3.1. Determination of Adhesion Rates of SH-SY5Y and HT22 Cells in the Presence of Extracellular Aβ42 or Its Aggregates by MTT Assay
To determine whether different species and forms of extracellular Aβ42 also have different effects on cell adhesion, and to elucidate the relationship of the anchoring effects of various extracellular Aβ42 aggregates on neural cell adhesion, the adhesion rates of SH-SY5Y and HT22 cells were first measured after they were seeded into 96-well plates at the same density and incubated for 12 h in the presence of the three typical Aβ42 species (monomers, oligomers, and fibrils) in soluble (or suspended) and deposited (or attached) forms. Meanwhile, scFv HT7 (or HT6) antibody was applied to further determine the independent effects of these extracellular Aβ42 aggregates on cell adhesion, since it can inhibit or neutralize the neurotoxicity of Aβ42 aggregates, especially Aβ42 oligomers [
26,
27]. The adhesion rates of SH-SY5Y and HT22 cell lines in the presence of the three Aβ42 species (monomers, oligomers, and fibrils) within 12 h are shown in
Figure 1.
Overall, different species and/or forms of extracellular Aβ42 affect the adhesion of SH-SY5Y cells to varying degrees in a concentration-dependent manner (
Figure 1A,B). In the Aβ42M group (
a–
g in
Figure 1A,B), the cell adhesion rates gradually decreased with the increase in extracellular Aβ42M concentration (from 0.02 to 2.0 μM), but there was no significant difference between the Aβ42M-treated group and the control group (
p > 0.05), except for the SAβ42M subgroup at high concentrations (2.0 μM; *
p < 0.05) (
d in
Figure 1A). However, with increasing duration of incubation with Aβ42M but without HT7 (or HT6) antibody, the cell adhesion rates in both the SAβ42M and DAβ42M subgroups, especially the SAβ42M subgroup, were further reduced (SAβ42M/DAβ42M subgroup vs the control group,
p < 0.05; SAβ42M vs DAβ42M subgroups at 2.0 μM,
p < 0.05) (data not shown).
Unlike extracellular Aβ42M, extracellular Aβ42O (
h–
n in
Figure 1A,B) and Aβ42F (
o–
u in
Figure 1A,B) caused a significant reduction in the cell adhesion rate within 12 h. In the Aβ42O group (
h–
n in
Figure 1A,B), both SAβ42O and DAβ42O significantly impaired cell adhesion in a concentration-dependent manner (*
p < 0.05, **
p < 0.01, ***
p < 0.001), and at the same concentration, extracellular SAβ42O caused a greater effect on cell adhesion than extracellular DAβ42O (SAβ42O group vs DAβ42O group, §
p < 0.05, §§
p <0.01) (
i,
k, and
n in
Figure 1A,B). This demonstrated that extracellular SAβ42O had a greater impact on cell adhesion than extracellular DAβ42O. This was similar to the case in the Aβ42M group, except that the difference in cell adhesion rates between the SAβ42O and DAβ42O subgroups was more significant. These results indicated that the adhesion of neural cells correlated negatively with the neurotoxicity of extracellular Aβ42O, since SAβ42O showed greater neurotoxicity than DAβ42O [
29]. Furthermore, the results from
h–
n in
Figure 1A,B indicated that the results shown from
a–
g of
Figure 1A,B should represent the combined effects of extracellular Aβ42M and Aβ42O (freshly formed) on SH-SY5Y cell adhesion, since Aβ42Ms gradually aggregate to Aβ42Os with increasing Aβ42M concentration during incubation at 37 °C. Thus, this also suggested that extracellular Aβ42M, either SAβ42M or DAβ42M, did not actually damage the adhesion of neural cells and might even enhance it.
Similarly, extracellular insoluble Aβ42F dose-dependently caused a significant reduction in cell adhesion (*
p < 0.05, **
p < 0.01, ***
p < 0.001) (
o–
u in
Figure 1A,B), and at the same concentration, extracellular SAβ42F showed a greater reduction in the cell adhesion rate than extracellular DAβ42F, especially at high Aβ42F concentration (§
p < 0.05), although extracellular Aβ42F showed slightly lower overall negative effects on cell adhesion than extracellular Aβ42O. This was apparently consistent with their correlation with neurotoxicity [
29].
