


Genetic Ablation of Ankrd1 Mitigates Cardiac Damage during Experimental Autoimmune Myocarditis in Mice
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Immunization
2.3. Echocardiography
2.4. Body, Heart, and Lung Weights
2.5. Histology
2.6. Quantitative RT-PCR
2.7. Protein Isolation and Western Blot Analysis
2.8. Statistical Analysis
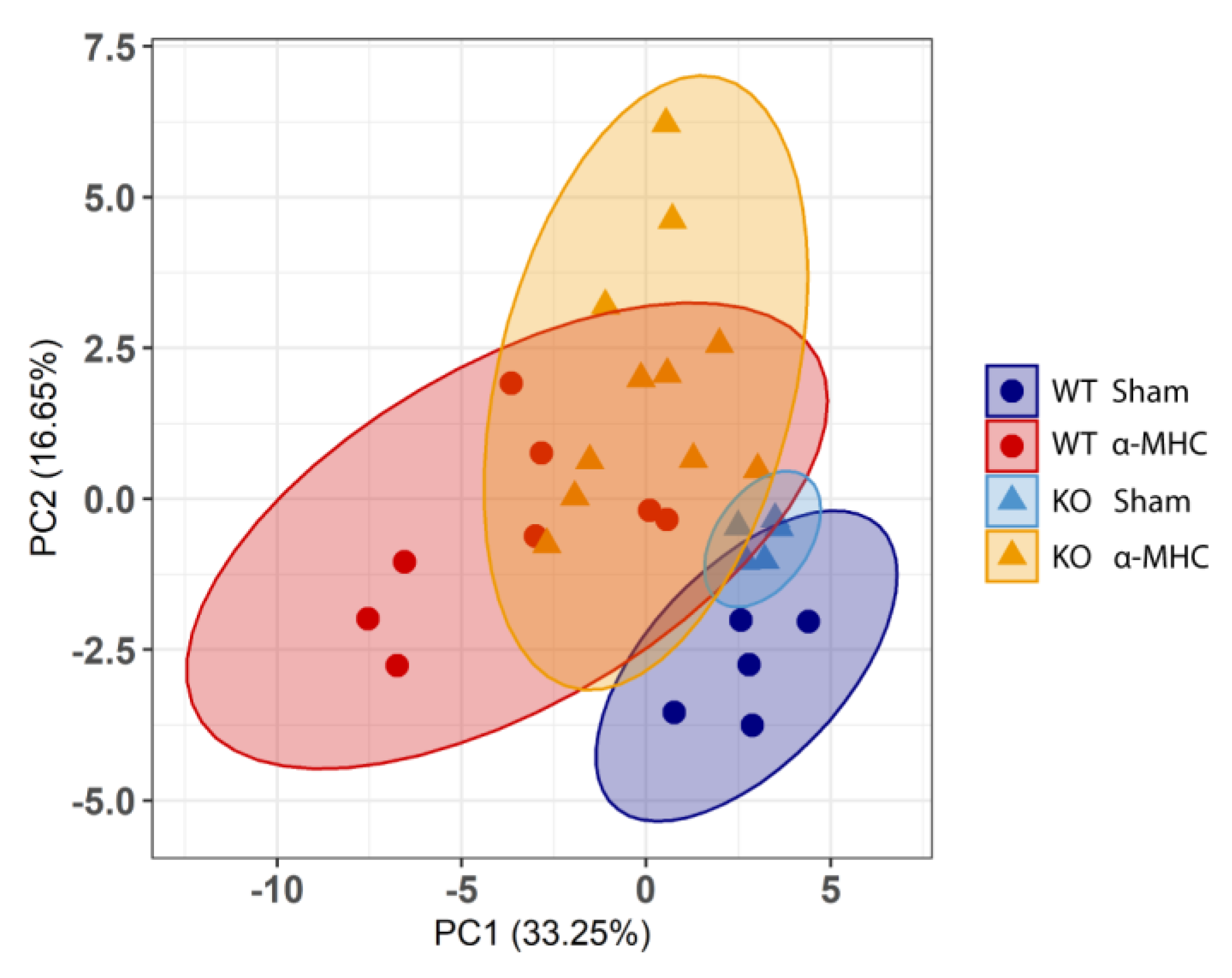
3. Results
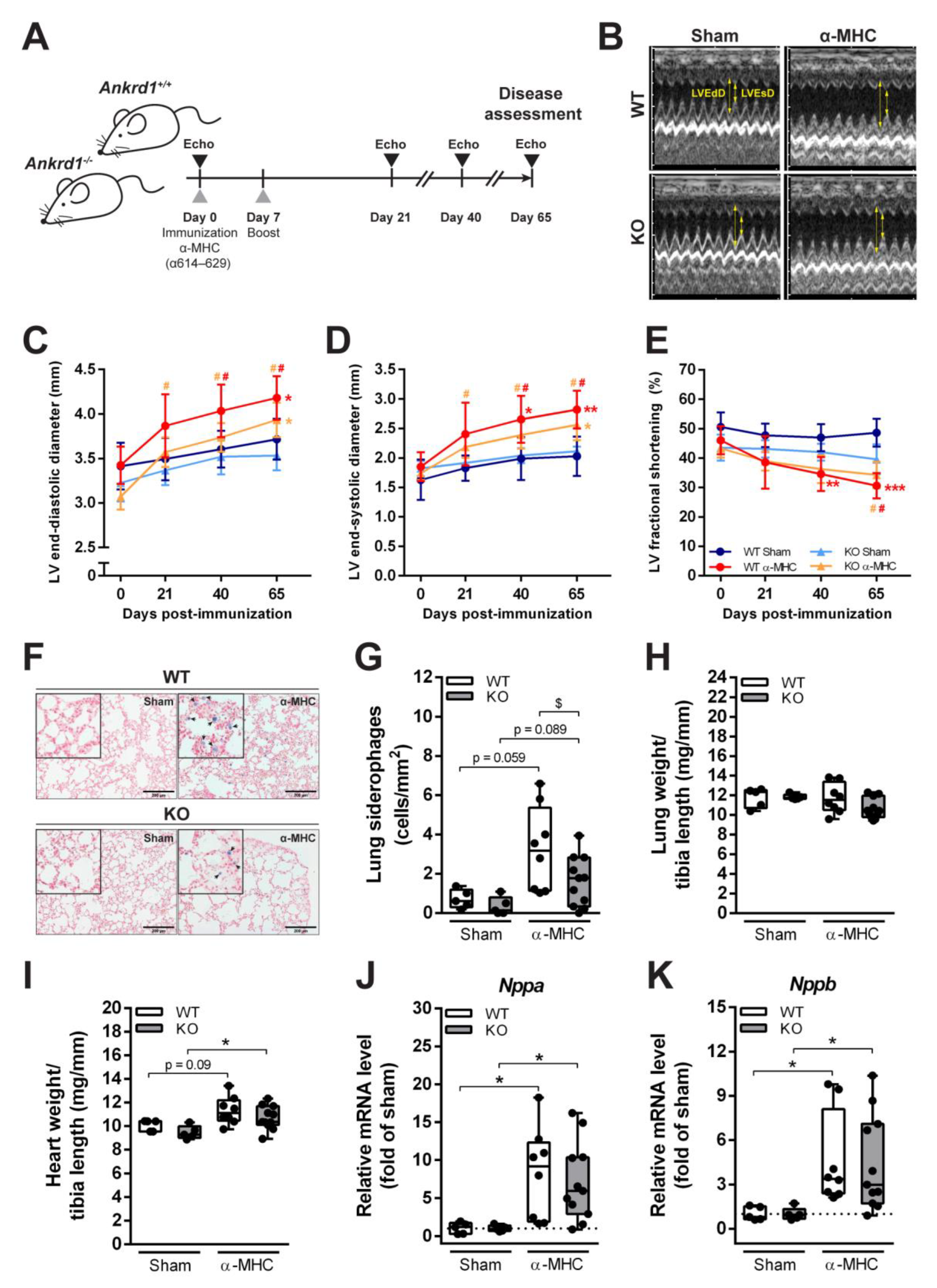
3.1. EAM Leads to HF in ANKRD1-Deficient Mice
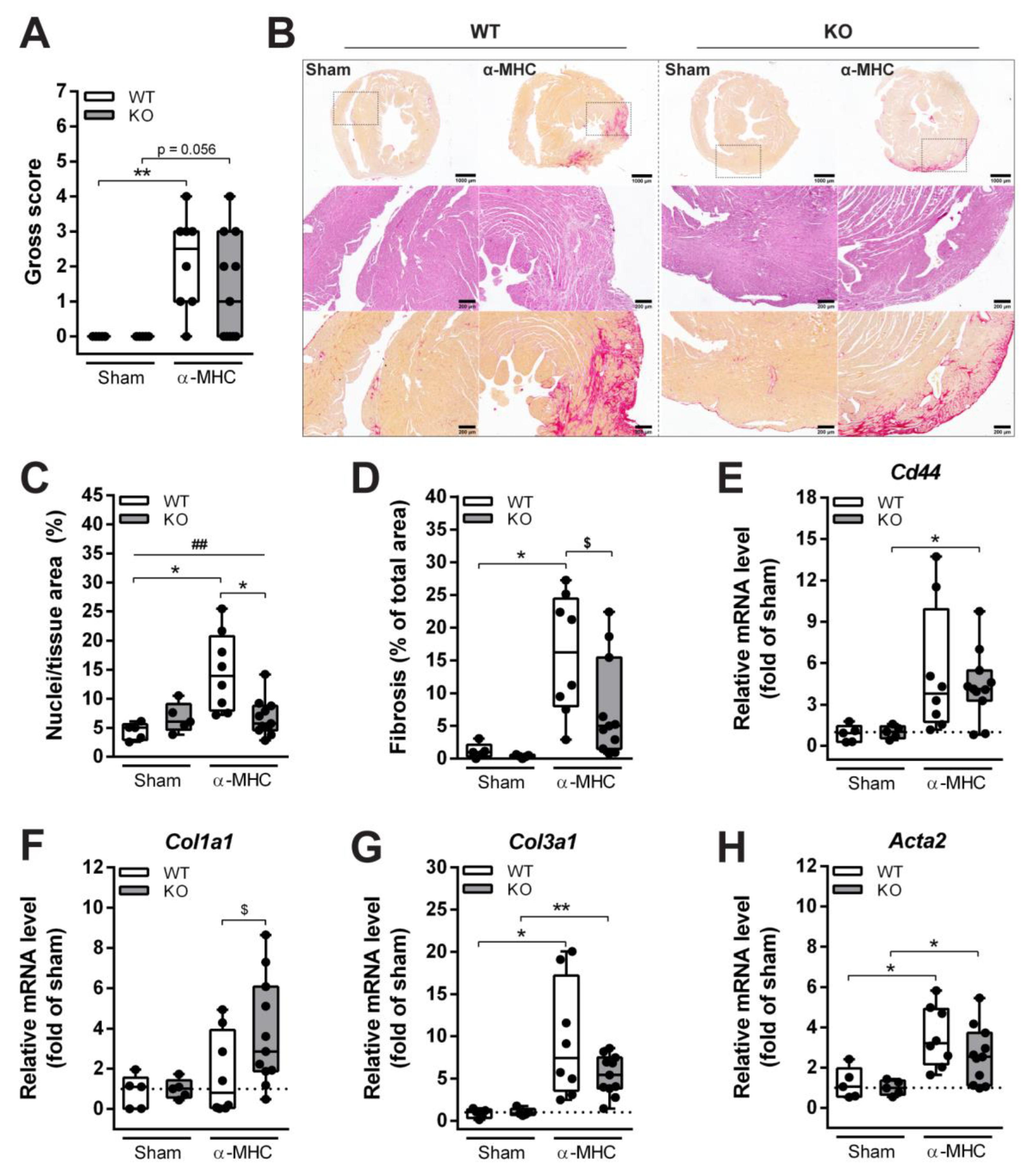
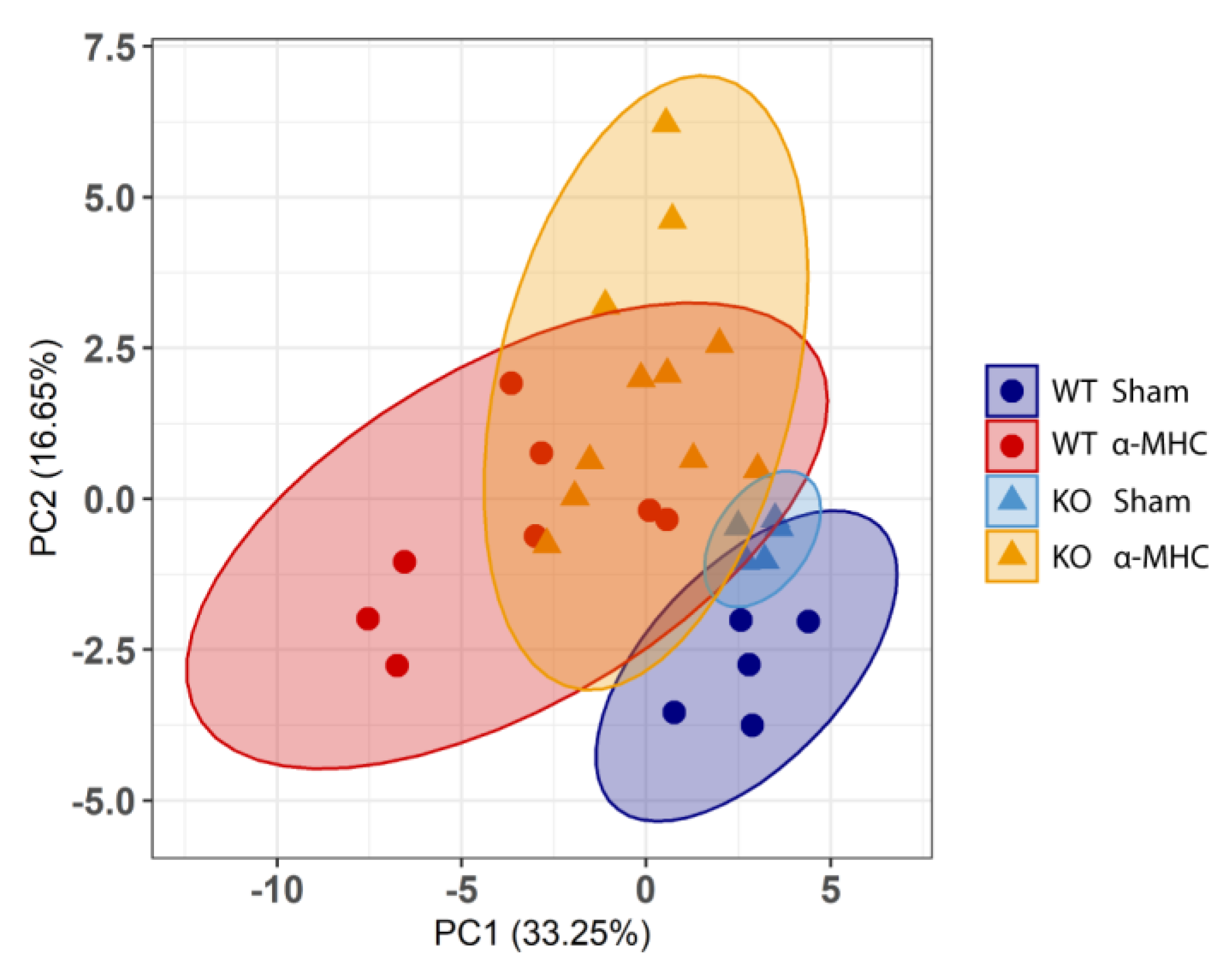
3.2. Loss of ANKRD1 Is Beneficial in the EAM-Induced DCM
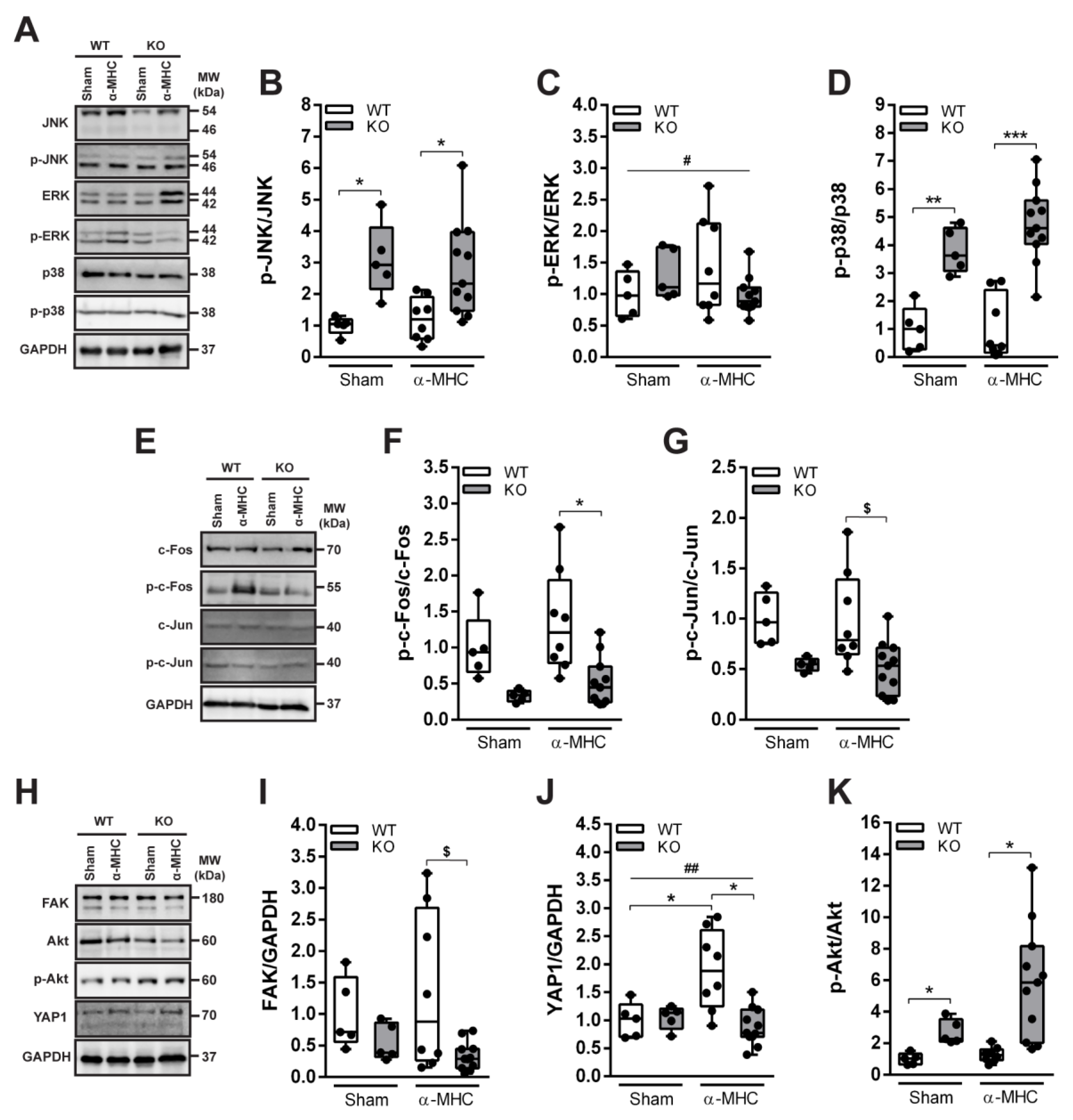
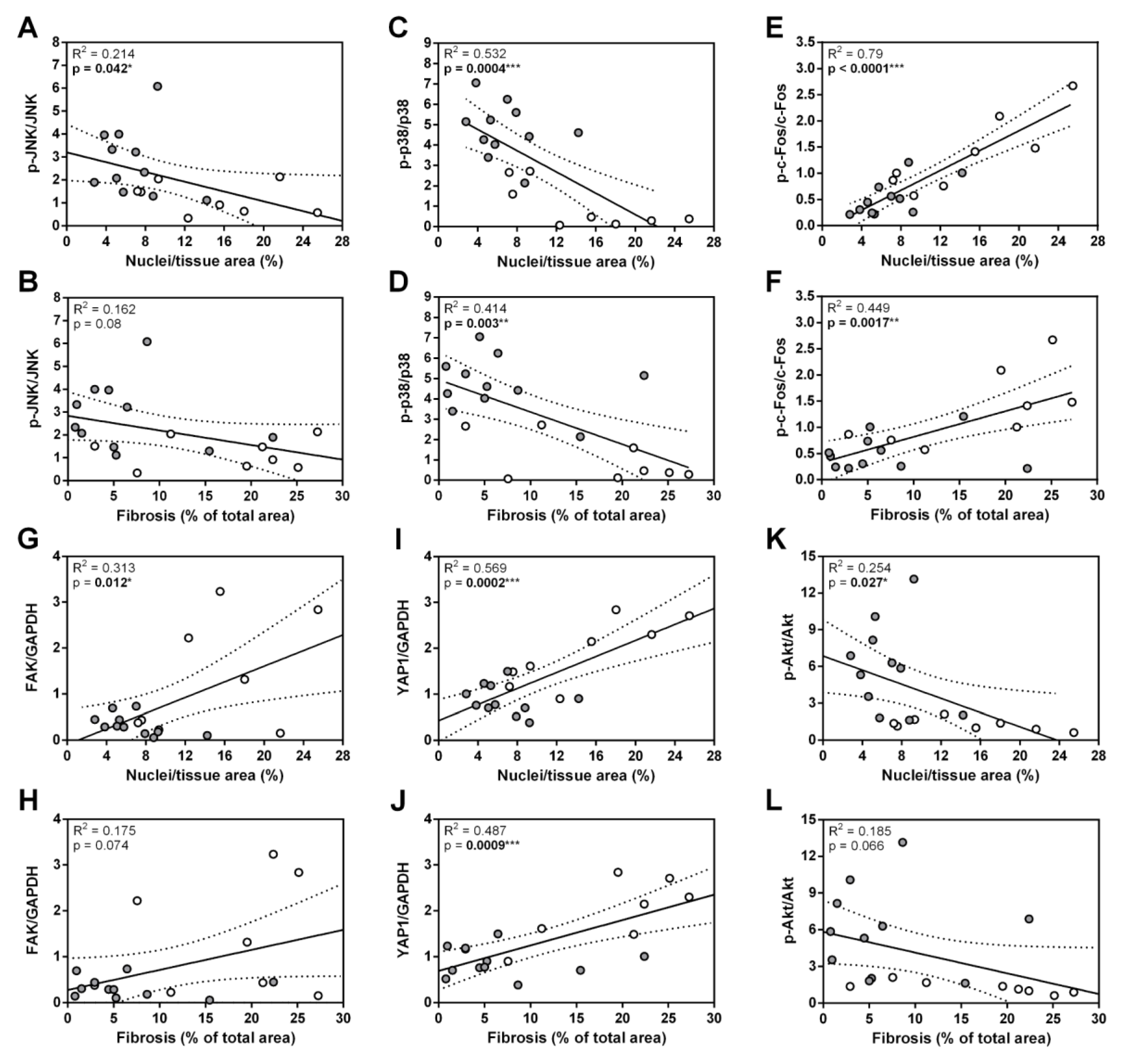
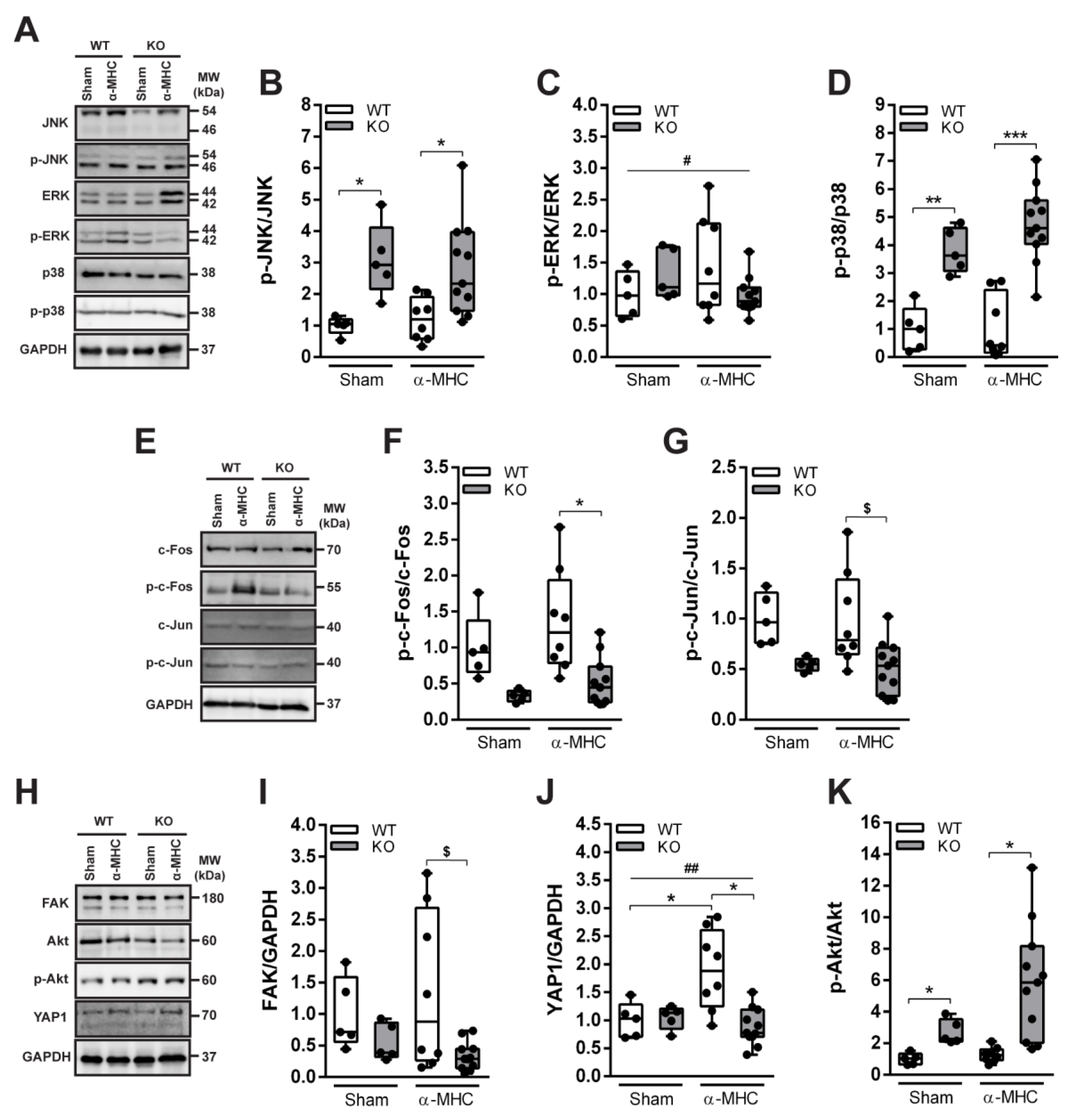
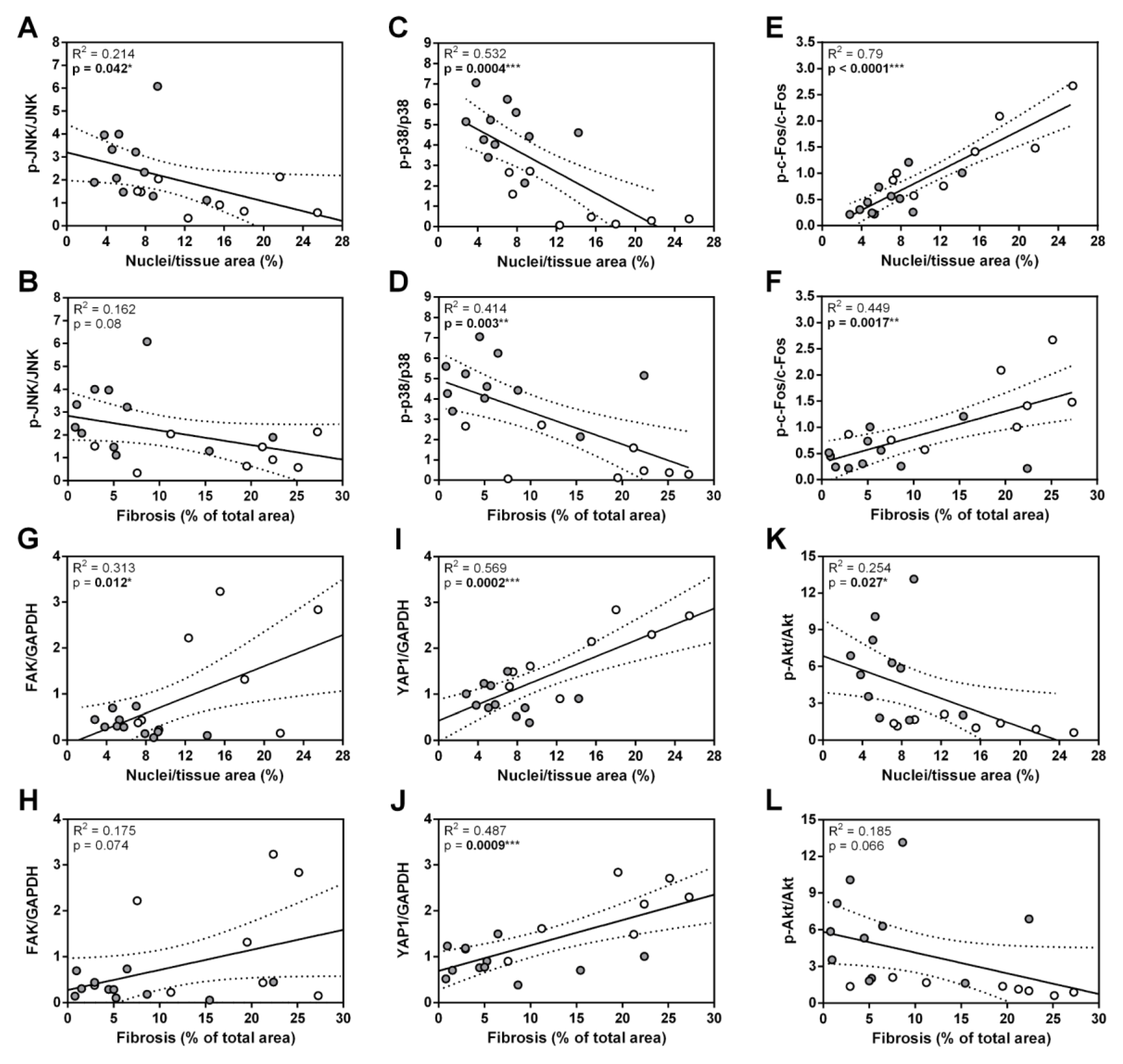
3.3. ANKRD1 - Deficiency Affects Overall Levels of MAPK/AP1 and the Response of Mechanosensing-Related Proteins in Post-EAM DCM
3.4. ANKRD1 Is Not a Major HF Mediator in Post-MC DCM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, A. The fate of acute myocarditis between spontaneous improvement and evolution to dilated cardiomyopathy: A review. Heart 2001, 85, 499–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschöpe, C.; Cooper, L.; Torre-Amione, G.; Van Linthout, S. Management of Myocarditis-Related Cardiomyopathy in Adults. Circ. Res. 2019, 124, 1568–1583. [Google Scholar] [CrossRef] [PubMed]
- Brüstle, K.; Heidecker, B. Checkpoint inhibitor induced cardiotoxicity: Managing the drawbacks of our newest agents against cancer. Oncotarget 2017, 8, 106165–106166. [Google Scholar] [CrossRef] [PubMed]
- Bruestle, K.; Hackner, K.; Kreye, G.; Heidecker, B. Autoimmunity in Acute Myocarditis: How Immunopathogenesis Steers New Directions for Diagnosis and Treatment. Curr. Cardiol. Rep. 2020, 22, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Buskiewicz, I.; Huber, S.; Fairweather, D. Chapter 12—Viral myocarditis and dilated cardiomyopathy: Mechanisms of cardiac injury, inflammation, and fibrosis. In Vascular Responses to Pathogens; Gavins, F.N.E., Stokes, K.Y., Eds.; Academic Press: Boston, MA, USA, 2016. [Google Scholar]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Ninh, V.K.; Brown, J.H. The contribution of the cardiomyocyte to tissue inflammation in cardiomyopathies. Curr. Opin. Physiol. 2020, 19, 129–134. [Google Scholar] [CrossRef]
