
The Amyloid Cascade Hypothesis in Alzheimer’s Disease: Should We Change Our Thinking?


 , , , and
, , , and 
{kind=link}
Abstract
1. Introduction
2. APP and Aβ Peptides
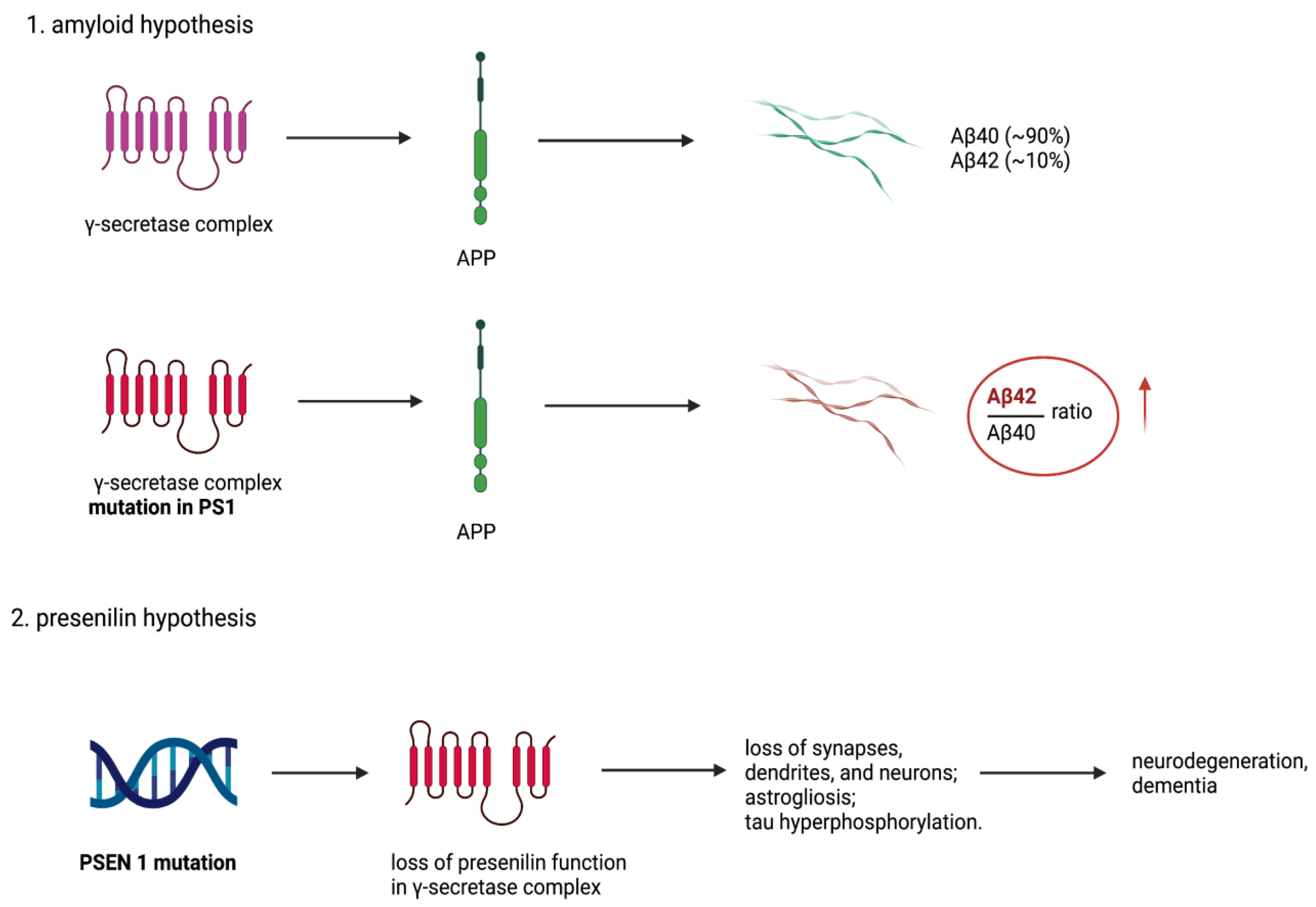
3. The Amyloid Cascade Hypothesis
4. AD Biomarkers
5. AD Drug Trials
6. The Presenilin Hypothesis
7. Synaptic Glutamate Signaling
8. Astrocyte Glutamate Transporter EAAT2
9. In Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The continuum of aging and age-related diseases: Common mechanisms but different rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef]
- Alzheimer, A. Uber eine eigenartige Erkrankung der Hirnrinde. Zentralbl. Nervenh. Psych. 1907, 18, 177–179. [Google Scholar]
- Bomilcar, I.; Bertrand, E.; Morris, R.G.; Mograbi, D.C. The seven selves of dementia. Front. Psychiatry 2021, 12, 646050. [Google Scholar] [CrossRef]
- Whitehouse, P.; George, D. The Myth of Alzheimer’s; St. Martin’s Press: New York, NY, USA, 2008. [Google Scholar]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Roses, A.D. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 1996, 47, 387–400. [Google Scholar] [CrossRef]
- Tanzi, R.E. A brief history of Alzheimer’s disease gene discovery. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S5–S13. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- Martens, Y.A.; Zhao, N.; Liu, C.-C.; Kanekiyo, T.; Yang, A.J.; Goate, A.M.; Holtzman, D.M.; Bu, G. ApoE Cascade Hypothesis in the pathogenesis of Alzheimer’s disease and related dementias. Neuron 2022, 110, 1304–1317. [Google Scholar] [CrossRef]
- International Alzheimer’s Disease Numbers of People with Dementia Worldwide. Available online: https://www.alzint.org/resource/world-alzheimer-report-2015/ (accessed on 18 February 2023).