Additionally, the results from
e–
g,
l–
n, and
s–
u of
Figure 1A,B showed that anti-oligomeric Aβ42 scFv HT7 (or HT6) antibody was able to dose-dependently defend the adhesion capability of SH-SY5Y cells (#
p < 0.05, ###
p < 0.001). This meant that by specifically targeting extracellular Aβ42 aggregates, especially Aβ42O, HT7 (or HT6) antibody could dose-dependently block their adverse effects on neural cell adhesion, in addition to protecting cell survival and motility [
26,
27,
29].
In addition to the SH-SY5Y cell line, the HT22 cell line is another in vitro model of neural cells. The HT22 cell line is derived from a primary mouse hippocampal neuronal culture and possesses more neuronal characteristics and phenotypes. To confirm the effect of extracellular SAβ42/DAβ42 or their aggregates on neuronal adhesion, the adhesion rate of HT22 cells was also measured under the same conditions as above. The results showed that overall, extracellular SAβ42/DAβ42 aggregates caused similar but greater decreases in the adhesion rate of HT22 cells than SH-SY5Y cells did.
Figure 1C,D shows the adhesion rates of HT22 cells in the Aβ42O group (data of Aβ42M and Aβ42F groups not shown). This indicated that HT22 cells might be more sensitive than SH-SY5Y cells to extracellular Aβ42 aggregates, at least in terms of adhesion.
Overall, the results of the SH-SY5Y/HT22 cells consistently demonstrated that different species and forms of extracellular Aβ42 had different effects on neural cell adhesion, and their effects were directly associated with their neurotrophicity or neurotoxicity; extracellular SAβ42 aggregates adversely affected neural cell adhesion more than extracellular DAβ42 aggregates, especially at higher concentrations. Clearly, the adverse effects of extracellular SAβ42 and DAβ42 aggregates on neural cell adhesion are inversely correlated with their anchoring effects, as reported by Zhang [
29].
3.2. Extracellular Aβ42 Aggregates Lead to Live De-Adhesion of SH-SY5Y Cells in Addition to Their Death
Given that the adhesion of SH-SY5Y/HT22 cells was positively correlated with the neurotrophicity of extracellular Aβ42M and negatively correlated with the neurotoxicity of Aβ42 aggregates, especially SAβ42O (
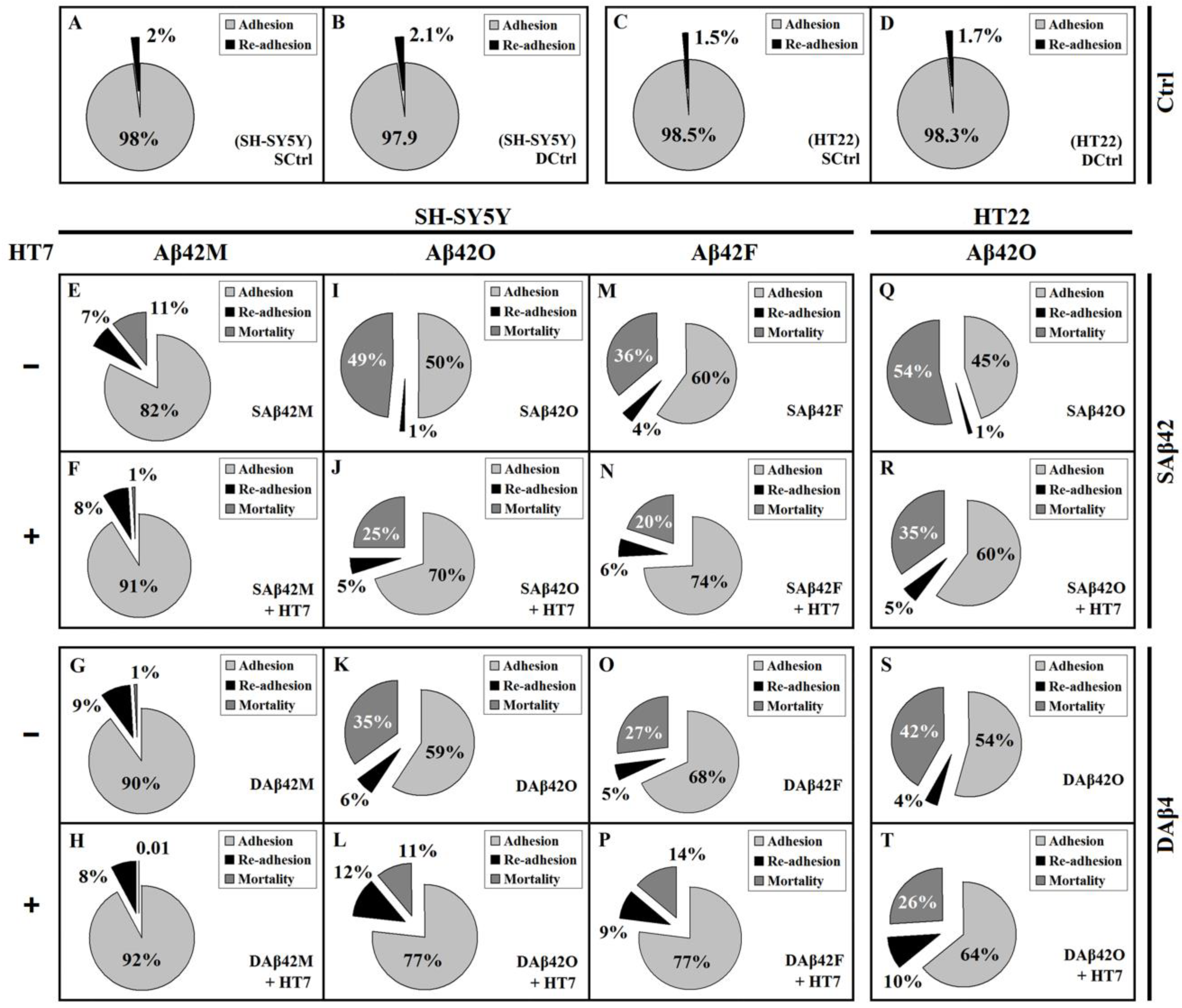