- Lun, A.S.; Chen, J.; Lange, S. Probing Muscle Ankyrin-Repeat Protein (MARP) Structure and Function. Anat. Rec. 2014, 297, 1615–1629. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Fleming, J.R.; Lange, S.; Hessel, A.L.; Bogomolovas, J.; Stronczek, C.; Grundei, D.; Ghassemian, M.; Biju, A.; Börgeson, E.; et al. Molecular Characterisation of Titin N2A and Its Binding of CARP Reveals a Titin/Actin Cross-Linking Mechanism. J. Mol. Biol. 2021, 433, 166901. [Google Scholar] [CrossRef]
- Bang, M.-L.; Bogomolovas, J.; Chen, J. Understanding the molecular basis of cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H181–H233. [Google Scholar] [CrossRef] [PubMed]
- Aihara, Y.; Kurabayashi, M.; Saito, Y.; Ohyama, Y.; Tanaka, T.; Takeda, S.-I.; Tomaru, K.; Sekiguchi, K.-I.; Arai, M.; Nakamura, T.; et al. Cardiac Ankyrin Repeat Protein Is a Novel Marker of Cardiac Hypertrophy. Hypertension 2000, 36, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogomolovas, J.; Brohm, K.; Čelutkienė, J.; Balčiūnaitė, G.; Bironaitė, D.; Bukelskienė, V.; Daunoravičus, D.; Witt, C.C.; Fielitz, J.; Grabauskienė, V.; et al. Induction of Ankrd1 in Dilated Cardiomyopathy Correlates with the Heart Failure Progression. BioMed Res. Int. 2015, 2015, 273936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrer, I.; Roselló-Lletí, E.; Rivera, M.; Molina-Navarro, M.M.; Tarazón, E.; Ortega, A.; Martínez-Dolz, L.; Triviño, J.C.; Lago, F.; González-Juanatey, J.R.; et al. RNA-sequencing analysis reveals new alterations in cardiomyocyte cytoskeletal genes in patients with heart failure. Lab. Investig. 2014, 94, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Cui, C.; Huang, Y.; Zhang, X.; Zhang, H.; Hu, S. Upregulated expression of cardiac ankyrin repeat protein in human failing hearts due to arrhythmogenic right ventricular cardiomyopathy. Eur. J. Heart Fail. 2009, 11, 559–566. [Google Scholar] [CrossRef]
