Abstract
Hundreds of proteins work together in microorganisms to coordinate and control normal activity in cells. Their degradation is not only the last step in the cell’s lifespan but also the starting point for its recycling. In recent years, protein degradation has been extensively studied in both eukaryotic and prokaryotic organisms. Understanding the degradation process is essential for revealing the complex regulatory network in microorganisms, as well as further artificial reconstructions and applications. This review will discuss several studies on protein quality-control family members Lon, FtsH, ClpP, the proteasome in Streptomyces, and a few classical model organisms, mainly focusing on their structure, recognition mechanisms, and metabolic influences.
1. Introduction
Protein homeostasis is an important prerequisite for bacteria to maintain normal activity. In a complex natural living environment, bacteria must deal with external stimuli, such as pressure, pH, temperature, and nutrient supply, and promptly remove the misfolded proteins caused by these factors. On the other hand, bacteria change the expression level of their proteins according to the needs of their own growth. They contain a simpler protein degradation system than eukaryotes. Usually, prokaryotes rely on four cytoplasmic ATP-dependent proteases—Lon, FtsH, Clp, and HslUV—while Streptomyces mainly contains the first three proteases. These proteases are part of the AAA+ protein family (ATPases associated with various cellular activities) []. Although they play a degrading function by recognizing different substrates, they have a similar structure and are composed of two parts, a peptidase (protease) and an unfoldase (ATPase) [,]. ATP hydrolysis by the unfoldase promotes the unfolding and translocation of the substrate protein into peptidase through the axial pore of the entire protease complex. Finally, the unfolded substrate is transferred to the proteolysis chamber, where peptide bond cleavage occurs []. In addition, proteins in bacteria can be specifically degraded through the proteasome. The proteasome was first discovered in actinomycetes [], and then it was extensively studied in Mycobacterium tuberculosis, where the important prokaryotic ubiquitin-like protein (Pup) was discovered []. Pup can be covalently labeled in a variety of functional proteins; therefore, the Pup-proteasome degradation pathway provides a reliable theoretical basis for the study of bacteria protein degradation.
Streptomyces is an indispensable member of the bacterial family, and its secondary metabolite library greatly enriches human clinical drug selection. This library includes antibiotics (such as daptomycin), immunosuppressants (such as rapamycin), antifungal drugs (such as amphotericin B), anticancer drugs (such as adriamycin), and antiparasitic drugs (such as ivermectin), etc., (the compounds described in this review are listed in Table 1) []. Their biosynthesis process is tightly controlled by regulatory pathways. In order to obtain more valuable metabolites, researchers have made lots of efforts to study the regulatory network, as well as gene cluster mining []. In recent years, the development of proteomics based on high-resolution mass spectrometry (MS) quantitatively revealed acylation types and modification sites. Characterizing the activities of key regulators in microorganisms is important to understand their function. Some researchers found that changes in protein activity could affect the biosynthesis process of Streptomyces []. Degradation is particularly a significant part of a protein’s lifespan. For the sake of convenience for follow-up research, we present a brief review that uses different types of proteases discussed in different points, combined with other model microorganisms, especially the protease function mechanism in Streptomyces.

Table 1.
Summary of sources and functions of twelve compounds.
2. Lon
Lon, as a member of the protein quality control family, was identified for the first time in Escherichia coli []. Early studies indicated that its C-terminal domain in E. coli contains a proteolytic active site formed by a serine-lysine catalytic dimer [] and may be involved in substrate recognition and binding [,]. Additionally, in further studies, researchers found that Lon’s N-terminal region also has a certain effect on degradation function. The deletion of N-terminal amino acids G91 and E226 in the Lon protease from Mycobacterium smegmatis both severely impaired the degradation of substrates α-casein in vitro and RcsA in vivo (Figure 1A) []. The full-length structure of Lon has always been a mystery. Until recently, it was analyzed by cryo–electron microscopy, which revealed a multilayered architecture featuring a tensegrity triangle complex, uniquely constructed by six long N-terminal helices (Figure 1B). However, the details of this are not described here []. In addition to its own structure region, a certain amino acid of Lon’s substrate is also closely related to degradation. For example, SulA, which functions in the stage of DNA damage, is a cell differentiation inhibitor and is degraded by the Lon protease in E. coli. Its affinity with Lon changes with the mutation of C-terminal histidine (Figure 1C) []. SoxS and MarA act as regulators of the redox process and antibiotic resistance enhancement in E. coli, respectively, and their half-life is prolonged due to the deletion of Lon. Moreover, inserting histidine at its N-terminus also increases Lon’s stability, suggesting that it may recognize the N-terminal domains of SoxS and MarA and play a role in degradation []. Nevertheless, more work is needed to further define the conserved sequences for Lon-mediated degradation.
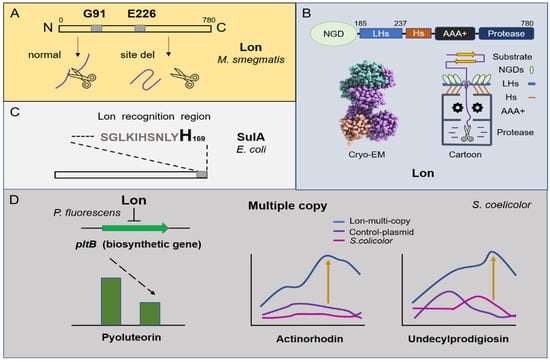
Figure 1.
Overview of the degradation mechanism of Lon protease and its regulation on metabolism. (A) N-terminal amino acids G91 and E226 of Lon protease from Mycobacterium smegmatis are the most important active sites. (B) The cartoon structure of Lon protease and the Cryo–EM structure of the N-terminal domain of Lon E. coli (The Cryo–EM structure was downloaded from the PDB database). The whole length of Lon contains N-terminal globular domains (NGDs), three-helix bundles (3Hs), interlocked long helices (LHs), an AAA+ domain, and a protease chamber. (C) The degradation of SulA by Lon protease depends on its C-terminal histidine. (D) Lon protease negatively regulates the production of pyoluteorin in Pseudomonas fluorescens Pf-5. The multiple copies of the lon gene in S. coelicolor cause an increase in the yield of undecylprodigiosin and actinorhodin.
Lon participates in different aspects of bacterial physiology, including cell differentiation, sporulation, pathogenicity, and survival under starvation conditions, and it can respond to external pressure stimuli. Studies show that NaCl (450 mM) can induce the expression of Lon and that ectoine treatment further enhances the expression level (5.2-fold) in Streptomyces sp., strain C-2012 []. The ATP-dependent Lon protease is also a heat shock protein. In Streptomyces coelicolor, the deletion of the DnaK chaperone machine in vivo results in negative feedback regulation in lon and clpB []. Subsequently, transcriptomics and translatomics showed that Lon and heat shock molecular chaperones (DnaK and GroES/GroEL1/GroEL2) are synergistically upregulated under heat stimulation []. In Streptomyces lividans, it is confirmed that Lon is a new stress response regulator and belongs to HAIR (HspR co-recognition sequence (CTTGAGT-N7-ACTCAAG)/HspR (heat shock) protein regulator). Briefly, its molecular chaperone interacts with unfolded proteins under heat induction, causing Lon (HspR) to function in a free form for derepression, which suggests that excessive production of Lon protease may be toxic to the bacteria [,]. Interestingly, the effect of Lon protease on microorganism metabolic function is relative. On the one hand, studies found that the mutation of Lon could damage spore germination under high temperatures, and Lon negatively regulated the antibiotic production of pyoluteorin in Pseudomonas fluorescens Pf-5 by influencing pltB biosynthetic gene transcription []. On the other hand, after inserting multiple copies of the lon gene in S. coelicolor, the production of actinorhodin increased by 34 times, and the production of undecylprodigiosin increased by nine times. Researchers believe that this was caused by Lon protease producing more antibiotic biosynthetic precursors through direct protein degradation (Figure 1D) [].