Figure 1), it was clear that any extracellular Aβ42 (including all species and forms) had consistent effects on the adhesion and viability of neural cells. To determine affiliation of adhesion of neural cells to their viability, the rates of re-adhesion of cells, i.e., the rates of de-adhesion of living cells, in all of the above groups were measured, and the corresponding mortality rates were calculated based on their adhesion and de-adhesion rates.
Figure 2 shows the relative adhesion, re-adhesion, and mortality rates of SH-SY5Y/HT22 cells in the presence of 2.0 μM extracellular Aβ42. The results shown in
Figure 2E–P demonstrated that there were a certain number of living cells, as well as dead cells, in the shed (or debonded) cells of each group, after exposure to various extracellular Aβ42 for 12 h. Apparently, these living cells (re-adherent cells) belonged to the cells that de-adhered alive, and represented those cells that were only slightly damaged, at least in adhesion, but were able to survive after being transferred to normal culture medium. Inevitably, in addition to the difference in adhesion rates, there were also significant differences in re-adhesion and mortality rates between the various Aβ42-treated groups.
In the Aβ42M group (
Figure 2E–H), the vast majority of shed cells were living cells rather than dead cells, except for the SAβ42M subgroup (
Figure 2E), where the slightly higher mortality rate was likely caused by a small amount of Aβ42O (freshly formed by Aβ42M aggregation). Apparently due to the beneficial support of Aβ42M and the protection of HT7 (or HT6) antibody, these de-adhered cells were able to stay alive for a period of time, such as 12 h, and re-adhere in order to grow. However, the case was quite different in the Aβ42O group (
Figure 2I–L), where the vast majority of shed cells belonged to dead rather than living cells (re-adherent cells), except for the DAβ42O plus HT7 subgroup (
Figure 2L), where the number of dead cells and number of re-adherent cells were nearly equal. The re-adhesion and lower mortality rates in the Aβ42F group (
Figure 2M–P) were similar to those in the Aβ42O group, which suggested that the neurotoxicity of extracellular Aβ42 aggregates was truly reflected in the actual mortality rate of neural cells, rather than in their total de-adhesion (or debonding) rate.
Similar results were obtained from HT22 cells; however, the mortality rate in the HT22 cell system was higher than that in the SH-SY5Y cell system (
p < 0.05 to
p < 0.001).