- Zolk, O.; Frohme, M.; Maurer, A.; Kluxen, F.-W.; Hentsch, B.; Zubakov, D.; Hoheisel, J.D.; Zucker, I.H.; Pepe, S.; Eschenhagen, T. Cardiac ankyrin repeat protein, a negative regulator of cardiac gene expression, is augmented in human heart failure. Biochem. Biophys. Res. Commun. 2002, 293, 1377–1382. [Google Scholar] [CrossRef]
- Zhang, N.; Ye, F.; Zhou, Y.; Zhu, W.; Xie, C.; Zheng, H.; Chen, H.; Chen, J.; Xie, X. Cardiac ankyrin repeat protein contributes to dilated cardiomyopathy and heart failure. FASEB J. 2021, 35, e21488. [Google Scholar] [CrossRef]
- Bang, M.-L.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Chien, K.R.; Chen, J. The Muscle Ankyrin Repeat Proteins CARP, Ankrd2, and DARP Are Not Essential for Normal Cardiac Development and Function at Basal Conditions and in Response to Pressure Overload. PLoS ONE 2014, 9, e93638. [Google Scholar] [CrossRef] [Green Version]
- Samaras, S.E.; Almodóvar-García, K.; Wu, N.; Yu, F.; Davidson, J.M. Global Deletion of Ankrd1 Results in a Wound-Healing Phenotype Associated with Dermal Fibroblast Dysfunction. Am. J. Pathol. 2014, 185, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Lange, S.; Gehmlich, K.; Lun, A.S.; Blondelle, J.; Hooper, C.; Dalton, N.D.; Alvarez, E.A.; Zhang, X.; Bang, M.-L.; Abassi, Y.A.; et al. MLP and CARP are linked to chronic PKCα signalling in dilated cardiomyopathy. Nat. Commun. 2016, 7, 12120. [Google Scholar] [CrossRef]
- Zhong, L.; Chiusa, M.; Cadar, A.G.; Lin, A.; Samaras, S.; Davidson, J.M.; Lim, C.C. Targeted inhibition of ANKRD1 disrupts sarcomeric ERK-GATA4 signal transduction and abrogates phenylephrine-induced cardiomyocyte hypertrophy. Cardiovasc. Res. 2015, 106, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čiháková, D.; Sharma, R.B.; Fairweather, D.; Afanasyeva, M.; Rose, N.R. Animal Models for Autoimmune Myocarditis and Autoimmune Thyroiditis. Methods Mol. Med. 2004, 102, 175–194. [Google Scholar] [CrossRef]
- Błyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neu, N.; Rose, N.R.; Beisel, K.W.; Herskowitz, A.; Gurri-Glass, G.; Craig, S.W. Cardiac myosin induces myocarditis in genetically predisposed mice. J. Immunol. 1987, 139, 3630–3636. [Google Scholar] [PubMed]
