
Tumor Suppressor Role of Wild-Type P53-Dependent Secretome and Its Proteomic Identification in PDAC


 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Drug
2.2. Cell Cultures
2.3. Transient Transfection Assay
2.4. Cell Proliferation Assay
2.5. Apoptosis Assay
2.6. Autophagosome Formation Assay
2.7. Wound-Closure Cell Migration Assay
2.8. Immunoblot Analysis
2.9. Protein Extraction from Conditioned Medium (CM)
2.10. Proteomic Analysis
2.11. Bioinformatics Evaluation of Proteomics Data
2.12. Statistical Analysis
3. Results
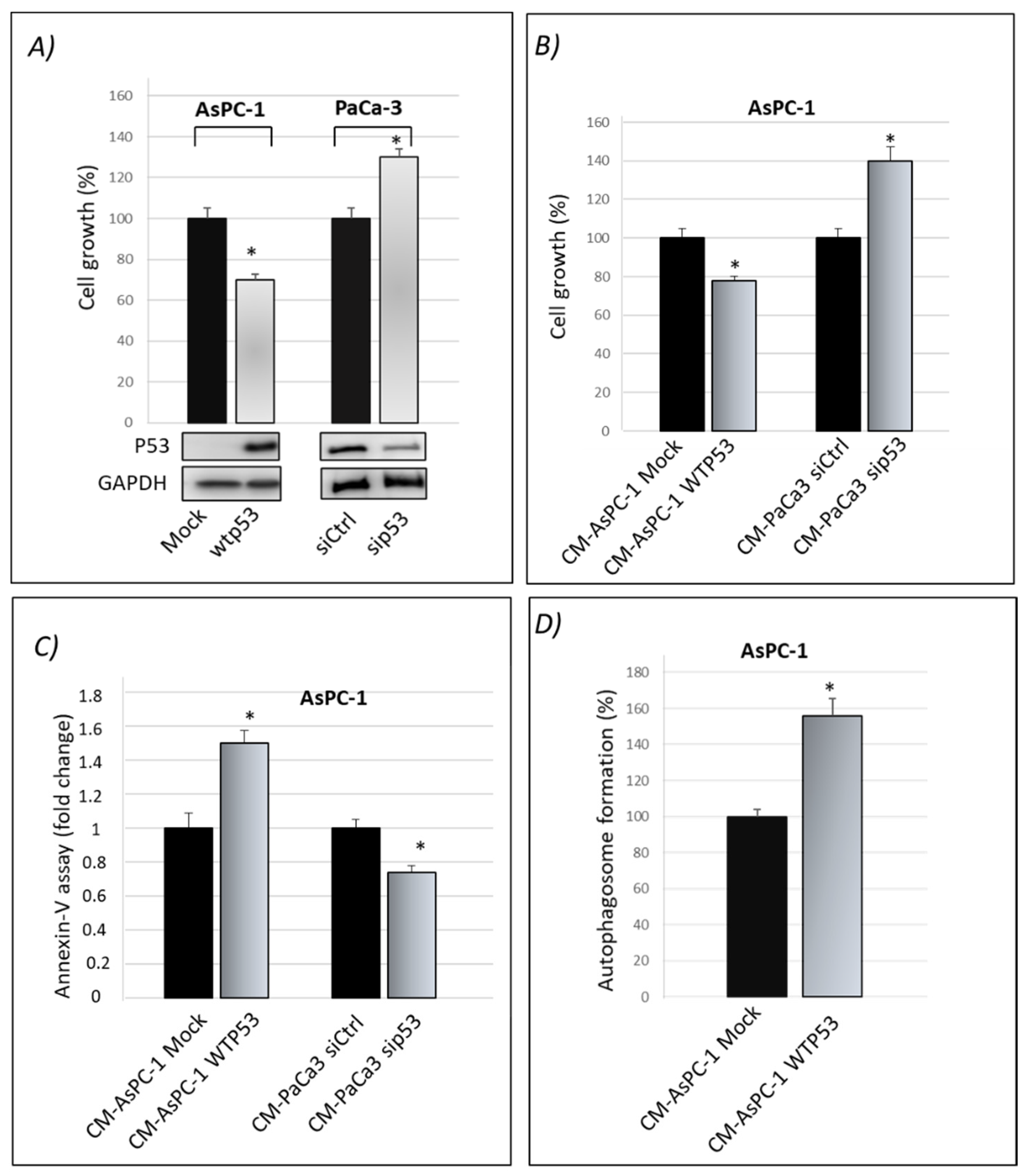
3.1. Cancer Cell Secretome of Wild-Type P53 PDAC Cells Exhibits Suppressor Roles
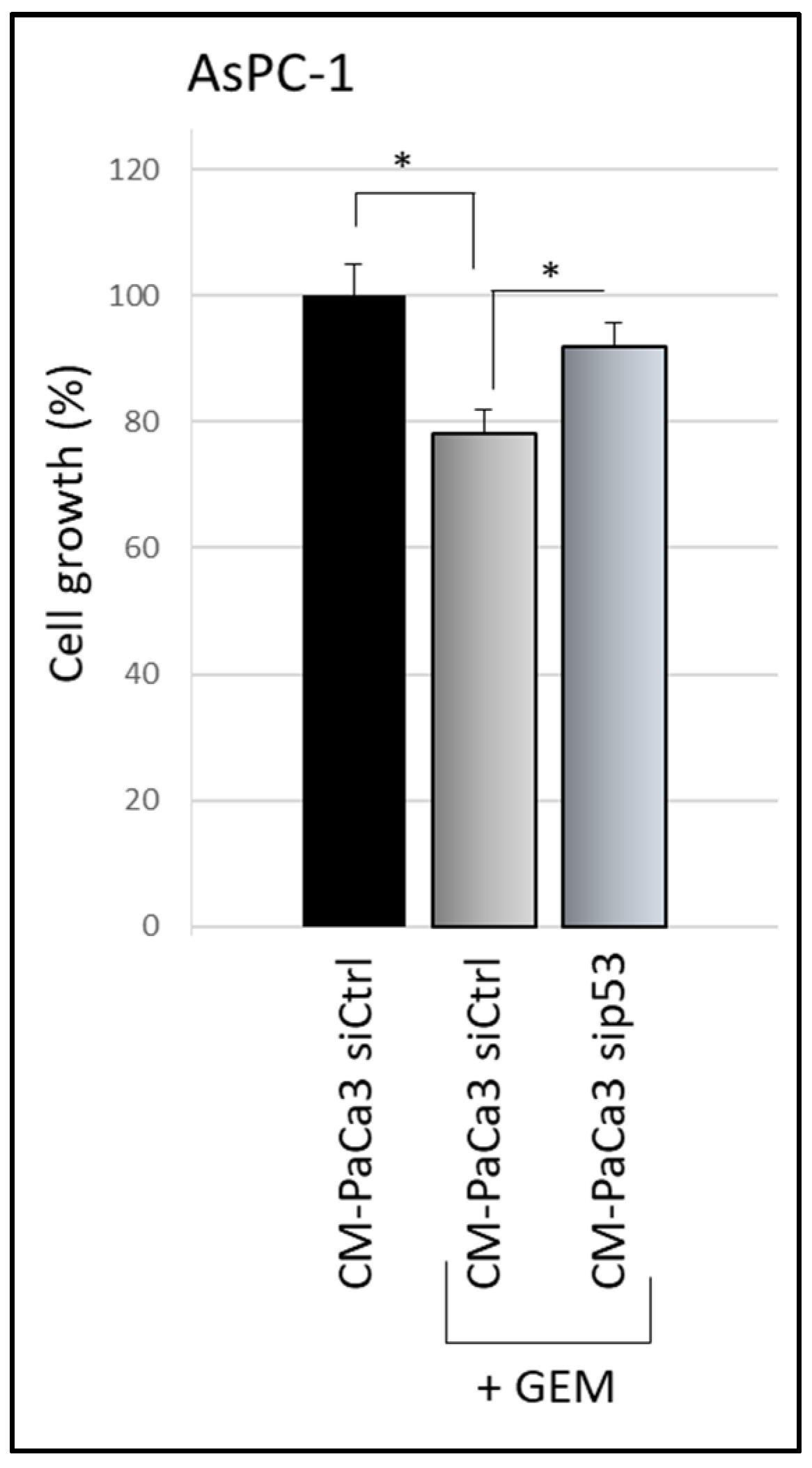
3.2. Wild-Type P53-Driven Secretome Counteracts Chemoresistance in PDAC Cells
3.3. Mutp53-Driven Secretome Stimulates Cancer Cell Migration
3.4. Identification of Secreted Proteins from Wild-Type P53-Driven Secretome
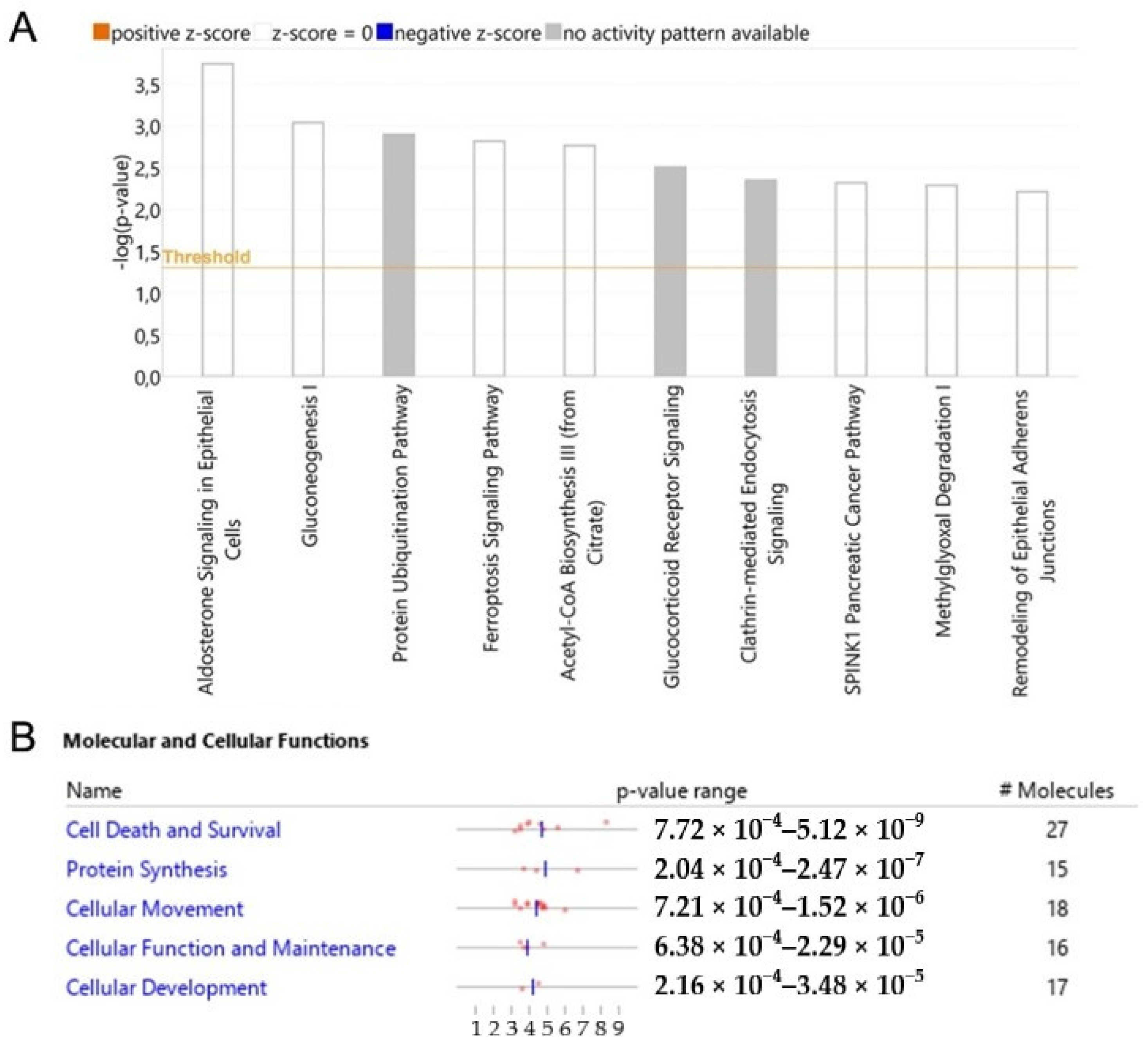
3.5. Dysregulated Pathways, Protein Interaction Networks and Upstream Regulators Related to Wtp53-Driven Secreted Proteins
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.K.; Chu, Q.D.; Tseng, J.F. Pancreatic Adenocarcinoma. In Surgical Oncology; Springer: New York, NY, USA, 2015; pp. 283–313. [Google Scholar]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, v56–v68. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B. Wild-Type p53: Tumors Can’t Stand It. Cell 2007, 128, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.-P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: An oncogenic transcription factor. Oncogene 2007, 26, 2212–2219. [Google Scholar] [CrossRef]
- Di Marco, M.; Astolfi, A.; Grassi, E.; Vecchiarelli, S.; Macchini, M.; Indio, V.; Casadei, R.; Ricci, C.; D’Ambra, M.; Taffurelli, G.; et al. Characterization of pancreatic ductal adenocarcinoma using whole transcriptome sequencing and copy number analysis by single-nucleotide polymorphism array. Mol. Med. Rep. 2015, 12, 7479–7484. [Google Scholar] [CrossRef][Green Version]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Park, C.; Na, D.; Han, J.Y.; Lee, J.; Park, O.-K.; Zhang, C.; Sung, C.O.; Moon, H.E.; Kim, Y.; et al. High prevalence of TP53 mutations is associated with poor survival and an EMT signature in gliosarcoma patients. Exp. Mol. Med. 2017, 49, e317. [Google Scholar] [CrossRef]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Butera, G.; Pacchiana, R.; Mullappilly, N.; Margiotta, M.; Bruno, S.; Conti, P.; Riganti, C.; Donadelli, M. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant p53 blocks SESN1/AMPK/PGC-1α/UCP2 axis increasing mitochondrial O2ˉ· production in cancer cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Cordani, M.; Padroni, C.; Blandino, G.; Di Agostino, S.; Donadelli, M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Butera, G.; Brandi, J.; Cavallini, C.; Scarpa, A.; Lawlor, R.T.; Scupoli, M.T.; Marengo, E.; Cecconi, D.; Manfredi, M.; Donadelli, M. The Mutant p53-Driven Secretome Has Oncogenic Functions in Pancreatic Ductal Adenocarcinoma Cells. Biomolecules 2020, 10, 884. [Google Scholar] [CrossRef]
- Cordani, M.; Pacchiana, R.; Butera, G.; D’Orazi, G.; Scarpa, A.; Donadelli, M. Mutant p53 proteins alter cancer cell secretome and tumour microenvironment: Involvement in cancer invasion and metastasis. Cancer Lett. 2016, 376, 303–309. [Google Scholar] [CrossRef]
- Brandi, J.; Manfredi, M.; Speziali, G.; Gosetti, F.; Marengo, E.; Cecconi, D. Proteomic approaches to decipher cancer cell secretome. Semin. Cell Dev. Biol. 2018, 78, 93–101. [Google Scholar] [CrossRef]
- Manfredi, M.; Martinotti, S.; Gosetti, F.; Ranzato, E.; Marengo, E. The secretome signature of malignant mesothelioma cell lines. J. Proteom. 2016, 145, 3–10. [Google Scholar] [CrossRef]
- Albanese, P.; Manfredi, M.; Re, A.; Marengo, E.; Saracco, G.; Pagliano, C. Thylakoid proteome modulation in pea plants grown at different irradiances: Quantitative proteomic profiling in a non-model organism aided by transcriptomic data integration. Plant J. 2018, 96, 786–800. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Manfredi, M.; Brandi, J.; Di Carlo, C.; Vita Vanella, V.; Barberis, E.; Marengo, E.; Patrone, M.; Cecconi, D. Marengo Mining cancer biology through bioinformatic analysis of proteomic data. Expert Rev. Proteom. 2019, 16, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003, 22, 7486–7495. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Markowitz, S.; Fearon, E.R.; Willson, J.K.V.; Vogelstein, B. Suppression of Human Colorectal Carcinoma Cell Growth by Wild-Type p53. Science 1990, 249, 912–915. [Google Scholar] [CrossRef]