3. ClpP
Casein hydrolase ClpP is a serine protease found in the bacterial kingdom. ClpP protease is a stable heptamer ring, and each subunit has a hydrolytic active site (Ser-His-Asp) called a catalytic triad []. Some bacteria have a single clpP gene, such as E. coli and Staphylococcus aureus. However, many pathogenic bacteria and actinomycetes encode two clpP alleles, including clpP1 and clpP2 [,], and some even contain clpP3, clpP4, and clpP5 []. There is a large family of Clp ATPases with different types of protein. The first type includes huge proteins with two ATP binding domains (ClpA, ClpB, ClpC, ClpD, and ClpE), and the second type includes smaller proteins with only one ATP binding domain (ClpM, ClpN, ClpX, and ClpY) [,,]. ClpP alone can only degrade short-chain peptides in vitro []. If larger substrates need to be degraded, cohort regulatory ATPases (ClpA, ClpC, ClpX) are needed to promote activation. Among these substrates, research on ClpX is more extensive, because ClpXP is one of the most common proteolytic complexes, found in actinomycetes and Nitrospira bacteria, and its structure is highly conserved (Figure 2A). ClpX has asymmetric hexameric rings, which can bind to the symmetric heptameric rings of ClpP and facilitate the transfer of the substrate to the protease chamber to complete the cleavage []. According to reams of research, ClpX contains an important IGF ring structure (Ile268-Gly269-Phe270) in E. coli, which is responsible for the pocket interaction between the interface and ClpP subunit []. The mutations of I268L, F270L, and V274A cause severe ClpP binding defects [].
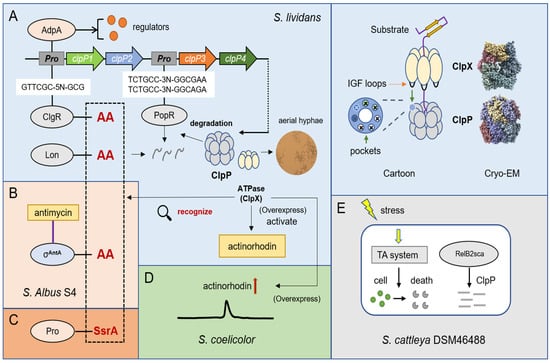
Figure 2.
Overview of the degradation mechanism of ClpP protease and its regulation on metabolism. (A) Cryo—EM structures and cartoon structures of ClpP protease and ClpX (Cryo—EM structures of E. coli were downloaded from PDB database). ClpP protease can degrade important regulators ClgR, Lon, and PopR in S. lividans. (B) Two alanines in the C-terminal of σAntA are essential for degradation by ClpP in S. albus S4. (C) ClpXP can specifically degrade SsrA-tagged proteins. (D) Overexpression of clpX can increase the production of actinorhodin in S. coelicolor. (E) The type II toxin—antitoxin (TA) system is regulated by ClpP protease in S. cattleya DSM46488 under osmotic pressure.
ClpP protease has various regulatory forms in Streptomyces. The absence of clpP1 and clpP2 produces a bald phenotype in S. lividans [], which implies that some undegraded proteins affect aerial hyphal growth. Additionally, the promoters of clpP1 and clpP2 are controlled by ClgR (a transcriptional activator), which is also confirmed in Bifidobacterium BreveUCC2003 []. ClgR binds to the promoter of clpP1 by recognizing the motif of GTTCGC-5N-GCG and can affect the expression of both clpC and lon in S. lividans []. Except for the two alleles of clpP1 and clpP2, the ClpP protease of S. lividans also includes clpP3 and clpP4, whose operons bind to PopR, a ClgR paralog. PopR activates the expression of clpP3 operon by binding a special region: TCTGCC-3N-GGCGAATCTGCC-3N-GGCAGA []. Intriguingly, clpP1 and clpP2 are subunits mainly responsible for the degradation of ClgR, PopR, Lon, and ClpC. In the degradation test of ClpP1, ClpP2, and their substrates, some researchers found that two alanines at the end of the carboxyl were very likely to form the major site for degradation recognition, and after the alanines of ClgR-AA and Lon-AA were mutated to aspartate acid (D), the abundance of protein in cells significantly increased (Figure 2A) [,]. Furthermore, other researchers confirmed that a special extracytoplasmic function (ECF) RNA polymerase sigma (σ) factor σAntA in Streptomyces albus S4, which regulates the production of antimycin, could also be degraded by ClpXP protease and the C-terminus contains these double alanines (Figure 2B) []. In addition, ClpXP can specifically degrade SsrA-tagged proteins (Figure 2C). Bacteria contain a so-called trans-translation system, which is based on a bifunctional translation messenger RNA, used to rescue stalled ribosomes. The incompletely synthesized peptides are marked as C-terminal degradation signals (−AAXXXXXALAA), which is the “SsrA label” [,,]. For example, the SigT protein involved in morphological differentiation and antibiotic synthesis was also degraded by ClpP/SsrA in S. coelicolor []. Combined with previous studies and reviews [,,,], there is no doubt that the substrate recognition of ClpXP mainly depends on ClpX. Additionally, in the degradation examples of Streptomyces, the recognition tags are actually very similar to those identified from proteomic experiments in E. coli. We listed five distinct identification tags, which show that ClpX is sensitive to double alanines and prefers positively charged amino acids, such as lysine, arginine, and histidine. This knowledge should prove helpful in further research on Streptomyces (Table 2).
Table 2.
Five distinct tags of ClpXP degradation substrate and similar motifs of FtsH degradation substrate identified from E. coli by proteomic experiments.
AdpA, as the central regulatory protein of Streptomyces, not only affects mycelial growth and spore germination but also plays an important role in the biosynthesis of secondary metabolites, such as streptomycin, daptomycin, and clavulanic acid Streptomyces griseus, Streptomyces roseosporus and Streptomyces clavuligerus, respectively [,,]. Studies confirmed that AdpA can also bind to the clpP1 and clpP2 operons in S. lividans (Figure 2A), and the lack of ClpP peptidase activity in the cell will decreases the number of AdpA, implying that they are involved in complex transcription and posttranscriptional interactions [,]. In addition, the overexpression of ClpX, an activator of ClpP, is able to activate the production of actinorhodin in S. lividans and increase the yield of actinorhodin in S. coelicolor (Figure 2D) []. The toxin—antitoxin (TA) system is a magic weapon used by bacteria to maintain an equilibrium state. It is well known that, under stress conditions such as nutrient deprivation, bacteria can degrade antitoxin proteins to produce more free toxin proteins that cause a programmed death. Some researchers first discovered the RelBE family protein of the type II toxin–antitoxin (TA) system in Streptomyces cattleya DSM46488, and the antitoxin protein RelB2sca can be degraded by ClpP protease to realize the response to the environment under osmotic pressure (Figure 2E) [].
