morris, A.P.; et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am. J. Hum. Genet. 2013, 92, 197–209. [Google Scholar] [CrossRef]
- Osaka, F.; Kawasaki, H.; Aida, N.; Saeki, M.; Chiba, T.; Kawashima, S.; Tanaka, K.; Kato, S. A new NEDD8-ligating system for cullin-4A. Genes Dev. 1998, 12, 2263–2268. [Google Scholar] [CrossRef]
- Whitby, F.G.; Xia, G.; Pickart, C.M.; Hill, C.P. Crystal Structure of the Human Ubiquitin-like Protein NEDD8 and Interactions with Ubiquitin Pathway Enzymes. J. Biol. Chem. 1998, 273, 34983–34991. [Google Scholar] [CrossRef]
- Morimoto, M.; Nishida, T.; Honda, R.; Yasuda, H. Modification of cullin-1 by ubiquitin-like protein Nedd8 enhances the activity of SCF(skp2) toward p27(kip1). Biochem. Biophys. Res. Commun. 2000, 270, 1093–1096. [Google Scholar] [CrossRef]
- Kawakami, T.; Chiba, T.; Suzuki, T.; Iwai, K.; Yamanaka, K.; Minato, N.; Suzuki, H.; Shimbara, N.; Hidaka, Y.; Osaka, F.; et al. NEDD8 recruits E2-ubiquitin to SCF E3 ligase. EMBO J. 2001, 20, 4003–4012. [Google Scholar] [CrossRef]
- Read, M.A.; Brownell, J.E.; Gladysheva, T.B.; Hottelet, M.; Parent, L.A.; Coggins, M.B.; Pierce, J.W.; Podust, V.N.; Luo, R.S.; Chau, V.; et al. Nedd8 Modification of Cul-1 Activates SCFbeta TrCP-Dependent Ubiquitination of Ikappa Balpha. Mol. Cell. Biol. 2000, 20, 2326–2333. [Google Scholar] [CrossRef]
- Wang, K.; Reichermeier, K.M.; Liu, X. Quantitative analyses for effects of neddylation on CRL2(VHL) substrate ubiquitination and degradation. Protein Sci. 2021, 30, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Deshaies, R.J. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol. Cell 2008, 32, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.M.; Borg, L.A.; Scott, D.C.; Hunt, H.W.; Hammel, M.; Schulman, B.A. Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell 2008, 134, 995–1006. [Google Scholar] [CrossRef]
- Baek, K.; Krist, D.T.; Prabu, J.R.; Hill, S.; Klugel, M.; Neumaier, L.M.; von Gronau, S.; Kleiger, G.; Schulman, B.A. NEDD8 nucleates a multivalent cullin-RING-UBE2D ubiquitin ligation assembly. Nature 2020, 578, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Pierce, N.W. Transfer of ubiquitin protein caught in the act. Nature 2020, 578, 372–373. [Google Scholar] [CrossRef]
- Kostrhon, S.; Prabu, J.R.; Baek, K.; Horn-Ghetko, D.; von Gronau, S.; Klugel, M.; Basquin, J.; Alpi, A.F.; Schulman, B.A. CUL5-ARIH2 E3-E3 ubiquitin ligase structure reveals cullin-specific NEDD8 activation. Nat. Chem. Biol. 2021, 17, 1075–1083. [Google Scholar] [CrossRef]
- Horn-Ghetko, D.; Krist, D.T.; Prabu, J.R.; Baek, K.; Mulder, M.P.C.; Klugel, M.; Scott, D.C.; Ovaa, H.; Kleiger, G.; Schulman, B.A. Ubiquitin ligation to F-box protein targets by SCF-RBR E3-E3 super-assembly. Nature 2021, 590, 671–676. [Google Scholar] [CrossRef]
- Scott, D.C.; Rhee, D.Y.; Duda, D.M.; Kelsall, I.R.; Olszewski, J.L.; Paulo, J.A.; de Jong, A.; Ovaa, H.; Alpi, A.F.; Harper, J.W.; et al. Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation. Cell 2016, 166, 1198–1214.e24. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Zou, J.; Ma, W.; Littlejohn, R.; Li, J.; Stansfield, B.K.; Kim, I.M.; Liu, J.; Zhou, J.; Weintraub, N.L.; Su, H. Transient inhibition of neddylation at neonatal stage evokes reversible cardiomyopathy and predisposes the heart to isoproterenol-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1406–H1416. [Google Scholar] [CrossRef]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Lyapina, S.; Cope, G.; Shevchenko, A.; Serino, G.; Tsuge, T.; Zhou, C.; Wolf, D.A.; Wei, N.; Shevchenko, A.; Deshaies, R.J. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science 2001, 292, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Serino, G.; Callis, J.; Crosby, W.L.; Lyapina, S.; Deshaies, R.J.; Gray, W.M.; Estelle, M.; Deng, X.W. Interactions of the COP9 signalosome with the E3 ubiquitin ligase SCFTIRI in mediating auxin response. Science 2001, 292, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Cope, G.A.; Suh, G.S.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 2002, 298, 608–611. [Google Scholar] [CrossRef]
- Pintard, L.; Kurz, T.; Glaser, S.; Willis, J.H.; Peter, M.; Bowerman, B. Neddylation and Deneddylation of CUL-3 Is Required to Target MEI-1/Katanin for Degradation at the Meiosis-to-Mitosis Transition in C. elegans. Curr. Biol. 2003, 13, 911–921. [Google Scholar] [CrossRef]
- Zhou, C.; Wee, S.; Rhee, E.; Naumann, M.; Dubiel, W.; Wolf, D.A. Fission Yeast COP9/Signalosome Suppresses Cullin Activity through Recruitment of the Deubiquitylating Enzyme Ubp12p. Mol. Cell 2003, 11, 927–938. [Google Scholar] [CrossRef]
- Wee, S.; Geyer, R.K.; Toda, T.; Wolf, D.A. CSN facilitates Cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat. Cell Biol. 2005, 7, 387–391. [Google Scholar] [CrossRef]
- He, Q.; Cheng, P.; He, Q.; Liu, Y. The COP9 signalosome regulates the Neurospora circadian clock by controlling the stability of the SCFFWD-1 complex. Genes Dev. 2005, 19, 1518–1531. [Google Scholar] [CrossRef]
- Pierce, N.W.; Lee, J.E.; Liu, X.; Sweredoski, M.J.; Graham, R.L.; Larimore, E.A.; Rome, M.; Zheng, N.; Clurman, B.E.; Hess, S.; et al. Cand1 Promotes Assembly of New SCF Complexes through Dynamic Exchange of F Box Proteins. Cell 2013, 153, 206–215. [Google Scholar] [CrossRef]
- Reitsma, J.M.; Liu, X.; Reichermeier, K.M.; Moradian, A.; Sweredoski, M.J.; Hess, S.; Deshaies, R.J. Composition and Regulation of the Cellular Repertoire of SCF Ubiquitin Ligases. Cell 2017, 171, 1326–1339.e14. [Google Scholar] [CrossRef]
- Liu, X.; Reitsma, J.M.; Mamrosh, J.L.; Zhang, Y.; Straube, R.; Deshaies, R.J. Cand1-Mediated Adaptive Exchange Mechanism Enables Variation in F-Box Protein Expression. Mol. Cell 2018, 69, 773–786.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Deshaies, R.J.; Liu, X. Assembly and Regulation of CRL Ubiquitin Ligases. Adv. Exp. Med. Biol. 2020, 1217, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Milic, J.; Tian, Y.; Bernhagen, J. Role of the COP9 Signalosome (CSN) in Cardiovascular Diseases. Biomolecules 2019, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Martin, D.S. The COP9 signalosome and cullin-RING ligases in the heart. Am. J. Cardiovasc. Dis. 2015, 5, 1–18. [Google Scholar] [PubMed]
- Lykke-Andersen, K.; Schaefer, L.; Menon, S.; Deng, X.W.; Miller, J.B.; Wei, N. Disruption of the COP9 signalosome Csn2 subunit in mice causes deficient cell proliferation, accumulation of p53 and cyclin E, and early embryonic death. Mol. Cell. Biol. 2003, 23, 6790–6797. [Google Scholar] [CrossRef]
- Yan, J.; Walz, K.; Nakamura, H.; Carattini-Rivera, S.; Zhao, Q.; Vogel, H.; Wei, N.; Justice, M.J.; Bradley, A.; Lupski, J.R. COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol. Cell. Biol. 2003, 23, 6798–6808. [Google Scholar] [CrossRef]
- Menon, S.; Chi, H.; Zhang, H.; Deng, X.W.; Flavell, R.A.; Wei, N. COP9 signalosome subunit 8 is essential for peripheral T cell homeostasis and antigen receptor-induced entry into the cell cycle from quiescence. Nat. Immunol. 2007, 8, 1236–1245. [Google Scholar] [CrossRef]