- Shaw, P.; Bovey, R.; Tardy, S.; Sahli, R.; Sordat, B.; Costa, J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc. Natl. Acad. Sci. USA 1992, 89, 4495–4499. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, E.; Stiewe, T. p53′s Extended Reach: The Mutant p53 Secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Solomon, H.; Molchadsky, A.; Jacob-Berger, A.; Goldfinger, N.; Rotter, V. Various stress stimuli rewire the profile of liver secretome in a p53-dependent manner. Cell Death Dis. 2018, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Banerjee, P.; Shi, L.; Xiao, G.-Y.; Rodriguez, B.L.; Grzeskowiak, C.L.; Liu, X.; Yu, J.; Gibbons, D.L.; Russell, W.K.; et al. p53 loss activates prometastatic secretory vesicle biogenesis in the Golgi. Sci. Adv. 2021, 7, eabf4885. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Shi, L.; Banerjee, P.; Liu, X.; Guo, H.-F.; Yu, J.; Bota-Rabassedas, N.; Rodriguez, B.L.; Gibbons, D.L.; Russell, W.K.; et al. A protumorigenic secretory pathway activated by p53 deficiency in lung adenocarcinoma. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, F.W.; Svoboda, P.; Reed, M.; Pohl, J.; Pyrzynska, B.; Van Meir, E.G. Proteomic identification of the wt-p53-regulated tumor cell secretome. Oncogene 2006, 25, 7650–7661. [Google Scholar] [CrossRef][Green Version]
- Zhu, C.-J.; Wang, Q.-Q.; Zhou, J.-L.; Liu, H.-Z.; Hua, F.; Yang, H.-Z.; Hu, Z.-W. The mineralocorticoid receptor-p38MAPK-NFκB or ERK-Sp1 signal pathways mediate aldosterone-stimulated inflammatory and profibrotic responses in rat vascular smooth muscle cells. Acta Pharmacol. Sin. 2012, 33, 873–878. [Google Scholar] [CrossRef]
- Bekasi, S.; Zalatnai, A. Overexpression of Glucocorticoid Receptor in Human Pancreatic Cancer and in Xenografts. An Immunohistochemical Study. Pathol. Oncol. Res. 2009, 15, 561–566. [Google Scholar] [CrossRef]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef]
- Yoshii, Y.; Furukawa, T.; Saga, T.; Fujibayashi, Y. Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: Overview and application. Cancer Lett. 2015, 356, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.M.; Seth, A.K. The ubiquitin-mediated protein degradation pathway in cancer: Therapeutic implications. Eur. J. Cancer 2004, 40, 2217–2229. [Google Scholar] [CrossRef] [PubMed]
- Casari, I.; Howard, J.A.; Robless, E.E.; Falasca, M. Exosomal integrins and their influence on pancreatic cancer progression and metastasis. Cancer Lett. 2021, 507, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Baum, B.; Georgiou, M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J. Cell Biol. 2011, 192, 907–917. [Google Scholar] [CrossRef]