ClpP protease is also a potential target of antibacterial drugs. In S. coelicolor, it was found that β-lactones have a lethal effect on strains lacking clpP3 and clpP4 genes. The mechanism of action has been proposed in previous studies of Mycobacterium tuberculosis. It has been suggested that an inhibitory effect can be made by forming a covalent bond between the serine active site of ClpP2 and β-lactones. Nevertheless, it needs to be further confirmed [].
4. FtsH
FtsH is the only membrane-anchored metalloprotease among the AAA+ proteases. FtsH protease is composed of three parts: the N-terminus (which contains two transmembrane regions), the AAA domain, and the C-terminus. Its molecular architecture consists of two rings where the protease domains possess an all-helical fold and form a flat hexagon that is covered by a toroid built by the AAA domains. The center of the complex hole is the active site for protease hydrolysis [,,]. When ATP is supplied, FtsH is able to bind to Zn2+ and function on the cell membrane. The conserved HEXXH motif (X = any amino acid) in the protease domain is essential for the coordinated catalysis of Zn2+ []. Studies in E. coli have shown that the most important function of FtsH is to regulate the optimal ratio between phospholipids (PL) and lipopolysaccharides (LPS) on the outer membrane by degrading the key enzyme LpxC in LPS biosynthesis []. The degradation of LpxC is related to its C-terminal sequence and length. Concretely, the C-terminus of degron contains the sequence LAXXXXXXAVLA (X = any amino acid), and the unstructured C-terminal tail must be at least 20 amino acids long, which is necessary for FtsH-mediated degradation [,]. The FtsH consensus sequence for substrate degradation is inconclusive for the other two well-known substrates: YfgM (a mediator of the cytoplasmic and extracytoplasmic stress responses) and RpoH (the master regulator of the heat-shock response). The degradation process of YfgM no longer requires the C-terminal sequence, but the first 14 amino acids (MEIYENENDQVEAV-) of the cytoplasmic N-terminal sequence []. Notably, label-free quantitative proteomics excavated similar motifs in E. coli, and it can be speculated that FtsH may be sensitive to glutamic acid and aspartic acid (Table 2) []. However, the degradation of RpoH depends on several scattered amino acids. The amino acid substitutions of L47, A50, and I54 affect the hydrolysis of RpoH [,,], and the replacement of A131, and K134 influences the stability of RpoH itself (Figure 3A) [].
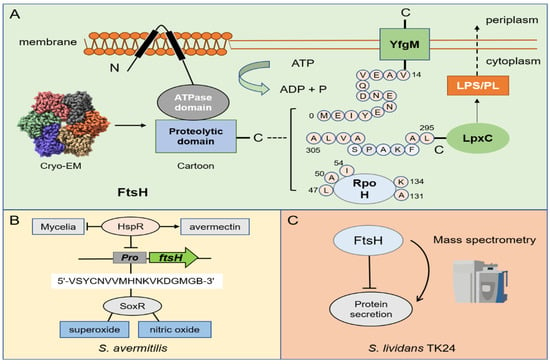
Figure 3.
Overview of the degradation mechanism of FtsH protease and its regulation on metabolism. (A) The cartoon structure of FtsH protease and its Cryo—EM structure of the cytoplasmic domain from E. coli (The Cryo—EM structure was downloaded from PDB database). (B) Three kinds of substrates (RpoH, YfgM, and LpxC) of FtsH protease. The pink sphere represents the key amino acid in the degradation process. (B) In S. avermitilis, HspR can bind to ftsHp to inhibit FtsH and negatively regulate its development. SoxR can specifically bind to ftsHp to mediate the resistance of superoxide and nitric oxide by recognizing 18-nt binding site (5′-VSYCNVVMHNKVKDGMGB-3′). (C) FtsH negatively regulates the protein secretion in S. lividans TK24.
FtsH is relatively less studied in Streptomyces compared with other proteases, so its molecular mechanism is not very clear in the regulation of Streptomyces. SoxR, which is a redox-sensitive transcriptional regulator, has been shown to mediate the resistance of superoxide and nitric oxide under oxidative stress in E. coli []. By comparing its target homologous gene of S. coelicolor with the ave gene in S. avermitilis, researchers predicted the 18-nt SoxR binding site (5′-VSYCNVVMHNKVKDGMGB-3′) in their promoter region (V = A/T/C; S = C/G; Y = T/C; N = A/G/C/T; M = A/C; H = A/T/C; K = G/T; B = G/C/T). Surprisingly, it is found that ftsHp meets this sequence feature and can specifically bind to SoxR. Researchers believe that although FtsH is not a key developmental gene of Streptomyces, it has a certain degree of effect on cell differentiation []. Later, studies on the heat shock-related inhibitor HspR in S. avermitilis found that HspR can bind to ftsHp to directly inhibit FtsH and negatively regulate its development. Nevertheless, this regulatory process is beneficial for the production of avermectin (Figure 3B) []. With the development of multiomics in recent years, some research groups have used transcriptomics and proteomics to explore the biological functions of FtsH protease in S. lividans. Seventeen proteins encoded by protease and protease inhibitor genes with a significant abundance have attracted attention with the change in the growth stage. When FtsH is deleted, the protein secretion capacity of S. lividans TK24 was significantly improved (Figure 3C). In comparison to other mutant strains, TK24ΔftsH also showed the most severe phenotypic changes. Therefore, FtsH is considered to be a major global regulator in S. lividans []. Similar research on FtsH was also conducted for other microorganisms. For example, the lack of FtsH strongly increases the abundance of ten cytoplasmic and membrane proteins in Corynebacterium glutamicum without affecting its growth []. Since secretion is a transmembrane cross process and FtsH functions on the cell membrane, we can speculate that Streptomyces is likely to be indirectly regulated by FtsH in the efflux and transport of antibiotics. Although it is greatly challenging to conduct research on membrane proteins, it is valuable for industrial applications.
5. Proteasome
Proteasomes are ubiquitous in actinomycetes. Their structures are the same as eukaryotic homologs, consisting of 28 subunits: two homoheptamer loops of β subunits containing the active site are sandwiched between the two homoheptamer loops ofα subunit, which prevents foreign proteins from entering the cylindrical proteolytic chamber in the absence of an activator (Figure 4A) []. In eukaryotic cells, some proteins are labeled with ubiquitin molecules and then recognized and degraded by the proteasome. In prokaryotic cells, the proteasome was first discovered in actinomycetes in the 1980s. However, a thorough understanding of the proteasome degradation mechanism was first achieved in 2008, when Pearce et al. discovered a protein with similar functions to ubiquitin in M. tuberculosis and named it prokaryotic ubiquitin-like protein (Pup) []. As a new degradation pathway, the Pup–proteasome system is a distinctive way to regulate intracellular protein levels and eliminate misfolded proteins and participates in regulating a variety of physiological functions in cells. The sequence K/RGGQ at the C-terminus of the Pup molecule contains the conserved Gly-Gly sequence in ubiquitin, which can specifically link with Lys to form a Gly-Gly-Lys isopeptide bond []. Generally, the carboxyl-terminal sequence of Pup is Gly-Gly-Gln, and Gln needs to be deamidated to Glu under the action of Dop (deamidase) to form covalent bonds []. Taking advantage of this feature, in 2009, Festa et al., used tandem affinity chromatography and mass spectrometry to systematically analyze the Pup–labeled protein in M. tuberculosis, according to whether the protein’s lysine site has the matching peptides of GGE (243 Da) to determine modification by Pup (Figure 4A) []. Of course, there are some prokaryotic cells that have Glu at the carboxyl end of Pup (Figure 4B). For example, when recombinant Pup, PafA (Pup ligase), and Dop were purified and expressed in vitro by Streptomyces hygroscopicus, it was found that although Dop is redundant as a deamidase when the tail of Pup is glutamate, it still has the ability to recycle Pup (a recycler of Pup by depupylation). Finally, these authors used the molecular mass of covalent modification to identify a Pup substrate (SHJG_3659) []. The depupylation function of Dop has also been confirmed in other microorganisms and is not explored further in this study [,]. When Pup is deamidated, it will hydrolyze ATP under the action of PafA and catalyze the phosphorylation of Glu [], forming an intermediate that is connected to the substrate Lys []. Then, Pup–protein will interact with Mpa (mycobacterial proteasome ATPase/the switch that regulates the “gate” of the proteasome α subunit) and be delivered to the proteasome for degradation (Figure 4A) [,,,].