- Zhao, R.; Yeung, S.C.; Chen, J.; Iwakuma, T.; Su, C.H.; Chen, B.; Qu, C.; Zhang, F.; Chen, Y.T.; Lin, Y.L.; et al. Subunit 6 of the COP9 signalosome promotes tumorigenesis in mice through stabilization of MDM2 and is upregulated in human cancers. J. Clin. Investig. 2011, 121, 851–865. [Google Scholar] [CrossRef]
- Lei, D.; Li, F.; Su, H.; Liu, J.; Wei, N.; Wang, X. Hepatic deficiency of COP9 signalosome subunit 8 induces ubiquitin-proteasome system impairment and Bim-mediated apoptosis in murine livers. PLoS ONE 2013, 8, e67793. [Google Scholar] [CrossRef]
- Su, H.; Li, J.; Menon, S.; Liu, J.; Kumarapeli, A.R.; Wei, N.; Wang, X. Perturbation of cullin deneddylation via conditional Csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ. Res. 2011, 108, 40–50. [Google Scholar] [CrossRef]
- Su, H.; Li, F.; Ranek, M.J.; Wei, N.; Wang, X. COP9 signalosome regulates autophagosome maturation. Circulation 2011, 124, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Li, J.; Zhang, H.; Ma, W.; Wei, N.; Liu, J.; Wang, X. COP9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity. Circ. Res. 2015, 117, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Su, H.B.; Li, J.; Osinska, H.; Li, F.Q.; Robbins, J.; Liu, J.B.; Wei, N.; Wang, X.J. The COP9 Signalosome Is Required for Autophagy, Proteasome-Mediated Proteolysis, and Cardiomyocyte Survival in Adult Mice. Circ. Heart Fail. 2013, 6, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Asare, Y.; Ommer, M.; Azombo, F.A.; Alampour-Rajabi, S.; Sternkopf, M.; Sanati, M.; Gijbels, M.J.; Schmitz, C.; Sinitski, D.; Tilstam, P.V.; et al. Inhibition of atherogenesis by the COP9 signalosome subunit 5 in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E2766–E2775. [Google Scholar] [CrossRef] [PubMed]
- Asare, Y.; Shagdarsuren, E.; Schmid, J.A.; Tilstam, P.V.; Grommes, J.; El Bounkari, O.; Schutz, A.K.; Weber, C.; de Winther, M.P.; Noels, H.; et al. Endothelial CSN5 impairs NF-kappaB activation and monocyte adhesion to endothelial cells and is highly expressed in human atherosclerotic lesions. Thromb. Haemost. 2013, 110, 141–152. [Google Scholar] [CrossRef]
- Dubiel, W.; Chaithongyot, S.; Dubiel, D.; Naumann, M. The COP9 Signalosome: A Multi-DUB Complex. Biomolecules 2020, 10, 1082. [Google Scholar] [CrossRef]
- Cavadini, S.; Fischer, E.S.; Bunker, R.D.; Potenza, A.; Lingaraju, G.M.; Goldie, K.N.; Mohamed, W.I.; Faty, M.; Petzold, G.; Beckwith, R.E.; et al. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 2016, 531, 598–603. [Google Scholar] [CrossRef]
- Emberley, E.D.; Mosadeghi, R.; Deshaies, R.J. Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-F-box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism. J. Biol. Chem. 2012, 287, 29679–29689. [Google Scholar] [CrossRef]
- Reitsma, J.M.; Liu, X.; Reichermeier, K.M.; Moradian, A.; Sweredoski, M.J.; Hess, S.; Deshaies, R.J. Nedd8 conjugation and Cand1-mediated exchange sustain a non-equilibrium cellular landscape of SCF E3 ubiquitin ligases. Cell 2017. accepted in principle. [Google Scholar]
- Lo, S.C.; Hannink, M. CAND1-mediated substrate adaptor recycling is required for efficient repression of Nrf2 by Keap1. Mol. Cell. Biol. 2006, 26, 1235–1244. [Google Scholar] [CrossRef]
- Reichermeier, K.M.; Straube, R.; Reitsma, J.M.; Sweredoski, M.J.; Rose, C.M.; Moradian, A.; den Besten, W.; Hinkle, T.; Verschueren, E.; Petzold, G.; et al. PIKES Analysis Reveals Response to Degraders and Key Regulatory Mechanisms of the CRL4 Network. Mol. Cell 2020, 77, 1092–1106.e9. [Google Scholar] [CrossRef]