- Jäättelä, M. Escaping Cell Death: Survival Proteins in Cancer. Exp. Cell Res. 1999, 248, 30–43. [Google Scholar] [CrossRef]
- Hershey, J.W. Regulation of protein synthesis and the role of eIF3 in cancer. Braz. J. Med. Biol. Res. 2010, 43, 920–930. [Google Scholar] [CrossRef]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef]
- Kurosu, T.; Fukuda, T.; Miki, T.; Miura, O. BCL6 overexpression prevents increase in reactive oxygen species and inhibits apoptosis induced by chemotherapeutic reagents in B-cell lymphoma cells. Oncogene 2003, 22, 4459–4468. [Google Scholar] [CrossRef]
- Pagan, J.K.; Arnold, J.; Hanchard, K.J.; Kumar, R.; Bruno, T.; Jones, M.J.K.; Richard, D.J.; Forrest, A.; Spurdle, A.; Verdin, E.; et al. A Novel Corepressor, BCoR-L1, Represses Transcription through an Interaction with CtBP. J. Biol. Chem. 2007, 282, 15248–15257. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-Cadherin Promotes Metastasis via Multiple Downstream Transcriptional Pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef]
- Schlichtholz, B.; Turyn, J.; Goyke, E.; Biernacki, M.; Jaskiewicz, K.; Sledzinski, Z.; Swierczynski, J. Enhanced Citrate Synthase Activity in Human Pancreatic Cancer. Pancreas 2005, 30, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M.; Hwang, S.; Seong, R. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem. Biophys. Res. Commun. 2016, 471, 373–379. [Google Scholar] [CrossRef]
- Ryschich, E.; Huszty, G.; Knaebel, H.; Hartel, M.; Büchler, M.; Schmidt, J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur. J. Cancer 2004, 40, 1418–1422. [Google Scholar] [CrossRef]
- Moniaux, N.; Chakraborty, S.; Yalniz, M.; Gonzalez, J.; Shostrom, V.K.; Standop, J.; Lele, S.M.; Ouellette, M.; Pour, P.M.; Sasson, A.R.; et al. Early diagnosis of pancreatic cancer: Neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br. J. Cancer 2008, 98, 1540–1547. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Pavlou, M.P.; Diamandis, E.P. Cancer secretomics reveal pathophysiological pathways in cancer molecular oncology. Mol. Oncol. 2010, 4, 496–510. [Google Scholar] [CrossRef]
- Schiarea, S.; Solinas, G.; Allavena, P.; Scigliuolo, G.M.; Bagnati, R.; Fanelli, R.; Chiabrando, C. Secretome Analysis of Multiple Pancreatic Cancer Cell Lines Reveals Perturbations of Key Functional Networks. J. Proteome Res. 2010, 9, 4376–4392. [Google Scholar] [CrossRef]