- Fontes, J.A.; Barin, J.G.; Talor, M.V.; Stickel, N.; Schaub, J.; Rose, N.R.; Čiháková, D. Complete Freund’s adjuvant induces experimental autoimmune myocarditis by enhancing IL-6 production during initiation of the immune response. Immun. Inflamm. Dis. 2017, 5, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Bogomolovas, J.; Šimoliūnas, E.; Rinkūnaitė, I.; Smalinskaitė, L.; Podkopajev, A.; Bironaitė, D.; Weis, C.-A.; Marx, A.; Bukelskienė, V.; Gretz, N.; et al. A Novel Murine Model of Parvovirus Associated Dilated Cardiomyopathy Induced by Immunization with VP1-Unique Region of Parvovirus B. BioMed Res. Int. 2016, 2016, 1627184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-Activated Protein Kinase Signaling in the Heart: Angels Versus Demons in a Heart-Breaking Tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Fan, G.; Zhao, H.; Wang, Z.; Li, F.; Zhang, P.; Zhang, J.; Wang, X.; Wang, W. Targeted inhibition of Focal Adhesion Kinase Attenuates Cardiac Fibrosis and Preserves Heart Function in Adverse Cardiac Remodeling. Sci. Rep. 2017, 7, 43146. [Google Scholar] [CrossRef] [Green Version]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated Protein Isoform 1 (Yap1) Promotes Cardiomyocyte Survival and Growth to Protect against Myocardial Ischemic Injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef] [Green Version]
- Sharifi-Sanjani, M.; Berman, M.; Goncharov, D.; Alhamaydeh, M.; Avolio, T.; Baust, J.; Chang, B.; Kobir, A.; Ross, M.; Croix, C.S.; et al. Yes-Associated Protein (Yap) is Up-Regulated in Heart Failure and Promotes Cardiac Fibroblast Proliferation. Int. J. Mol. Sci. 2021, 22, 6164. [Google Scholar] [CrossRef]
- Samarel, A.M. Focal adhesion signaling in heart failure. Pflügers Arch.—Eur. J. Physiol. 2014, 466, 1101–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wang, W.; Wang, X.; Wang, X.; Song, Y.; Zhang, J.; Zhao, H. Focal adhesion kinase mediates atrial fibrosis via the AKT/S6K signaling pathway in chronic atrial fibrillation patients with rheumatic mitral valve disease. Int. J. Cardiol. 2013, 168, 3200–3207. [Google Scholar] [CrossRef]