Figure 2Q–T shows the relative adhesion, re-adhesion, and mortality rates of HT22 cells in the presence of extracellular SAβ42O/DAβ42O at 2.0 μM with or without HT7 (or HT6) antibody (data of HT22 cells in the presence of extracellular Aβ42M and Aβ42F not shown). The similar re-adhesion rate and higher mortality rate in the HT22 cell system than in the SH-SY5Y cell system indicated that HT22 cells are more susceptible to the toxic damage of extracellular Aβ42O, which was consistent with the results shown from
h–
n of
Figure 1A,B and from
a–
g of
Figure 1C,D.
The results of
Figure 2 showed that, overall, both adhesion and re-adhesion rates of neural cells negatively correlated with the neurotoxicity of extracellular Aβ42 aggregates, and the actual mortality rate of neural cells positively correlated with their neurotoxicity. Furthermore, the results of
Figure 2 demonstrated again that extracellular SAβ42 aggregates had a greater adverse effect on neural cell adhesion than extracellular DAβ42 aggregates. By comparing the effects of extracellular SAβ42 and DAβ42 aggregates on the adhesion, re-adhesion, and mortality rates of target cells, we found that the greater the neurotoxicity of the extracellular Aβ42 species, the more significant the differences in their damage to neural cell adhesion between their SAβ42 and DAβ42 forms. The presence of re-adherent cells also demonstrated that the damage of neural cells by extracellular toxic Aβ42 aggregates was progressive. Therefore, the higher the toxic activity of the extracellular Aβ42 aggregates, the shorter the survival duration of the cells that were de-adhered alive and the faster they progressed towards death, which inevitably led to a higher mortality rate of the target cells. Additionally, the presence of re-adherent cells also implied that the damage of the neurotoxic Aβ42 aggregates to the cytoskeleton system of neural cells might be more initial or more direct than their damage to the overall cell metabolism. Evidently, the proportion of re-adherent cells shown in
Figure 2 represented the proportion of the cells that might be healed or repaired after being damaged (at least in terms of adhesion capacity) by extracellular toxic Aβ42 aggregates. Inevitably, the ultimate fate of these re-adherent cells depended on numerous factors, both inside and outside of the cells.
In addition, it was found that extracellular DAβ42F-HT7 always showed a slightly greater negative effect on the de-adhered cells (lower re-adhesion rate and higher mortality rate) than extracellular DAβ42O-HT7, which might be because the large DAβ42F/DAβ42F-HT7 units physically blocked normal interactions between neural cells and ECM more than the small DAβ42O/DAβ42O-HT7 units, which was directly reflected in their effect on neural cell adhesion(HT7/HT6 alone did not affect the cell adhesion rate,
Figure 1). Furthermore, comparisons of the results between
h–
n of
Figure 1A,B and
a–
g of
Figure 1C,D and between
Figure 2E–H and M–P implied that neuronal cells, especially those in the hippocampus, might be more sensitive to the neurotoxicity of extracellular Aβ42O, which was consistent with our previous report [
29].
3.3. Effects of Extracellular Aβ42 or Its Aggregates on Neurite Formation and Extension in Differentiated SH-SY5Y Cells
In addition to regulating neural cell adhesion, ECM also regulates neural cell morphology, including neurite outgrowth (neuritogenesis) [
31]; meanwhile, neural cell adhesion also directly affects neurite outgrowth. Given that different species and forms of extracellular Aβ42 exerted different effects on the adhesion of SH-SY5Y/HT22 cells, in order to elucidate the correlation between changes in neurite outgrowth and cell adhesion in the presence of extracellular Aβ42 or its aggregates, it was necessary to further investigate the effects of extracellular Aβ42 or its aggregates on neurite outgrowth. This required the use of appropriately differentiated model cells to accurately represent what may be occurring in neurites in vivo. The differentiated SH-SY5Y cells are a useful experimental model for the study of neurite outgrowth because they undergo differentiation to a neuron-like phenotype. The appropriately differentiated SH-SY5Y cells were first prepared and their typical neuronal morphology (with numerous long, branching neurites) was examined by preliminary experiments. Following this, Aβ42-treated differentiated SH-SY5Y cells were imaged, and the length and number of their neurites were quantitatively and statistically analyzed. The representative whole images of differentiated SH-SY5Y cells after 24 h of incubation with various extracellular SAβ42 and DAβ42 species for 24 h are shown in
Figure S1, and their enlarged partial images and the quantitative results of the length and number of neurites of the cells are shown in
Figure 3.