- Hwang, J.-H.; Lee, S.H.; Lee, K.H.; Lee, K.Y.; Kim, H.; Ryu, J.K.; Yoon, Y.B.; Kim, Y.-T. Cathepsin B is a target of Hedgehog signaling in pancreatic cancer. Cancer Lett. 2009, 273, 266–272. [Google Scholar] [CrossRef]
- Wu, Y.; Lee, Y.; Li, W.; Hsu, W.; Lin, H.; Chang, L.; Huang, A.; Jhan, J.; Wu, W.; Li, C.; et al. High Transaldolase 1 expression predicts poor survival of patients with upper tract urothelial carcinoma. Pathol. Int. 2021, 71, 463–470. [Google Scholar] [CrossRef]
- Brandi, J.; Pozza, E.D.; Dando, I.; Biondani, G.; Robotti, E.; Jenkins, R.; Elliott, V.; Park, K.; Marengo, E.; Costello, E.; et al. Secretome protein signature of human pancreatic cancer stem-like cells. J. Proteom. 2016, 136, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Meng, T.; Zheng, X.; Liu, Y.; Hao, R.; Yan, Y.; Chen, S.; You, H.; Xing, J.; Dong, Y. Transgelin 2 Promotes Paclitaxel Resistance, Migration, and Invasion of Breast Cancer by Directly Interacting with PTEN and Activating PI3K/Akt/GSK-3β Pathway. Mol. Cancer Ther. 2019, 18, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Berge, G.; Mælandsmo, G.M. Evaluation of potential interactions between the metastasis-associated protein S100A4 and the tumor suppressor protein p53. Amino Acids 2010, 41, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Orre, L.M.; Pernemalm, M.; Lengqvist, J.; Lewensohn, R.; Lehtiö, J. Up-regulation, Modification, and Translocation of S100A6 Induced by Exposure to Ionizing Radiation Revealed by Proteomics Profiling. Mol. Cell. Proteom. 2007, 6, 2122–2131. [Google Scholar] [CrossRef]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496.e6. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Seo, Y.R.; Kelley, M.R.; Smith, M.L. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 14548–14553. [Google Scholar] [CrossRef]
- Lin, C.-W.; Lai, G.-M.; Chen, K.-C.; Lin, T.-H.; Fan, J.-J.; Hsu, R.-L.; Chou, C.-M.; Lin, C.-M.; Kandaswami, C.C.; Lee, M.-T.; et al. RPS12 increases the invasiveness in cervical cancer activated by c-Myc and inhibited by the dietary flavonoids luteolin and quercetin. J. Funct. Foods 2015, 19, 236–247. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, X. Roles of nucleolin. Saudi Med. J. 2016, 37, 1312–1318. [Google Scholar] [CrossRef]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef]