- Shiraishi, S.; Zhou, C.; Aoki, T.; Sato, N.; Chiba, T.; Tanaka, K.; Yoshida, S.; Nabeshima, Y.; Nabeshima, Y.; Tamura, T.A. TBP-interacting protein 120B (TIP120B)/cullin-associated and neddylation-dissociated 2 (CAND2) inhibits SCF-dependent ubiquitination of myogenin and accelerates myogenic differentiation. J. Biol. Chem. 2007, 282, 9017–9028. [Google Scholar] [CrossRef] [PubMed]
- Sinner, M.F.; Tucker, N.R.; Lunetta, K.L.; Ozaki, K.; Smith, J.G.; Trompet, S.; Bis, J.C.; Lin, H.; Chung, M.K.; Nielsen, J.B.; et al. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation 2014, 130, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Song, J.; Xu, M.; Lv, L.; Liu, C.; Shen, J.; Huang, Y. NEURL rs6584555 and CAND2 rs4642101 contribute to postoperative atrial fibrillation: A prospective study among Chinese population. Oncotarget 2016, 7, 42617–42624. [Google Scholar] [CrossRef] [PubMed]
- Gregers, E.; Ahlberg, G.; Christensen, T.; Jabbari, J.; Larsen, K.O.; Herfelt, C.B.; Henningsen, K.M.; Andreasen, L.; Thiis, J.J.; Lund, J.; et al. Deep sequencing of atrial fibrillation patients with mitral valve regurgitation shows no evidence of mosaicism but reveals novel rare germline variants. Heart Rhythm 2017, 14, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Koskimaki, J.; Zhang, D.; Li, Y.; Saadat, L.; Moore, T.; Lightle, R.; Polster, S.P.; Carrion-Penagos, J.; Lyne, S.B.; Zeineddine, H.A.; et al. Transcriptome clarifies mechanisms of lesion genesis versus progression in models of Ccm3 cerebral cavernous malformations. Acta Neuropathol. Commun. 2019, 7, 132. [Google Scholar] [CrossRef]
- Weng, L.C.; Hall, A.W.; Choi, S.H.; Jurgens, S.J.; Haessler, J.; Bihlmeyer, N.A.; Grarup, N.; Lin, H.; Teumer, A.; Li-Gao, R.; et al. Genetic Determinants of Electrocardiographic P-Wave Duration and Relation to Atrial Fibrillation. Circ. Genom. Precis. Med. 2020, 13, 387–395. [Google Scholar] [CrossRef]
- Moksnes, M.R.; Rosjo, H.; Richmond, A.; Lyngbakken, M.N.; Graham, S.E.; Hansen, A.F.; Wolford, B.N.; Gagliano Taliun, S.A.; LeFaive, J.; Rasheed, H.; et al. Genome-wide association study of cardiac troponin I in the general population. Hum. Mol. Genet. 2021, 30, 2027–2039. [Google Scholar] [CrossRef]
- Christophersen, I.E.; Rienstra, M.; Roselli, C.; Yin, X.; Geelhoed, B.; Barnard, J.; Lin, H.; Arking, D.E.; Smith, A.V.; Albert, C.M.; et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat. Genet. 2017, 49, 946–952. [Google Scholar] [CrossRef]
- Gorska, A.A.; Sandmann, C.; Riechert, E.; Hofmann, C.; Malovrh, E.; Varma, E.; Kmietczyk, V.; Olschlager, J.; Jurgensen, L.; Kamuf-Schenk, V.; et al. Muscle-specific Cand2 is translationally upregulated by mTORC1 and promotes adverse cardiac remodeling. EMBO Rep. 2021, 22, e52170. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz, S.; Wang, K.; Sjögren, B.; Liu, X. Roles of Cullin-RING Ubiquitin Ligases in Cardiovascular Diseases. Biomolecules 2022, 12, 416. https://doi.org/10.3390/biom12030416
Diaz S, Wang K, Sjögren B, Liu X. Roles of Cullin-RING Ubiquitin Ligases in Cardiovascular Diseases. Biomolecules. 2022; 12(3):416. https://doi.org/10.3390/biom12030416
Chicago/Turabian StyleDiaz, Stephanie, Kankan Wang, Benita Sjögren, and Xing Liu. 2022. "Roles of Cullin-RING Ubiquitin Ligases in Cardiovascular Diseases" Biomolecules 12, no. 3: 416. https://doi.org/10.3390/biom12030416
APA StyleDiaz, S., Wang, K., Sjögren, B., & Liu, X. (2022). Roles of Cullin-RING Ubiquitin Ligases in Cardiovascular Diseases. Biomolecules, 12(3), 416. https://doi.org/10.3390/biom12030416