It can be seen from
Figure S1 that, overall, the length and/or number of neurites in each neuron were shortened and/or reduced to varying degrees after 24 h of treatment with various extracellular aggregates. The enlarged partial images shown in
Figure 3A are representative of the morphology of the cells in each group. During this period, the control cells formed numerous long neurites and showed normal differentiation patterns (
a,
b in
Figure 3A), and their shape closely resembled the morphology of neurons in vivo. Compared with the control cells, some, although not all, Aβ42-treated cells (
c,
d,
g,
h, and
k–
l in
Figure 3A) exhibited dramatically shortened neurites and/or reduced number of neurites, indicating impaired neurite outgrowths; however, the presence of HT7 significantly mitigated these impairments. Specifically, in the Aβ42M group (
c–
f in
Figure 3A), the length and number of neurites did not appear to be significantly different from those in the control group (
a,
b in
Figure 3A), especially in the presence of HT7 antibody (
e–
f in
Figure 3A). However, in the Aβ42O (
g–
j in
Figure 3A) and Aβ42F groups (
k–
n in
Figure 3A), the length and number of neurites were significantly reduced, especially in the absence of HT7 (
g,
h and
k,
l in
Figure 3A). Furthermore, we noticed that the impairment of neurites occurred mainly in their length in the SAβ42 group, and in their number in the DAβ42 group, especially in the Aβ42O group.
Figure 3B,C shows the quantitative data of the length and number of neurites in the corresponding groups shown in
Figure 3A, based on all images obtained from five replicate experiments with different batches of cells. As shown from
a–
d in
Figure 3B,C, after 24 h of cell culture in the presence of extracellular Aβ42M with or without HT7 antibody, there were no significant differences in neurite length and number between most Aβ42M-treated and control (100%) groups, except for in the SAβ42M-treated group (*
p < 0.05). Similarly, extracellular Aβ42M presumably did not affect neurite occurrence and extension, and the slight reductions in both neurite length and number in the SAβ42M subgroups (
a in
Figure 3B,C), and perhaps in the DAβ42M subgroup (
b in
Figure 3B,C), were presumably caused by a small amount of Aβ42O, newly formed by Aβ42M aggregation during the 24-h incubation period, which could be demonstrated by the significant differences between
a and
c in
Figure 3B,C (#
p < 0.05). Anti-oligomeric Aβ42 antibody HT7 (or HT6) has been reported to exert neuroprotective efficacy by specifically targeting Aβ42O [
26,
27]. Therefore, this also suggested that extracellular Aβ42O might have a direct blocking or inhibitory effect on the neuritogenesis of neural cells, and extracellular SAβ42O was likely to have a greater negative effect on neurite occurrence and extension than extracellular DAβ42O, although there was no significant difference between
a and
b in
Figure 3B and C during the 24-h period.
However, the quantitative data from
e–
h of
Figure 3B,C showed that most of the cells treated with either extracellular SAβ42O/DAβ42O and SAβ42F/DAβ42F failed to achieve a normal length and/or number of neurites, indicating that extracellular Aβ42 aggregates caused a dramatic decline in neurite length and number (*
p < 0.05, **
p < 0.01, ***
p < 0.001); similarly, the damage effects of these extracellular Aβ42 aggregates could be inhibited or blocked to some extent by the anti-oligomeric Aβ42 antibody HT7 (#
p < 0.05). Evidently, the damage effects of the extracellular Aβ42 aggregates on neurite occurrence and extension were positively correlated with their neurotoxicity. On the other hand, within the same period (e.g., 12 h), the protective efficacy on neurite outgrowth of HT7 (or HT6) antibody at equimolar levels to extracellular Aβ42 appeared to be limited compared to its protective efficacy on cell adhesion (###
p < 0.001, #
p < 0.05,
k and
n, r and
u of
Figure 1A,B), which suggested that, in addition to the biological effects of extracellular Aβ42 aggregates, their physical barrier effects might also be responsible for blocking or disrupting neurite outgrowth. Further, the results from
e–
j of
Figure 3B showed that there was a significant difference in neurite length between the SAβ42 and DAβ42 subgroups in these three pairs (§
p < 0.05). This suggested that extracellular SAβ42 aggregates significantly affected neurite extension (or growth) in particular. In contrast, there was no significant difference in the number of neurites between these subgroups (
Figure 3C). Since the cell adhesion rate in the SAβ42O/SAβ42F subgroup was significantly lower than that in the DAβ42O/DAβ42F subgroup (
k and
r in
Figure 1A,B,
d in
Figure 1C,D), the average number of neurites per neural cell in the DAβ42O/DAβ42F subgroup should actually be lower than that in the SAβ42O/SAβ42F subgroup.