- New, M.; Van Acker, T.; Sakamaki, J.-I.; Jiang, M.; Saunders, R.E.; Long, J.; Wang, V.M.-Y.; Behrens, A.; Cerveira, J.; Sudhakar, P.; et al. MDH1 and MPP7 Regulate Autophagy in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 1884–1898. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Tyagi, R.; Chowdhury, S.; Reilly, L.; Fu, C.; Xu, R.; Resnick, A.C.; Snyder, S.H. IPMK Mediates Activation of ULK Signaling and Transcriptional Regulation of Autophagy Linked to Liver Inflammation and Regeneration. Cell Rep. 2019, 26, 2692–2703.e7. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Le, N.; Alotaibi, M.; Gewirtz, D.A. Cytotoxic Autophagy in Cancer Therapy. Int. J. Mol. Sci. 2014, 15, 10034–10051. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot ID | Uniprot Accession Name | Protein Name | Gene Name | FC (KD/wt p53) | p-Value |
|---|---|---|---|---|---|
| O00410 | IPO5_HUMAN | Importin-5 | IPO5 | 4.7 | 4.31 × 10−4 |
| O75874 | IDHC_HUMAN | Isocitrate dehydrogenase | IDH1 | 3.0 | 3.56 × 10−4 |
| P01008 | ANT3_HUMAN | Antithrombin-III | SERPINC1 | 1.6 | 2.66 × 10−4 |
| P02786 | TFR1_HUMAN | Transferrin receptor protein 1 | TFRC | 3.8 | 8.59 × 10−4 |
| P04792 | HSPB1_HUMAN | Heat shock protein beta-1 | HSPB1 | 1.8 | 1.72 × 10−2 |
| P04908 | H2A1_HUMAN | Histone H2A type 1-B/E | HIST1 | 2.1 | 2.39 × 10−3 |
| P07478 | TRY2_HUMAN | Trypsin-2 | PRSS2 | 1.3 | 4.27 × 10−2 |
| P07858 | CATB_HUMAN | Cathepsin B | CTSB | 2.6 | 3.33 × 10−3 |
| P14136 | K2C8_HUMAN | Glial fibrillary acidic protein | GFAP | 5.5 | 1.49 × 10−2 |
| P14555 | PA2GA_HUMAN | Phospholipase A2 | PLA2G2A | 2.4 | 1.86 × 10−4 |
| P19338 | NUCL_HUMAN | Nucleolin | NCL | 1.4 | 9.66 × 10−4 |
| P22692 | IBP4_HUMAN | Insulin-like growth factor-binding protein 4 | IGFBP4 | 1.9 | 9.48 × 10−3 |
| P25398 | RS12_HUMAN | 40S ribosomal protein S12 | RPS12 | 1.6 | 2.92 × 10−2 |
| P37837 | TALDO_HUMAN | Transaldolase | TALDO1 | 1.4 | 4.35 × 10−3 |
| P40925 | MDHC_HUMAN | Malate dehydrogenase | MDH1 | 1.8 | 9.26 × 10−3 |
| P53396 | ACLY_HUMAN | ATP-citrate synthase | ACLY | 2.9 | 6.18 × 10−4 |
| P54652 | HSP7C_HUMAN | Heat shock-related 70 kDa protein 2 | HSPA2 | 1.7 | 6.79 × 10−3 |
| P62158 | CALM_HUMAN | Calmodulin | CALM1 | 1.5 | 1.31 × 10−2 |
| P80188 | NGAL_HUMAN | Neutrophil gelatinase-associated lipocalin | LCN2 | 2.0 | 7.20 × 10−5 |
| Q01105 | SET_HUMAN | Protein SET | SET | 1.6 | 1.55 × 10−2 |