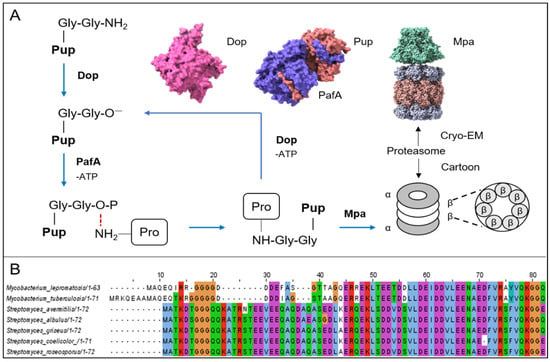
Figure 4.
Overview of the degradation mechanism of Pup–proteasome and different types of Pup. (A) Degradation diagram of the interaction between the proteasome, Pup, Dop, PafA, and Mpa. The pink Cryo—EM structure of Dop is identified from Acidothermus cellulolyticus. The Cryo—EM structure of Pup-PafA complex is identified from Corynebacterium glutamicum. The purple area represents PafA, and the orange area represents Pup. The Cryo—EM structure of the Mpa—proteasome complex is identified from Mycobacterium tuberculosis (the Cryo—EM structures were all downloaded from the PDB database). (B) The alignment of Pup amino acid sequences between M. lepromatosis, M. tuberculosis, S. avermitilis, S. albulus, S. griseus, S. coelicolor, and S. roseosporus (amino acid sequences were downloaded from the NCBI database).
The 20S proteasome is composed of two types of subunits (PrcA and PrcB) in S. coelicolor and has a chymotrypsin-like activity in synthetic substrates []. The most significant change caused by the loss of proteasome in S. coelicolor is the clear increase in the level of nonheme chloroperoxidase SCO0465, which is consistent with the increase in resistance to hydrogen peroxide []. In addition, the PafA deletion mutant has spore-producing defects, and the deletion of Pup shows greater changes than the proteasome mutants [], including the significantly reduced production of the secondary metabolites, undecylprodigiosin and actinorhodin []. This implies that Pup has other functions aside from directing protein targeting proteasome degradation in S. coelicolor. Notably, subsequent studies in other microorganisms have verified this finding. Researchers have found that pupylation is not limited to protein degradation. In the case of oxidative stress, and carbon and nitrogen starvation, Corynebacterium glutamicum, which lacks proteasomes, opens up this kind of protein modification and causes the phenomenon of ferritin release []. One study focused on the proteasome in S. lividans and found that the proteasome deletion strain had no significant impact on phenotypes compared with the wild-type strains; however, a high yield can be achieved for soluble human tumor necrosis factor receptor II (shuTNFRII) and salmon calcitonin (sCT) []. The same situation also occurs in S. roseosporus. Although the deletion of the proteasome causes a bald phenotype and makes the bacteria lose the production capacity of red pigment and daptomycin, it is a better host for expressing the protein than WT []. This result indicated that the disruption of proteasome genes might help to efficiently produce heterologous proteins, which provides a new strategy for heterologous expression. In prokaryotes, in addition to interaction with Pup molecules, the proteasome may also interact with the Cdc48-like protein (Cpa) of actinomycetes to achieve the degradation of certain proteins. It has been proven that Cpa can interact with 20S core particles in vitro to form ring stacks and collinear complexes, but no degradation substrate has been discovered thus far [,]. Additionally, tunicamycin, a fermentation product of Streptomyces hygroscopicus, can inhibit the ubiquitin-proteasome system in eukaryotic cells rather than its own proteasome degradation system []. This is a vital discovery regarding the application of the proteasome, but its mechanism remains unclear. It has been a very effective drug development strategy by using ubiquitin ligase to induce ubiquitination and subsequently transport the target protein to the 26S proteasome for degradation. However, it is still limited to eukaryotes. To date, some researchers have found that the recognition ligand linked to arginine (B3A) and protected by tert-butyl carbamate (Boc3) can induce ligand degradation. This process requires the proteasome but does not involve the ubiquitination of the target protein. The B3A ligand stimulates the activity of the purified 20S proteasome, which allows the tag to directly bind to the 20S proteasome [], providing a novel strategy for degradation in prokaryotes. Based on this, it is possible to control the metabolic pathways to benefit antibiotic production during the growth of actinomycetes, which is achieved by inducing the degradation of certain proteins.
6. Cross Talk
Protease and proteasome together constitute a protein quality control system in bacteria, which sometimes creates a more complex regulation network for understanding a single protein degradation process. Since they are able to function alternately, they pose a huge challenge for further research.
An early study on the protease of E. coli discovered that the mutagenic protein UmuD’ and its homodimers-UmuD were involved in a complex proteolytic pathway. As the precursor of UmuD’, UmuD has no mutagenic activity and can be degraded by Lon protease in vivo. However, UmuD’ is not affected by Lon protease and is degraded by the ClpXP protease [,]. Intriguingly, the N-terminal tail is proven to be the key recognition site for UmuD to participate in the degradation of Lon, and UmuD’ is also targeted to ClpXP through the 24 amino acids of the N-terminus of UmuD, which is specifically degraded []. In addition to E. coli, examples of ClpXP and Lon synergistic action are also found in Caulobacter crescentus. CtrA, a positive global regulator, promotes the expression of DNA methyltransferase CcrM to complete cell division. ccrM transcription stops just before cell division when the CtrA is degraded by ClpXP. Soon after, CcrM is continuously degraded by Lon [,].
In S. coelicolor, the SigT protein responsible for the negative regulation of morphological differentiation is in a stable state under the binding of anti-σ factor RstA, which protects itself from independent proteasome degradation. However, if RstA is degraded, the exposed SigT will bind to the promoter regions of clpP1 and clpP2 and induce the production of ClpP protein so that it can be degraded itself. Surprisingly, undecylprodigiosin and actinorhodin can regulate the binding of SigT and clpP1/2 in this degradation process. The dual regulation of ClpP and proteasome indicates that it has a complex and delicate degradation system in S. coelicolor. In addition, after the overexpression of the two pathway-specific genes redD and actII-orf4 in the proteasome-deficient strain, compared with the original strain, it increased the yield of undecylprodigiosin by 3–5 times, and the yield of actinorhodin by 9–30 times, indicating that the production of antibiotics is closely related to the degradation system [,].
S. roseosporus is the producer of daptomycin. Due to the special feature of its precursor, adding capric acid during fermentation is considered a necessary means to increase its production. Unfortunately, capric acid (>2.5 mM) is toxic to S. roseosporus. Under this kind of exogenous stimulation, transcriptomics surprisingly found that the genes encoding proteasome and ClpB protease were significantly upregulated, responding to oxidative stress by degrading misfolded proteins []. Unfortunately, there is a lack of research that explicitly focuses on this mechanism, but it provides the basis to address the toxicity of decanoic acid from the perspective of protein degradation.