The images and quantitative data in
Figure 3 demonstrate that extracellular Aβ42 aggregates have complex and diverse adverse effects on neurite outgrowth through their biological effects and perhaps their physical barrier effects, in which SAβ42 aggregates appeared to mainly affect neurite extension, while DAβ42 aggregates appeared to mainly affect neurite occurrence. This implied that extracellular SAβ42 and DAβ42 aggregates adversely affect neurite outgrowth through similar but not identical mechanisms, and thus, damaged neurites in different ways. As a result, however, damage to either of these two aspects would eventually damage the neurite network.
3.4. Dynamic Changes in Levels and Subcellular Localization of Scaffold Protein Palladin
- (1)
Determination of intracellular palladin level in differentiated and undifferentiated SH-SY5Y by dot blot assay
Since intracellular palladin is directly involved in the assembly and construction of the neuronal actin cytoskeleton and directly regulates actin dynamics, it was necessary to determine whether intracellular palladin is affected by extracellular Aβ42 or its aggregates, and if so, how it relates to the detrimental effects of extracellular Aβ42 aggregates on cell adhesion (
Figure 1 and
Figure 2) and neurite outgrowth (
Figure 3). Therefore, we next analyzed the levels of intracellular palladin in differentiated SH-SY5Y cells after 24 h of culture in the presence of extracellular SAβ42 and DAβ42 and their aggregates with or without anti-oligomeric Aβ42 antibody HT7 (or HT6) by dot blot assay (
Figure 4) and examined its subcellular localization by IF staining (
Figure 5).
As shown in
Figure 4A, various species and forms of extracellular Aβ42 caused varying degrees of changes in the levels of intracellular palladin within 24 h. Semiquantitative analysis (
Figure 4B) of these dot images by grayscale scanning showed that, surprisingly, extracellular SAβ42 and DAβ42 induced almost opposite changes in the levels of palladin compared to the controls (100%). Clearly, the levels of palladin in the SAβ42O/SAβ42F subgroups were significantly up-regulated (
e and
i in
Figure 4B), whereas those in the DAβ42O/DAβ42F subgroups were significantly down-regulated (
f and
j in
Figure 4B) (**
p < 0.01, ***
p < 0.001 between Aβ42O/F groups vs control). Consequently, there were significant differences in palladin levels between each pair of DAβ42O/DAβ42F and SAβ42O/SAβ42F groups (§§
p < 0.01). These seemingly contradictory results might be the intracellular manifestation of the different effects of extracellular SAβ42 and DAβ42 aggregates on target cells. This indicated that the soluble (or suspended) and deposited (or attached) forms of extracellular Aβ42 aggregates induced different aspects of target cells and facilitated distinct intracellular palladin responses over 24 h. This implied that the mechanisms by which extracellular SAβ42 and DAβ42 aggregates affected neural cells might be very different; after all, the SAβ42 aggregates in ECM exerted their toxic matrix effect through targeting a more spatial neural cell surface, whereas the DAβ42 aggregates in ECM exerted their toxic matrix effect only through targeting a limited neural cell surface. Likewise, similar but slight up-regulation and down-regulation in palladin levels occurred in the SAβ42M and DAβ42M subgroups, respectively (
a and
b in
Figure 4B), although there was no statistically significant difference between the subgroups or between Aβ42M and the control groups. Similarly, the slight increase or decrease in palladin levels in the SAβ42M or DAβ42M subgroups was presumably induced by a small amount of SAβ42O or DAβ42O (freshly formed by Aβ42M aggregation), respectively.
However, in the presence of HT7 (or HT6) antibodies, both up- and down-regulation of palladin levels in the Aβ42O/Aβ42F-treated groups (
g,
h,
k, and
l in
Figure 4B) were significantly attenuated, and the levels of palladin in these groups were not statistically significantly different from that in the control group. This also demonstrated that extracellular Aβ42O/Aβ42F indeed induced changes (up- or down-regulation) in intracellular palladin levels, but induction could be inhibited or blocked by anti-oligomeric Aβ42 scFv HT7 (or HT6) antibodies.