| Q04828 | AK1C1_HUMAN | Aldo-keto reductase family 1 member C1 | AKR1C1 | 1.5 | 8.20 × 10−3 |
| Q15582 | BGH3_HUMAN | Transforming growth factor-beta-induced protein ig-h3 | TGFBI | 1.4 | 5.93 × 10−3 |
| Q32P51 | RA1L2_HUMAN | Heterogeneous nuclear ribonucleoprotein A1-like 2 | HNRNPA1L2 | 1.3 | 7.76 × 10−3 |
| Q5H9F3 | BCORL_HUMAN | BCL-6 corepressor-like protein 1 | BCORL1 | 24.3 | 5.74 × 10−5 |
| Q6UWE0 | LRSM1_HUMAN | E3 ubiquitin-protein ligase LRSAM1 | LRSAM1 | 2.2 | 6.48 × 10−3 |
| Q86TI0 | TBCD1_HUMAN | TBC1 domain family member 1 | TBC1D1 | 2.5 | 8.13 × 10−5 |
| Q92598 | HS105_HUMAN | Heat shock protein 105 kDa | HSPH1 | 1.9 | 1.65 × 10−2 |
| Q96HU8 | DIRA2_HUMAN | GTP-binding protein Di-Ras2 | DIRAS2 | 36.4 | 4.02 × 10−4 |
| Q96QV6 | H2A1A_HUMAN | Histone H2A type 1-A | HIST1H2AA | 1.3 | 1.29 × 10−3 |
| A8MST6 | CEP78_HUMAN | Centrosomal protein of 78 kDa | CEP78 | 23.7 | 2.99 × 10−3 |
| B2RPK0 | HGB1A_HUMAN | Putative high mobility group protein B1-like 1 | HMGB1P1 | 0.6 | 8.43 × 10−3 |
| O14556 | G3P_HUMAN | Glyceraldehyde-3-phosphate dehydrogenase | GAPDHS | 0.5 | 2.66 × 10−2 |
| P01034 | CYTC_HUMAN | Cystatin-C | CST3 | 0.6 | 4.58 × 10−3 |
| P02538 | K2C6B_HUMAN | Keratin, type II cytoskeletal 6A | KRT6A | 0.5 | 1.53 × 10−3 |
| P02768 | ALBU_HUMAN | Serum albumin | ALB | 0.6 | 4.06 × 10−4 |
| P07737 | PROF1_HUMAN | Profilin-1 | PFN1 | 0.5 | 9.28 × 10−6 |
| P08238 | HS90B_HUMAN | Heat shock protein HSP 90-beta | HSP90AB1 | 0.6 | 1.14 × 10−2 |
| P14324 | FPPS_HUMAN | Farnesyl pyrophosphate synthase | FDPS | 0.3 | 9.93 × 10−4 |
| P23284 | PPIB_HUMAN | Peptidyl-prolyl cis-trans isomerase B | PPIB | 0.7 | 1.40 × 10−2 |
| P26447 | S10A4_HUMAN | Protein S100-A4 | S100A4 | 0.3 | 1.72 × 10−4 |
| P37802 | TAGL2_HUMAN | Transgelin-2 | TAGLN2 | 0.3 | 2.18 × 10−3 |
| P60709 | ACTB_HUMAN | Actin, cytoplasmic 1 | ACTB | 0.6 | 5.29 × 10−5 |
| Q04760 | LGUL_HUMAN | Lactoylglutathione lyase | GLO1 | 0.5 | 5.79 × 10−4 |
| Q13885 | TBB5_HUMAN | Tubulin beta-3 chain | TUBB3 | 0.3 | 2.70 × 10−3 |
| Q58FG1 | HS90A_HUMAN | Putative heat shock protein HSP 90-alpha A4 | HSP90AA4P | 0.3 | 2.16 × 10−3 |
| Q92688 | AN32B_HUMAN | Acidic leucine-rich nuclear phosphoprotein 32 family member B | ANP32B | 0.4 | 5.73 × 10−3 |
| Q9Y536 | PPIA_HUMAN | Peptidyl-prolyl cis-trans isomerase A-like 4A | PPIAL4A | 0.4 | 3.52 × 10−3 |
| Diseases and Functions | ||
| Leukocytes movement | ||