- Lin, Z.; Zhou, P.; von Gise, A.; Gu, F.; Ma, Q.; Chen, J.; Guo, H.; van Gorp, P.R.; Wang, D.-Z.; Pu, W.T. Pi3kcb Links Hippo-YAP and PI3K-AKT Signaling Pathways to Promote Cardiomyocyte Proliferation and Survival. Circ. Res. 2015, 116, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Cao, L.; Massey, I.Y. Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol. Cell. Biochem. 2021, 476, 4045–4059. [Google Scholar] [CrossRef] [PubMed]
- Moulik, M.; Vatta, M.; Witt, S.H.; Arola, A.M.; Murphy, R.T.; McKenna, W.J.; Boriek, A.M.; Oka, K.; Labeit, S.; Bowles, N.E.; et al. ANKRD1, the Gene Encoding Cardiac Ankyrin Repeat Protein, is a Novel Dilated Cardiomyopathy Gene. J. Am. Coll. Cardiol. 2009, 54, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempton, A.; Cefalu, M.; Justice, C.; Baich, T.; Derbala, M.; Canan, B.; Janssen, P.M.; Mohler, P.J.; Smith, S.A. Altered regulation of cardiac ankyrin repeat protein in heart failure. Heliyon 2018, 4, e00514. [Google Scholar] [CrossRef] [Green Version]
- Muslin, A.J. MAPK signalling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin. Sci. 2008, 115, 203–218. [Google Scholar] [CrossRef] [Green Version]
- Weckbach, L.T.; Schweizer, L.; Kraechan, A.; Bieber, S.; Ishikawa-Ankerhold, H.; Hausleiter, J.; Massberg, S.; Straub, T.; Klingel, K.; Grabmaier, U.; et al. Association of Complement and MAPK Activation with SARS-CoV-2—Associated Myocardial Inflammation. JAMA Cardiol. 2022, 7, 286. [Google Scholar] [CrossRef]
- Opavsky, M.A.; Martino, T.; Rabinovitch, M.; Penninger, J.; Richardson, C.; Petric, M.; Trinidad, C.; Butcher, L.; Chan, J.; Liu, P.P. Enhanced ERK-1/2 activation in mice susceptible to coxsackievirus-induced myocarditis. J. Clin. Investig. 2002, 109, 1561–1569. [Google Scholar] [CrossRef]
- Takeishi, Y.; Huang, Q.; Abe, J.-I.; Che, W.; Lee, J.-D.; Kawakatsu, H.; Hoit, B.D.; Berk, B.C.; Walsh, R.A. Activation of mitogen-activated protein kinases and p90 ribosomal S6 kinase in failing human hearts with dilated cardiomyopathy. Cardiovasc. Res. 2002, 53, 131–137. [Google Scholar] [CrossRef]