With reference to previous reports [
20], palladin levels are found to rapidly increase in the astrocytes located closest to the wound edge; thus, we speculated that the over-elevated palladin levels in the SAβ42O/SAβ42F groups (
e and
i in
Figure 4B) might represent a stress response of neural cells to extracellular SAβ42 aggregates, but this stress response did not manifest in the DAβ42O/DAβ42F groups (
f and
j in
Figure 4B). Although the reason for this difference was not understood here, it at least indicated that the stress-dependent up-regulation of palladin was related to extracellular SAβ42 aggregates. However, this excessive up-regulation of palladin levels in the SAβ42O/F groups apparently failed to truly achieve compensatory improvement of cytoskeletal organization in the target cells; thus, neither neural cell adhesion (
h–
u in
Figure 1A,C) nor neurite outgrowth (
e and
i in
Figure 3B,C) were compensatively secured or repaired. This indicated that although palladin plays a critical role in maintaining the integrity of cytoskeleton organization, it is only necessary, not sufficient.
In contrast, down-regulation of palladin levels induced by extracellular DAβ42O/DAβ42F (
f and
j in
Figure 4B) leads to the decline in the actin cytoskeleton organization, according to previous reports [
19,
32] that down-regulation of palladin may reduce neuronal adhesion and retard neuronal growth cone formation and neurite outgrowth, which inevitably causes the impairment of cell adhesion and neurite outgrowth. Thus, the down-regulation of palladin levels in the DAβ42O/DAβ42F groups should be largely, if not entirely, responsible for the decrease in cell adhesion (
h–
u in
Figure 1B,D) and neurite outgrowth (both in length and number) (
f and
j in
Figure 3B,C). Furthermore, the down-regulation of palladin levels might be one of the manifestations of the toxic effects of extracellular DAβ42O/DAβ42F on neural cells.
To further determine whether these changes in palladin levels in
Figure 4B were related to the duration of extracellular Aβ42 action, time-course experiments from 4 to 72 h were performed by dot blot assay for all of the above groups, and the semiquantitative analysis for dot images obtained by grayscale scanning is shown in
Figure 4C (dot images not shown). We found that the overall pattern of changes in palladin levels at other time points was almost identical to that at 24 h (
Figure 4B), but palladin levels in most groups gradually decreased over time after 24 h. In addition, the palladin levels in the groups with SAβ42F/DAβ42F plus the HT7 antibody (small white/black triangle,
Figure 4C) were almost the same after 48 h, which might be related to the easier deposition of large SAβ42F-HT7 particles. Moreover, all of the above analyses of palladin levels were again performed using undifferentiated SH-SY5Y cells, and almost identical results were obtained (data not shown).
Taken together, the results in
Figure 4 demonstrated that extracellular Aβ42 aggregates induced changes in the level of intracellular palladin; further, there was a specific or distinct correspondence between the species and form of extracellular Aβ42 aggregates and the resulting intracellular palladin levels, as follows: extracellular SAβ42 aggregates tended to induce an excessive up-regulation of intracellular palladin levels, whereas extracellular DAβ42 aggregates tended to induce a down-regulation of intracellular palladin levels, both of which might affect the integrity of the intracellular cytoskeleton and result in a degenerate and abnormal cytoskeleton system, thereby leading to impairment of neural cell adhesion (
Figure 1 and
Figure 2) and neurite outgrowth (both in number and length) (
Figure 3). Thus, either abnormally elevated or reduced levels of intracellular palladin was detrimental to both neural cell adhesion and neurite outgrowth.
- (2)
Analysis of subcellular localization of intracellular palladin in differentiated SH-SY5Y cells and HT22 cells by IF staining.
To investigate differences in the subcellular distribution of palladin in the presence of extracellular Saβ42 and Daβ42 and their aggregates, IF staining with fluorescence-labeled anti-palladin antibody and confocal imaging were performed using differentiated SH-SY5Y cells and HT22 cells. After incubating SH-SY5Y cells with various extracellular Aβ42 for 24 h, we observed palladin (green) in the cells, including in the cell body and neurites, by confocal laser scanning microscopy. The representative whole images of SH-SY5Y and HT22 cells are shown in
Figure S2A, and their enlarged partial images are shown in
Figure 5.