7. Conclusions
Despite the progress of protein degradation thus far, our knowledge of microbial proteostasis remains quite limited, the association between protein degradation and the primary/secondary metabolism or morphological differentiation is undeniable, yet more investigations are still essential. In particular, the following areas warrant a thorough investigation. (i) Degradation mechanisms need to be dissected, especially the featured degradation signals of target proteins. Although some degradation signals, such as the N-terminus for Lon and FtsH, SsrA tag for ClpXP are important, the key amino acid residues or conserved motifs and characteristic features seem to be unclear. Furthermore, how the signals are accurately recognized and the time when the degradation of target proteins is switched on and off, need to be resolved. (ii) The regulatory mechanism of protein degradation must be considered. In a few cases, we know that some regulators, such as ClgR for ClpP and HspR for FtsH, are involved in the regulation of protein degradation. However, generally, the cascade regulatory network is complex, thus the regulatory pathways of protein degradation remain indistinct. The many global or pleiotropic regulators that participate in proteostasis and how the consortium of regulators work together to affect protein synthesis and degradation must be systematically revealed. Considering the complexity of regulation, integrating multi-omics, such as transcriptomics, proteomics, and even translatomics would be an efficient strategy to comb the regulatory network of proteostasis. (iii) The temporal regulation of protein degradation is pivotal for the practical applications of proteostasis for improving the performance of microbial cells and enhancing the production of value-added compounds, such as polypeptides, antibiotics, or enzymes. The abundance of proteins, particularly key or rate-limiting enzymes, is closely related to the biosynthesis of target compounds. Proteases serve important functions in protein quality control, which is also a dynamic process. Some studies suggest that engineering protease-targeted proteins can stabilize them against degradation. More importantly, the development of synthetic biology endows us with the ability to engineer controllable protein degradation. Therefore, based on the degradation and regulatory mechanisms of proteostasis, we can perform the dynamic quality control of target proteins by regulating proteases or the modification of target proteins for facilitating the production of target products. Here, we emphasized the importance of Streptomyces in microbial drugs and summarized the development of protein degradation and antibiotic biosynthesis. In Streptomyces, the time-course metabolic shift from primary to secondary metabolism especially requires the temporal regulation of different proteins, which assist with the growth in the growth stage and secondary metabolism in the stationary stage. In particular, the stability and availability of the enzymes directly involved in antibiotic biosynthesis in the stationary stage are very important for drug production. Apart from overexpression, genetic modification or the fine-tuning of protein degradation are alternative methods for stabilizing these against degradation. (iv) Proteostasis advances the development of cell-free protein synthesis systems. Based on the extended knowledge of protein degradation, we can develop more efficient cell-free systems by engineering protease-deficient hosts or stabilizing target proteins. In E. coli, protease-deficient mutants were constructed to reduce protein degradation and used for cell-free protein synthesis. Although many Streptomyces-based cell-free protein synthesis systems were developed and showed a considerable advantage, a large number of proteases in Streptomyces limit their extensive applications and the expensive addition of a protease inhibitor is usually required. In future research, we can thoroughly explore protein degradation and delete all active proteases in Streptomyces to develop highly efficient cell-free protein synthesis systems.
In summary, due to the robustness of bacteria, numerous endogenous proteins are degraded by proteases or proteasomes and cannot be conveniently captured or studied. Therefore, understanding the process of protein degradation is an indispensable part of analyzing the complex regulatory network and performing rational strain engineering to improve the production of value-added compounds in microorganisms. Continually revealing the interaction of proteases or proteasomes with other regulators not only helps medicinal bacteria, such as Streptomyces to serve better as producers of antibiotics, but it may also provide a new theoretical basis for research on other pathogens.
Author Contributions
W.X.; writing—original draft preparation, investigation, project administration, resources, visualization. W.G.; writing—original draft preparation, resources, visualization. Q.B.; writing—original draft preparation, investigation, visualization. Y.L.; writing—review and editing, supervision, project administration, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (grant number 31730002) and the National Key R&D Program of China (grant number 2019YFA09005400).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.C.; Trame, C.B.; Tsuruta, H.; Wilbanks, S.M.; Reddy, V.S.; McKay, D.B. Crystal and solution structures of an HslUV protease-chaperone complex. Cell 2000, 103, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Wickner, S.; Maurizi, M.R.; Gottesman, S. Posttranslational quality control: Folding, refolding, and degrading proteins. Science 1999, 286, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Nagy, I.; Tamura, T.; Vanderleyden, J.; Baumeister, W.; De Mot, R. The 20S proteasome of Streptomyces coelicolor. J. Bacteriol. 1998, 180, 5448–5453. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.J.; Mintseris, J.; Ferreyra, J.; Gygi, S.P.; Darwin, K.H. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 2008, 322, 1104–1107. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Challis, G.L. Mining microbial genomes for new natural products and biosynthetic pathways. Microbiology 2008, 154, 1555–1569. [Google Scholar] [CrossRef]
- Tucker, A.C.; Escalante-Semerena, J.C. Acetoacetyl-CoA synthetase activity is controlled by a protein acetyltransferase with unique domain organization in Streptomyces lividans. Mol. Microbiol. 2013, 87, 152–167. [Google Scholar] [CrossRef]
- Swamy, K.H.; Goldberg, A.L. E. coli contains eight soluble proteolytic activities, one being ATP dependent. Nature 1981, 292, 652–654. [Google Scholar] [CrossRef]
- Ebel, W.; Skinner, M.M.; Dierksen, K.P.; Scott, J.M.; Trempy, J.E. A conserved domain in Escherichia coli Lon protease is involved in substrate discriminator activity. J. Bacteriol. 1999, 181, 2236–2243. [Google Scholar] [CrossRef]