- Kyoi, S.; Otani, H.; Matsuhisa, S.; Akita, Y.; Tatsumi, K.; Enoki, C.; Fujiwara, H.; Imamura, H.; Kamihata, H.; Iwasaka, T. Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc. Res. 2006, 69, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Romero-Becerra, R.; Santamans, A.M.; Folgueira, C.; Sabio, G. p38 MAPK Pathway in the Heart: New Insights in Health and Disease. Int. J. Mol. Sci. 2020, 21, 7412. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.; Ren, X.; Qian, X.; Ye, P.; Luo, J.; Gao, X.; Zhang, J.; Chen, S. Inhibition of JNK and p38 MAPK-mediated inflammation and apoptosis by ivabradine improves cardiac function in streptozotocin-induced diabetic cardiomyopathy. J. Cell. Physiol. 2018, 234, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Blythe, N.M. Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. J. Cardiovasc. Dev. Dis. 2019, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Kojic, S.; Nestorovic, A.; Rakicevic, L.; Belgrano, A.; Stankovic, M.; Divac, A.; Faulkner, G. A novel role for cardiac ankyrin repeat protein Ankrd1/CARP as a co-activator of the p53 tumor suppressor protein. Arch. Biochem. Biophys. 2010, 502, 60–67. [Google Scholar] [CrossRef]
- Hill, C.; Würfel, A.; Heger, J.; Meyering, B.; Schlüter, K.-D.; Weber, M.; Ferdinandy, P.; Aronheim, A.; Schulz, R.; Euler, G. Inhibition of AP-1 signaling by JDP2 overexpression protects cardiomyocytes against hypertrophy and apoptosis induction. Cardiovasc. Res. 2013, 99, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Windak, R.; Müller, J.; Felley, A.; Akhmedov, A.; Wagner, E.F.; Pedrazzini, T.; Sumara, G.; Ricci, R. The AP-1 Transcription Factor C-Jun Prevents Stress-Imposed Maladaptive Remodeling of the Heart. PLoS ONE 2013, 8, e73294. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; He, B.; Goldstein, S.; Ge, J.; Wang, Z.; Ruiz, G. The expression and significance of protooncogene c-fos in viral myocarditis. Virol. J. 2010, 7, 285. [Google Scholar] [CrossRef] [Green Version]
- Windak, R. The AP-1 Transcription Factor C-Jun, but Not C-Fos, Prevents Pathologic Cardiac Remodeling. Ph.D. Thesis, ETH Zurich, Zurich, Switzerland, 2009. [Google Scholar]