As can been seen from the images in
Figure 5, there were some differences in the subcellular distribution of palladin in the different groups. Images of control cells (
a and
b in
Figure 5) showed that palladin was not uniformly distributed to each region in the cells. Specifically, palladin was concentrated at the growth cone-like or neurite branch sites (middle and right arrows), while it was also intermittently distributed at the periphery of cytoplasmic regions and in puncta along the length of neurites (left arrows;
a and
b in
Figure 5), consistent with previous reports [
19]. This indicated that the distribution of palladin within neural cells had regional characteristics. In the Aβ42M group (
d,
e, and
f in
Figure 5), the subcellular localization of palladin was basically the same as that in the control group, except for that in the SAβ42M subgroup, where palladin was mainly distributed in the cytoplasm and only slightly concentrated at the growth cone-like region (
c in
Figure 5). By relating this subcellular localization of palladin with the corresponding results in
d of
Figure 1A and in
a of
Figure 3B,C, it followed that extracellular Aβ42 oligomers (freshly formed) induced an aberrant change in the subcellular localization of palladin in neural cells, but that this induction could be inhibited or blocked by anti-oligomeric Aβ42 antibodies, such as HT7 (
d and
f in
Figure 5).
However, in the Aβ42O (
g–
j in
Figure 5) and Aβ42F (
k–
n in
Figure 5) groups, the difference in the subcellular localization of palladin between the SAβ42O/SAβ42F and DAβ42O/DAβ42F subgroups or between the Aβ42O/Aβ42F subgroups with and without HT7 (or HT6) appeared to be very significant. As shown in the SAβ42O/F subgroups (
g and
k of
Figure 5), high levels of palladin were present in irregular growth cone-like regions and throughout their disorganized extensions (arrows). Combined with the results in
i–
k and
p–
r of
Figure 1A, in
b–
d of
Figure 1C and in
e and
i of
Figure 3B,C, the results of the SAβ42O/SAβ42F subgroups in
Figure 5 suggested that the excessive up-regulation of palladin and chaotic distribution of palladin in neural cells might cause a collapse response in neurite outgrowth, which should be one of the reasons for the decreased cell adhesion and neurite outgrowth. However, in the DAβ42O/DAβ42F subgroups (
i and
m of
Figure 5), much less palladin was observed in slightly irregular growth cone-like regions and their disorganized extension areas, which also corresponded to the declines in cell adhesion (
i–
k and
p–
r of
Figure 1B,
b–
d of
Figure 1D) and neurites (
f and
j of
Figure 3B,C). These declines should be caused by defective cytoskeletal organization due to reduced palladin. In contrast, the distributions of palladin in the Aβ42O/Aβ42F plus HT7 subgroups (
h,
j,
l, and
n in
Figure 5) appeared to be modified somewhat; that is, the palladin levels appeared to be more similar to those in the control groups (
a and
b in
Figure 5).
The results shown in
Figure 5 demonstrated that excessive up-regulation of palladin resulted in difficulty in forming efficient growth cones at the appropriate sites, possibly due to cytoskeletal disturbances, resulting in the failure of neurites to grow properly. Conversely, palladin deficiency also resulted in a reduction in available growth cones, resulting in a reduction in the number of efficient long neurites. Additionally, overall, the results in
Figure 5 confirmed an increase in the palladin level in the SAβ42O/SAβ42F subgroups and a decrease in the palladin level in the DAβ42O/DAβ42F subgroups, as shown in
Figure 4. Apparently, the results of
Figure 5 were consistent with those of
Figure 1,
Figure 2,
Figure 3 and
Figure 4. These consistencies, taken together with previous reports [
19], demonstrated that extracellular Aβ42 aggregates might induce an abnormal change in the level and subcellular distribution of intracellular palladin in neural cells, which could lead to a range of consequences, including the impairment of neural cell adhesion and neurite outgrowth. To examine the relationship of palladin with neurite extension in neuronal cells, the above IF experiments were also performed with the HT22 neuronal cell line. In parallel experiments, almost identical results were obtained in HT22 cells, and the representative IF images of the Aβ42O group are shown in
Figure S2B. Taken together, the results in
Figure 4 and
Figure 5 provided evidence that, at least, optimal intracellular palladin level and subcellular localization were required for the normal adhesion and neuritogenesis of neural cells.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}