- Amerik, A.K.; Antonov, V.K.; Gorbalenya, A.E.; Kotova, S.A.; Rotanova, T.V.; Shimbarevich, E.V. Site-directed mutagenesis of La protease. A catalytically active serine residue. FEBS. Lett. 1991, 287, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Glockshuber, R. A point mutation within the ATP-binding site inactivates both catalytic functions of the ATP-dependent protease La (Lon) from Escherichia coli. FEBS. Lett. 1994, 356, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Roudiak, S.G.; Shrader, T.E. Functional role of the N-terminal region of the Lon protease from Mycobacterium smegmatis. Biochemistry 1998, 37, 11255–11263. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hsieh, K.Y.; Kuo, C.I.; Lee, S.H.; Pintilie, G.D.; Zhang, K.; Chang, C.I. Complete three-dimensional structures of the Lon protease translocating a protein substrate. Sci. Adv. 2021, 7, eabj7835. [Google Scholar] [CrossRef]
- Ishii, Y.; Amano, F. Regulation of SulA cleavage by Lon protease by the C-terminal amino acid of SulA, histidine. Biochem. J. 2001, 358, 473–480. [Google Scholar] [CrossRef]
- Griffith, K.L.; Shah, I.M.; Wolf, R.E., Jr. Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol. Microbiol. 2004, 51, 1801–1816. [Google Scholar] [CrossRef]
- Sadeghi, A.; Soltani, B.M.; Jouzani, G.S.; Karimi, E.; Nekouei, M.K.; Sadeghizadeh, M. Taxonomic study of a salt tolerant Streptomyces sp. strain C-2012 and the effect of salt and ectoine on lon expression level. Microbiol. Res. 2014, 169, 232–238. [Google Scholar] [CrossRef]
- Bucca, G.; Brassington, A.M.; Hotchkiss, G.; Mersinias, V.; Smith, C.P. Negative feedback regulation of dnaK, clpB and lon expression by the DnaK chaperone machine in Streptomyces coelicolor, identified by transcriptome and in vivo DnaK-depletion analysis. Mol. Microbiol. 2003, 50, 153–166. [Google Scholar] [CrossRef]
- Bucca, G.; Pothi, R.; Hesketh, A.; Möller-Levet, C.; Hodgson, D.A.; Laing, E.E.; Stewart, G.R.; Smith, C.P. Translational control plays an important role in the adaptive heat-shock response of Streptomyces coelicolor. Nucleic. Acids. Res. 2018, 46, 5692–5703. [Google Scholar] [CrossRef]
- Tsilibaris, V.; Maenhaut-Michel, G.; Van Melderen, L. Biological roles of the Lon ATP-dependent protease. Res. Microbiol. 2006, 157, 701–713. [Google Scholar] [CrossRef]
- Sobczyk, A.; Bellier, A.; Viala, J.; Mazodier, P. The lon gene, encoding an ATP-dependent protease, is a novel member of the HAIR/HspR stress-response regulon in actinomycetes. Microbiology 2002, 148, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Whistler, C.A.; Stockwell, V.O.; Loper, J.E. Lon protease influences antibiotic production and UV tolerance of Pseudomonas fluorescens Pf-5. Appl. Environ. Microbiol. 2000, 66, 2718–2725. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Demir, Z.; Bayraktar, A.; Tunca, S. One extra copy of lon gene causes a dramatic increase in actinorhodin production by Streptomyces coelicolor A3(2). Curr. Microbiol. 2019, 76, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Gribun, A.; Kimber, M.S.; Ching, R.; Sprangers, R.; Fiebig, K.M.; Houry, W.A. The ClpP double ring tetradecameric protease exhibits plastic ring-ring interactions, and the N termini of its subunits form flexible loops that are essential for ClpXP and ClpAP complex formation. J. Biol. Chem. 2005, 280, 16185–16196. [Google Scholar] [CrossRef] [PubMed]
- Akopian, T.; Kandror, O.; Raju, R.M.; Unnikrishnan, M.; Rubin, E.J.; Goldberg, A.L. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO. J. 2012, 31, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- De Crécy-Lagard, V.; Servant-Moisson, P.; Viala, J.; Grandvalet, C.; Mazodier, P. Alteration of the synthesis of the Clp ATP-dependent protease affects morphological and physiological differentiation in Streptomyces. Mol. Microbiol. 1999, 32, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Gominet, M.; Seghezzi, N.; Mazodier, P. Acyl depsipeptide (ADEP) resistance in Streptomyces. Microbiology 2011, 157, 2226–2234. [Google Scholar] [CrossRef]
- Derré, I.; Rapoport, G.; Msadek, T. CtsR, a novel regulator of stress and heat shock response, controls clp and molecular chaperone gene expression in gram-positive bacteria. Mol. Microbiol. 1999, 31, 117–131. [Google Scholar] [CrossRef]
- Wawrzynow, A.; Wojtkowiak, D.; Marszalek, J.; Banecki, B.; Jonsen, M.; Graves, B.; Georgopoulos, C.; Zylicz, M. The ClpX heat-shock protein of Escherichia coli, the ATP-dependent substrate specificity component of the ClpP-ClpX protease, is a novel molecular chaperone. EMBO. J. 1995, 14, 1867–1877. [Google Scholar] [CrossRef]
- Wickner, S.; Gottesman, S.; Skowyra, D.; Hoskins, J.; McKenney, K.; Maurizi, M.R. A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc. Natl. Acad. Sci. USA 1994, 91, 12218–12222. [Google Scholar] [CrossRef]
- Maurizi, M.R.; Clark, W.P.; Katayama, Y.; Rudikoff, S.; Pumphrey, J.; Bowers, B.; Gottesman, S. Sequence and structure of ClpP, the proteolytic component of the ATP-dependent Clp protease of Escherichia coli. J. Biol. Chem. 1990, 265, 12536–12545. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. eLife 2020, 9, e52774. [Google Scholar] [CrossRef]
- Martin, A.; Baker, T.A.; Sauer, R.T. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol. Cell 2007, 27, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Amor, A.J.; Schmitz, K.R.; Baker, T.A.; Sauer, R.T. Roles of the ClpX IGF loops in ClpP association, dissociation, and protein degradation. Protein Sci. 2019, 28, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Zhang, Z.; Cronin, M.; Canchaya, C.; Kenny, J.G.; Fitzgerald, G.F.; van Sinderen, D. The ClgR protein regulates transcription of the clpP operon in Bifidobacterium breve UCC 2003. J. Bacteriol. 2005, 187, 8411–8426. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bellier, A.; Mazodier, P. ClgR, a novel regulator of clp and lon expression in Streptomyces. J. Bacteriol. 2004, 186, 3238–3248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Viala, J.; Rapoport, G.; Mazodier, P. The clpP multigenic family in Streptomyces lividans: Conditional expression of the clpP3 clpP4 operon is controlled by PopR, a novel transcriptional activator. Mol. Microbiol. 2000, 38, 602–612. [Google Scholar] [CrossRef]
- Viala, J.; Mazodier, P. ClpP-dependent degradation of PopR allows tightly regulated expression of the clpP3 clpP4 operon in Streptomyces lividans. Mol. Microbiol. 2002, 44, 633–643. [Google Scholar] [CrossRef][Green Version]
- Bellier, A.; Gominet, M.; Mazodier, P. Post-translational control of the Streptomyces lividans ClgR regulon by ClpP. Microbiology 2006, 152, 1021–1027. [Google Scholar] [CrossRef][Green Version]
- Bilyk, B.; Kim, S.; Fazal, A.; Baker, T.A.; Seipke, R.F. Regulation of antimycin biosynthesis is controlled by the ClpXP protease. mSphere 2020, 5, e00144-20. [Google Scholar] [CrossRef]