| Protein | Regulation | Function |
| HSPB1 | Up-regulated | Decrease movement of leucocytes |
| PPIB | Down-regulated | Increase movement of leucocytes |
| ACTB | Down-regulated | Increase movement of leucocytes |
| HS90B | Down-regulated | Increase movement of leucocytes |
| ALB | Down-regulated | Increase movement of leucocytes |
| S10A4 | Down-regulated | Increase movement of leucocytes |
| Carcinoma | ||
| Protein | Regulation | Function |
| CATB | Up-regulated | Increase carcinoma |
| ALB | Down-regulated | Decrease carcinoma |
| CYTC | Down-regulated | Decrease carcinoma |
| SAA4 | Down-regulated | Decrease carcinoma |
| Advanced malignant tumour | ||
| Protein | Regulation | Function |
| CATB | Up-regulated | Increase advanced malignant tumour |
| BGH3 | Up-regulated | Increase advanced malignant tumour |
| ACTB | Down-regulated | Decrease advanced malignant tumour |
| CYTC | Down-regulated | Decrease advanced malignant tumour |
| SAA4 | Down-regulated | Increase advanced malignant tumour |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butera, G.; Manfredi, M.; Fiore, A.; Brandi, J.; Pacchiana, R.; De Giorgis, V.; Barberis, E.; Vanella, V.; Galasso, M.; Scupoli, M.T.; et al. Tumor Suppressor Role of Wild-Type P53-Dependent Secretome and Its Proteomic Identification in PDAC. Biomolecules 2022, 12, 305. https://doi.org/10.3390/biom12020305
Butera G, Manfredi M, Fiore A, Brandi J, Pacchiana R, De Giorgis V, Barberis E, Vanella V, Galasso M, Scupoli MT, et al. Tumor Suppressor Role of Wild-Type P53-Dependent Secretome and Its Proteomic Identification in PDAC. Biomolecules. 2022; 12(2):305. https://doi.org/10.3390/biom12020305
Chicago/Turabian StyleButera, Giovanna, Marcello Manfredi, Alessandra Fiore, Jessica Brandi, Raffaella Pacchiana, Veronica De Giorgis, Elettra Barberis, Virginia Vanella, Marilisa Galasso, Maria Teresa Scupoli, and et al. 2022. "Tumor Suppressor Role of Wild-Type P53-Dependent Secretome and Its Proteomic Identification in PDAC" Biomolecules 12, no. 2: 305. https://doi.org/10.3390/biom12020305
APA StyleButera, G., Manfredi, M., Fiore, A., Brandi, J., Pacchiana, R., De Giorgis, V., Barberis, E., Vanella, V., Galasso, M., Scupoli, M. T., Marengo, E., Cecconi, D., & Donadelli, M. (2022). Tumor Suppressor Role of Wild-Type P53-Dependent Secretome and Its Proteomic Identification in PDAC. Biomolecules, 12(2), 305. https://doi.org/10.3390/biom12020305