- Torsoni, A.S.; Constancio, S.S.; Nadruz, W., Jr.; Hanks, S.K.; Franchini, K.G. Focal Adhesion Kinase Is Activated and Mediates the Early Hypertrophic Response to Stretch in Cardiac Myocytes. Circ. Res. 2003, 93, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Hernández, A.E.D.R. FAK controls the mechanical activation of YAP, a transcriptional regulator required for durotaxis. FASEB J. 2018, 32, 1099–1107. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Luo, J.; Hou, N. Molecular Mechanism of Hippo-YAP1/TAZ Pathway in Heart Development, Disease, and Regeneration. Front. Physiol. 2020, 11, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, N.; Wen, Y.; Yuan, X.; Xu, H.; Wang, X.; Li, F.; Ye, B. Activation of Yap1/Taz signaling in ischemic heart disease and dilated cardiomyopathy. Exp. Mol. Pathol. 2017, 103, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Iyengar, R.; Koshman, Y.E.; Kim, T.; Russell, B.; Martin, J.L.; Heroux, A.L.; Robia, S.L.; Samarel, A.M. Serine-910 phosphorylation of focal adhesion kinase is critical for sarcomere reorganization in cardiomyocyte hypertrophy. Cardiovasc. Res. 2011, 92, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.P.D.; Clemente, C.F.; Carvalho, H.F.; Carvalheira, J.B.; Nadruz, W.; Franchini, K.G. FAK mediates the activation of cardiac fibroblasts induced by mechanical stress through regulation of the mTOR complex. Cardiovasc. Res. 2009, 86, 421–431. [Google Scholar] [CrossRef]
- Taniyama, Y.; Walsh, K. Elevated Myocardial Akt Signaling Ameliorates Doxorubicin-Induced Congestive Heart Failure and Promotes Heart Growth. J. Mol. Cell. Cardiol. 2002, 34, 1241–1247. [Google Scholar] [CrossRef]
- Yao, H.; Han, X.; Han, X. The Cardioprotection of the Insulin-Mediated PI3K/Akt/mTOR Signaling Pathway. Am. J. Cardiovasc. Drugs 2014, 14, 433–442. [Google Scholar] [CrossRef]
- Mohamed, J.S.; Boriek, A.M. Loss of desmin triggers mechanosensitivity and up-regulation of Ankrd1 expression through Akt-NF-κB signaling pathway in smooth muscle cells. FASEB J. 2011, 26, 757–765. [Google Scholar] [CrossRef]
- Ling, S.S.M.; Chen, Y.-T.; Wang, J.; Richards, A.M.; Liew, O.W. Ankyrin Repeat Domain 1 Protein: A Functionally Pleiotropic Protein with Cardiac Biomarker Potential. Int. J. Mol. Sci. 2017, 18, 1362. [Google Scholar] [CrossRef] [Green Version]