- Levchenko, I.; Grant, R.A.; Wah, D.A.; Sauer, R.T.; Baker, T.A. Structure of a delivery protein for an AAA+ protease in complex with a peptide degradation tag. Mol. Cell 2003, 12, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Baker, T.A.; Sauer, R.T. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of SsrA-tagged substrates. Mol. Cell 2008, 29, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, S.; Roche, E.; Zhou, Y.; Sauer, R.T. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998, 12, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.M.; Sun, N.; Wang, F.; Luo, S.; Zhou, Z.; Feng, W.H.; Huang, F.L.; Li, Y.Q. Dual positive feedback regulation of protein degradation of an extra-cytoplasmic function sigma factor for cell differentiation in Streptomyces coelicolor. J. Biol. Chem. 2013, 288, 31217–31228. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef]
- Levchenko, I.; Seidel, M.; Sauer, R.T.; Baker, T.A. A specificity-enhancing factor for the ClpXP degradation machine. Science 2000, 289, 2354–2356. [Google Scholar] [CrossRef]
- Flynn, J.M.; Neher, S.B.; Kim, Y.I.; Sauer, R.T.; Baker, T.A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 2003, 11, 671–683. [Google Scholar] [CrossRef]
- Flynn, J.M.; Levchenko, I.; Seidel, M.; Wickner, S.H.; Sauer, R.T.; Baker, T.A. Overlapping recognition determinants within the SsrA degradation tag allow modulation of proteolysis. Proc. Natl. Acad. Sci. USA 2001, 98, 10584–10589. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Yamazaki, H.; Kato, J.Y.; Tomono, A.; Horinouchi, S. AdpA, a central transcriptional regulator in the A-factor regulatory cascade that leads to morphological development and secondary metabolism in Streptomyces griseus. Biosci. Biotechnol. Biochem. 2005, 69, 431–439. [Google Scholar] [CrossRef]
- López-García, M.T.; Santamarta, I.; Liras, P. Morphological differentiation and clavulanic acid formation are affected in a Streptomyces clavuligerus adpA-deleted mutant. Microbiology 2010, 156, 2354–2365. [Google Scholar] [CrossRef]
- Zheng, Y.; Sun, C.F.; Fu, Y.; Chen, X.A.; Li, Y.Q.; Mao, X.M. Dual regulation between the two-component system PhoRP and AdpA regulates antibiotic production in Streptomyces. J. Ind. Microbiol. Biotechnol. 2019, 46, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Guyet, A.; Gominet, M.; Benaroudj, N.; Mazodier, P. Regulation of the clpP1clpP2 operon by the pleiotropic regulator AdpA in Streptomyces lividans. Arch. Microbiol. 2013, 195, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Guyet, A.; Benaroudj, N.; Proux, C.; Gominet, M.; Coppée, J.Y.; Mazodier, P. Identified members of the Streptomyces lividans AdpA regulon involved in differentiation and secondary metabolism. BMC Microbiol. 2014, 14, 81. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, P.; Tai, C.; Deng, Z.; Gan, J.; Oggioni, M.R.; Ou, H.Y. Identification and characterization of chromosomal relBE toxin-antitoxin locus in Streptomyces cattleya DSM46488. Sci. Rep. 2016, 6, 32047. [Google Scholar] [CrossRef]
- Compton, C.L.; Schmitz, K.R.; Sauer, R.T.; Sello, J.K. Antibacterial activity of and resistance to small molecule inhibitors of the ClpP peptidase. ACS. Chem. Biol. 2013, 8, 2669–2677. [Google Scholar] [CrossRef]
- Krzywda, S.; Brzozowski, A.M.; Verma, C.; Karata, K.; Ogura, T.; Wilkinson, A.J. The crystal structure of the AAA domain of the ATP-dependent protease FtsH of Escherichia coli at 1.5 A resolution. Structure 2002, 10, 1073–1083. [Google Scholar] [CrossRef]
- Langklotz, S.; Baumann, U.; Narberhaus, F. Structure and function of the bacterial AAA protease FtsH. Biochim. Biophys. Acta 2012, 1823, 40–48. [Google Scholar] [CrossRef]
- Bieniossek, C.; Schalch, T.; Bumann, M.; Meister, M.; Meier, R.; Baumann, U. The molecular architecture of the metalloprotease FtsH. Proc. Natl. Acad. Sci. USA 2006, 103, 3066–3071. [Google Scholar] [CrossRef]
- Ogura, T.; Inoue, K.; Tatsuta, T.; Suzaki, T.; Karata, K.; Young, K.; Su, L.H.; Fierke, C.A.; Jackman, J.E.; Raetz, C.R.; et al. Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease FtsH (HflB) in Escherichia coli. Mol. Microbiol. 1999, 31, 833–844. [Google Scholar] [CrossRef]
- Fuhrer, F.; Langklotz, S.; Narberhaus, F. The C-terminal end of LpxC is required for degradation by the FtsH protease. Mol. Microbiol. 2006, 59, 1025–1036. [Google Scholar] [CrossRef]
- Fuhrer, F.; Muller, A.; Baumann, H.; Langklotz, S.; Kutscher, B.; Narberhaus, F. Sequence and length recognition of the C-terminal turnover element of LpxC, a soluble substrate of the membrane-bound FtsH protease. J. Mol. Biol. 2007, 372, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Bittner, L.M.; Westphal, K.; Narberhaus, F. Conditional proteolysis of the membrane protein YfgM by the FtsH protease depends on a novel N-terminal degron. J. Biol. Chem. 2015, 290, 19367–19378. [Google Scholar] [CrossRef]
- Arends, J.; Thomanek, N.; Kuhlmann, K.; Marcus, K.; Narberhaus, F. In vivo trapping of FtsH substrates by label-free quantitative proteomics. Proteomics 2016, 16, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, M.; Yura, T.; Tsuchimoto, S.; Fukumori, Y.; Kanemori, M. Conserved region 2.1 of Escherichia coli heat shock transcription factor sigma32 is required for modulating both metabolic stability and transcriptional activity. J. Bacteriol. 2004, 186, 7474–7480. [Google Scholar] [CrossRef] [PubMed]
- Obrist, M.; Narberhaus, F. Identification of a turnover element in region 2.1 of Escherichia coli sigma32 by a bacterial one-hybrid approach. J. Bacteriol. 2005, 187, 3807–3813. [Google Scholar] [CrossRef][Green Version]
- Yura, T.; Guisbert, E.; Poritz, M.; Lu, C.Z.; Campbell, E.; Gross, C.A. Analysis of sigma32 mutants defective in chaperone-mediated feedback control reveals unexpected complexity of the heat shock response. Proc. Natl. Acad. Sci. USA 2007, 104, 17638–17643. [Google Scholar] [CrossRef] [PubMed]
- Obrist, M.; Langklotz, S.; Milek, S.; Fuhrer, F.; Narberhaus, F. Region C of the Escherichia coli heat shock sigma factor RpoH (sigma32) contains a turnover element for proteolysis by the FtsH protease. FEMS. Microbiol. Lett. 2009, 290, 199–208. [Google Scholar] [CrossRef]
- Hidalgo, E.; Demple, B. An iron-sulfur center essential for transcriptional activation by the redox-sensing SoxR protein. EMBO J. 1994, 13, 138–146. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, X.; Yang, H.; Yan, H.; Wen, Y. Redox-sensitive transcriptional regulator SoxR directly controls antibiotic production, development and thiol-oxidative stress response in Streptomyces avermitilis. Microb. Biotechnol. 2022, 15, 561–576. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Q.; Yang, M.; Chen, Z.; Li, J.; Wen, Y. Heat shock repressor HspR directly controls avermectin production, morphological development, and H2O2 stress response in Streptomyces avermitilis. Appl. Environ. Microbiol. 2021, 87, e0047321. [Google Scholar] [CrossRef]