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Abdalla, N.; Kawas, C.H.; Corrada, M.M. Forecasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimers Dement. 2018, 14, 121–129. [Google Scholar] [CrossRef] [PubMed]
- NIH Bypass Budget Proposal for Fiscal Year 2021. Available online: https://www.nia.nih.gov/sites/default/files/2019-07/FY21-bypass-budget-report-508.pdf (accessed on 18 February 2023).
- Yuan, X.-Z.; Sun, S.; Tan, C.-C.; Yu, J.-T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Hedskog, L.; Wiehager, B.; Ankarcrona, M. Amyloid-β peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 2015, 43, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Ben Halima, S.; Mishra, S.; Raja, K.M.P.; Willem, M.; Baici, A.; Simons, K.; Brüstle, O.; Koch, P.; Haass, C.; Caflisch, A.; et al. Specific inhibition of β-secretase processing of the Alzheimer disease amyloid precursor protein. Cell Rep. 2016, 14, 2127–2141. [Google Scholar] [CrossRef]
- Xia, W. γ-Secretase and its modulators: Twenty years and beyond. Neurosci. Lett. 2019, 701, 162–169. [Google Scholar] [CrossRef]
- Liu, L.; Ding, L.; Rovere, M.; Wolfe, M.S.; Selkoe, D.J. A cellular complex of BACE1 and γ-secretase sequentially generates Aβ from its full-length precursor. J. Cell Biol. 2019, 218, 644–663. [Google Scholar] [CrossRef]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Järemo, P.; Jejcic, A.; Jelic, V.; Shahnaz, T.; Oweling, M.; Winblad, B.; Behbahani, H. erythrocyte amyloid beta peptide isoform distributions in alzheimer and mild cognitive impairment. Curr. Alzheimer Res. 2019, 16, 1050–1054. [Google Scholar] [CrossRef]
- Bush, A.I.; Martins, R.N.; Rumble, B.; Moir, R.; Fuller, S.; Milward, E.; Currie, J.; Ames, D.; Weidemann, A.; Fischer, P. The amyloid precursor protein of Alzheimer’s disease is released by human platelets. J. Biol. Chem. 1990, 265, 15977–15983. [Google Scholar] [CrossRef]
- Mönning, U.; König, G.; Prior, R.; Mechler, H.; Schreiter-Gasser, U.; Masters, C.L.; Beyreuther, K. Synthesis and secretion of Alzheimer amyloid beta A4 precursor protein by stimulated human peripheral blood leucocytes. FEBS Lett. 1990, 277, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis”. FEBS J. 2017, 284, 1040–1044. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef]
- Rosenberg, R.N.; Lambracht-Washington, D.; Yu, G.; Xia, W. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016, 73, 867–874. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef]
- Vemuri, P.; Weigand, S.D.; Przybelski, S.A.; Knopman, D.S.; Smith, G.E.; Trojanowski, J.Q.; Shaw, L.M.; Decarli, C.S.; Carmichael, O.; Bernstein, M.A.; et al. Cognitive reserve and Alzheimer’s disease biomarkers are independent determinants of cognition. Brain 2011, 134, 1479–1492. [Google Scholar] [CrossRef]
- Rosenberg, R.N. Defining amyloid pathology in persons with and without dementia syndromes: Making the right diagnosis. JAMA 2015, 313, 1913–1914. [Google Scholar] [CrossRef]
- Mountz, J.M.; Laymon, C.M.; Cohen, A.D.; Zhang, Z.; Price, J.C.; Boudhar, S.; McDade, E.; Aizenstein, H.J.; Klunk, W.E.; Mathis, C.A. Comparison of qualitative and quantitative imaging characteristics of [11C]PiB and [18F]flutemetamol in normal control and Alzheimer’s subjects. NeuroImage. Clin. 2015, 9, 592–598. [Google Scholar] [CrossRef]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Knopman, D.S.; Parisi, J.E.; Salviati, A.; Floriach-Robert, M.; Boeve, B.F.; Ivnik, R.J.; Smith, G.E.; Dickson, D.W.; Johnson, K.A.; Petersen, L.E.; et al. Neuropathology of cognitively normal elderly. J. Neuropathol. Exp. Neurol. 2003, 62, 1087–1095. [Google Scholar] [CrossRef]
- Snowdon, D.A. Aging and Alzheimer’s disease: Lessons from the Nun Study. Gerontologist 1997, 37, 150–156. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. Addendum: The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2017, 546, 564. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.; Lemere, C.; Atri, A.; Sabbagh, M.; Salloway, S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimers Res. Ther. 2021, 13, 98. [Google Scholar] [CrossRef]