- Bin, L.; Li, X.; Richers, B.; Streib, J.E.; Hu, J.W.; Taylor, P.; Leung, D.Y. Ankyrin repeat domain 1 regulates innate immune responses against herpes simplex virus 1: A potential role in eczema herpeticum. J. Allergy Clin. Immunol. 2018, 141, 2085–2093.e1. [Google Scholar] [CrossRef] [Green Version]
- Kong, M.; Ma, T.; Xiang, B. ANKRD1 and SPP1 as diagnostic markers and correlated with immune infiltration in biliary atresia. Medicine 2021, 100, e28197. [Google Scholar] [CrossRef]
- Liu, X.-H.; Bauman, W.A.; Cardozo, C. ANKRD1 modulates inflammatory responses in C2C12 myoblasts through feedback inhibition of NF-κB signaling activity. Biochem. Biophys. Res. Commun. 2015, 464, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Almodóvar-García, K.; Kwon, M.; Samaras, S.E.; Davidson, J.M. ANKRD1 Acts as a Transcriptional Repressor of MMP13 via the AP-1 Site. Mol. Cell. Biol. 2014, 34, 1500–1511. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinkūnaitė, I.; Šimoliūnas, E.; Alksnė, M.; Bartkutė, G.; Labeit, S.; Bukelskienė, V.; Bogomolovas, J. Genetic Ablation of Ankrd1 Mitigates Cardiac Damage during Experimental Autoimmune Myocarditis in Mice. Biomolecules 2022, 12, 1898. https://doi.org/10.3390/biom12121898
Rinkūnaitė I, Šimoliūnas E, Alksnė M, Bartkutė G, Labeit S, Bukelskienė V, Bogomolovas J. Genetic Ablation of Ankrd1 Mitigates Cardiac Damage during Experimental Autoimmune Myocarditis in Mice. Biomolecules. 2022; 12(12):1898. https://doi.org/10.3390/biom12121898
Chicago/Turabian StyleRinkūnaitė, Ieva, Egidijus Šimoliūnas, Milda Alksnė, Gabrielė Bartkutė, Siegfried Labeit, Virginija Bukelskienė, and Julius Bogomolovas. 2022. "Genetic Ablation of Ankrd1 Mitigates Cardiac Damage during Experimental Autoimmune Myocarditis in Mice" Biomolecules 12, no. 12: 1898. https://doi.org/10.3390/biom12121898
APA StyleRinkūnaitė, I., Šimoliūnas, E., Alksnė, M., Bartkutė, G., Labeit, S., Bukelskienė, V., & Bogomolovas, J. (2022). Genetic Ablation of Ankrd1 Mitigates Cardiac Damage during Experimental Autoimmune Myocarditis in Mice. Biomolecules, 12(12), 1898. https://doi.org/10.3390/biom12121898