- Busche, T.; Tsolis, K.C.; Koepff, J.; Rebets, Y.; Ruckert, C.; Hamed, M.B.; Bleidt, A.; Wiechert, W.; Lopatniuk, M.; Yousra, A.; et al. Multi-omics and targeted approaches to determine the role of cellular proteases in Streptomyces protein secretion. Front. Microbiol. 2018, 9, 1174. [Google Scholar] [CrossRef] [PubMed]
- Ludke, A.; Kramer, R.; Burkovski, A.; Schluesener, D.; Poetsch, A. A proteomic study of Corynebacterium glutamicum AAA+ protease FtsH. BMC Microbiol. 2007, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Kahne, S.C.; Darwin, K.H. Structural determinants of regulated proteolysis in pathogenic bacteria by ClpP and the proteasome. Curr. Opin. Struct. Biol. 2021, 67, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef]
- Striebel, F.; Imkamp, F.; Sutter, M.; Steiner, M.; Mamedov, A.; Weber-Ban, E. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat. Struct. Mol. Biol. 2009, 16, 647–651. [Google Scholar] [CrossRef]
- Festa, R.A.; McAllister, F.; Pearce, M.J.; Mintseris, J.; Burns, K.E.; Gygi, S.P.; Darwin, K.H. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis. PLoS ONE 2010, 5, e8589. [Google Scholar] [CrossRef]
- Xu, X.; Niu, Y.; Liang, K.; Shen, G.; Cao, Q.; Yang, Y. Analysis of pupylation of Streptomyces hygroscopicus 5008 in vitro. Biochem. Biophys. Res. Commun. 2016, 474, 126–130. [Google Scholar] [CrossRef]
- Elharar, Y.; Roth, Z.; Hecht, N.; Rotkopf, R.; Khalaila, I.; Gur, E. Posttranslational regulation of coordinated enzyme activities in the Pup-proteasome system. Proc. Natl. Acad. Sci. USA 2016, 113, E1605–E1614. [Google Scholar] [CrossRef]
- Zerbib, E.; Schlussel, S.; Hecht, N.; Bagdadi, N.; Eichler, J.; Gur, E. The prokaryotic ubiquitin-like protein presents poor cleavage sites for proteasomal degradation. Cell Rep. 2021, 36, 109428. [Google Scholar] [CrossRef]
- Iyer, L.M.; Burroughs, A.M.; Aravind, L. Unraveling the biochemistry and provenance of pupylation: A prokaryotic analog of ubiquitination. Biol. Direct. 2008, 3, 45. [Google Scholar] [CrossRef]
- Guth, E.; Thommen, M.; Weber-Ban, E. Mycobacterial ubiquitin-like protein ligase PafA follows a two-step reaction pathway with a phosphorylated pup intermediate. J. Biol. Chem. 2011, 286, 4412–4419. [Google Scholar] [CrossRef] [PubMed]
- Darwin, K.H.; Lin, G.; Chen, Z.; Li, H.; Nathan, C.F. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol. Microbiol. 2005, 55, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, H.; Wang, T.; Pan, H.; Lin, G.; Li, H. Structural basis for the assembly and gate closure mechanisms of the Mycobacterium tuberculosis 20S proteasome. EMBO J. 2010, 29, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.J.; Arora, P.; Festa, R.A.; Butler-Wu, S.M.; Gokhale, R.S.; Darwin, K.H. Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J. 2006, 25, 5423–5432. [Google Scholar] [CrossRef]
- Wang, T.; Li, H.; Lin, G.; Tang, C.; Li, D.; Nathan, C.; Darwin, K.H.; Li, H. Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure 2009, 17, 1377–1385. [Google Scholar] [CrossRef]
- De Mot, R.; Schoofs, G.; Nagy, I. Proteome analysis of Streptomyces coelicolor mutants affected in the proteasome system reveals changes in stress-responsive proteins. Arch. Microbiol. 2007, 188, 257–271. [Google Scholar] [CrossRef]
- Compton, C.L.; Fernandopulle, M.S.; Nagari, R.T.; Sello, J.K. Genetic and Proteomic Analyses of Pupylation in Streptomyces coelicolor. J. Bacteriol. 2015, 197, 2747–2753. [Google Scholar] [CrossRef]
- Boubakri, H.; Seghezzi, N.; Duchateau, M.; Gominet, M.; Kofronova, O.; Benada, O.; Mazodier, P.; Pernodet, J.L. The absence of pupylation (prokaryotic ubiquitin-like protein modification) affects morphological and physiological differentiation in Streptomyces coelicolor. J. Bacteriol. 2015, 197, 3388–3399. [Google Scholar] [CrossRef]
- Kuberl, A.; Polen, T.; Bott, M. The pupylation machinery is involved in iron homeostasis by targeting the iron storage protein ferritin. Proc. Natl. Acad. Sci. USA 2016, 113, 4806–4811. [Google Scholar] [CrossRef]
- Hong, B.; Wang, L.; Lammertyn, E.; Geukens, N.; Van Mellaert, L.; Li, Y.; Anne, J. Inactivation of the 20S proteasome in Streptomyces lividans and its influence on the production of heterologous proteins. Microbiology 2005, 151, 3137–3145. [Google Scholar] [CrossRef][Green Version]
- Xu, W.F.; Fang, J.L.; Bu, Q.T.; Lyu, Z.Y.; Zhu, C.Y.; Sun, C.F.; Zhao, Q.W.; Li, Y.Q. A novel strategy of gene screen based on multi-omics in Streptomyces roseosporus. Appl. Microbiol. Biotechnol. 2022, 106, 3103–3112. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.U.; Weber-Ban, E. The bacterial proteasome at the core of diverse degradation pathways. Front. Mol. Biosci. 2019, 6, 23. [Google Scholar] [CrossRef]
- Ziemski, M.; Jomaa, A.; Mayer, D.; Rutz, S.; Giese, C.; Veprintsev, D.; Weber-Ban, E. Cdc48-like protein of actinobacteria (Cpa) is a novel proteasome interactor in mycobacteria and related organisms. eLife 2018, 7, e34055. [Google Scholar] [CrossRef] [PubMed]
- Rivo, Y.B.; Alkarimah, A.; Ramadhani, N.N.; Cahyono, A.W.; Laksmi, D.A.; Winarsih, S.; Fitri, L.E. Metabolite extract of Streptomyces hygroscopicus Hygroscopicus inhibit the growth of Plasmodium berghei through inhibition of ubiquitin-proteasome system. Trop. Biomed. 2013, 30, 291–300. [Google Scholar] [PubMed]
- Frank, E.G.; Ennis, D.G.; Gonzalez, M.; Levine, A.S.; Woodgate, R. Regulation of SOS mutagenesis by proteolysis. Proc. Natl. Acad. Sci. USA 1996, 93, 10291–10296. [Google Scholar] [CrossRef]
- Gonzalez, M.; Rasulova, F.; Maurizi, M.R.; Woodgate, R. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: In vitro degradation and identification of residues required for proteolysis. Genes Dev. 1998, 12, 3889–3899. [Google Scholar] [CrossRef]
- Gonzalez, M.; Rasulova, F.; Maurizi, M.R.; Woodgate, R. Subunit-specific degradation of the UmuD/D' heterodimer by the ClpXP protease: The role of trans recognition in UmuD' stability. EMBO J. 2000, 19, 5251–5258. [Google Scholar] [CrossRef]
- Wright, R.; Stephens, C.; Zweiger, G.; Shapiro, L.; Alley, M.R. Caulobacter Lon protease has a critical role in cell-cycle control of DNA methylation. Genes Dev. 1996, 10, 1532–1542. [Google Scholar] [CrossRef]
- Jenal, U.; Fuchs, T. An essential protease involved in bacterial cell-cycle control. EMBO J. 1998, 17, 5658–5669. [Google Scholar] [CrossRef]
- Mao, X.M.; Ren, N.N.; Sun, N.; Wang, F.; Zhou, R.C.; Tang, Y.; Li, Y.Q. Proteasome involvement in a complex cascade mediating SigT degradation during differentiation of Streptomyces coelicolor. FEBS Lett. 2014, 588, 608–613. [Google Scholar] [CrossRef]
- Liao, G.; Liu, Q.; Xie, J. Transcriptional analysis of the effect of exogenous decanoic acid stress on Streptomyces roseosporus. Microb. Cell Fact. 2013, 12, 19. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).