- Chia, C.W.; Egan, J.M.; Ferrucci, L. Age-Related Changes in Glucose Metabolism, Hyperglycemia, and Cardiovascular Risk. Circ. Res. 2018, 123, 886–904. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Dekundy, A.; Pulte, I. Memantine and cholinesterase inhibitors: Complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox. Res. 2013, 24, 358–369. [Google Scholar] [CrossRef]
- Khoury, R.; Rajamanickam, J.; Grossberg, G.T. An update on the safety of current therapies for Alzheimer’s disease: Focus on rivastigmine. Ther. Adv. drug Saf. 2018, 9, 171–178. [Google Scholar] [CrossRef]
- Gąsiorowski, K.; Brokos, J.B.; Sochocka, M.; Ochnik, M.; Chojdak-Łukasiewicz, J.; Zajączkowska, K.; Fułek, M.; Leszek, J. Current and Near-Future Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2022, 20, 1144–1157. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. The Past and the Future of Alzheimer’s Disease Fluid Biomarkers. J. Alzheimers Dis. 2018, 62, 1125–1140. [Google Scholar] [CrossRef]
- Lloret, A.; Esteve, D.; Lloret, M.-A.; Cervera-Ferri, A.; Lopez, B.; Nepomuceno, M.; Monllor, P. When Does Alzheimer’s Disease Really Start? The Role of Biomarkers. Int. J. Mol. Sci. 2019, 20, 5536. [Google Scholar] [CrossRef]
- Hrubešová, K.; Fousková, M.; Habartová, L.; Fišar, Z.; Jirák, R.; Raboch, J.; Setnička, V. Search for biomarkers of Alzheimer’s disease: Recent insights, current challenges and future prospects. Clin. Biochem. 2019, 72, 39–51. [Google Scholar] [CrossRef]
- Yu, J.-T.; Xu, W.; Tan, C.-C.; Andrieu, S.; Suckling, J.; Evangelou, E.; Pan, A.; Zhang, C.; Jia, J.; Feng, L.; et al. Evidence-based prevention of Alzheimer’s disease: Systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1201–1209. [Google Scholar] [CrossRef]
- Kivipelto, M.; Mangialasche, F.; Snyder, H.M.; Allegri, R.; Andrieu, S.; Arai, H.; Baker, L.; Belleville, S.; Brodaty, H.; Brucki, S.M.; et al. World-Wide FINGERS Network: A global approach to risk reduction and prevention of dementia. Alzheimers Dement. 2020, 16, 1078–1094. [Google Scholar] [CrossRef]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet. Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef]
- Jack, C.R.J.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Ray, S.; Britschgi, M.; Herbert, C.; Takeda-Uchimura, Y.; Boxer, A.; Blennow, K.; Friedman, L.F.; Galasko, D.R.; Jutel, M.; Karydas, A.; et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat. Med. 2007, 13, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Leszek, J.; Malyszczak, K.; Bartys, A.; Staniszewska, M.; Gamian, A. Analysis of serum of patients with Alzheimer’s disease for the level of advanced glycation end products. Am. J. Alzheimers Dis. Other Demen. 2006, 21, 360–365. [Google Scholar] [CrossRef]
- Staniszewska, M.; Bronowicka-Szydełko, A.; Gostomska-Pampuch, K.; Szkudlarek, J.; Bartyś, A.; Bieg, T.; Gamian, E.; Kochman, A.; Picur, B.; Pietkiewicz, J.; et al. The melibiose-derived glycation product mimics a unique epitope present in human and animal tissues. Sci. Rep. 2021, 11, 2940. [Google Scholar] [CrossRef]
- Morris, J.C.; Aisen, P.S.; Bateman, R.J.; Benzinger, T.L.S.; Cairns, N.J.; Fagan, A.M.; Ghetti, B.; Goate, A.M.; Holtzman, D.M.; Klunk, W.E.; et al. Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network. Clin. Investig. 2012, 2, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.C.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.S.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef]
- Bateman, R.J.; Benzinger, T.L.; Berry, S.; Clifford, D.B.; Duggan, C.; Fagan, A.M.; Fanning, K.; Farlow, M.R.; Hassenstab, J.; McDade, E.M.; et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement. 2017, 13, 8–19. [Google Scholar] [CrossRef]
- Lopez Lopez, C.; Tariot, P.N.; Caputo, A.; Langbaum, J.B.; Liu, F.; Riviere, M.-E.; Langlois, C.; Rouzade-Dominguez, M.-L.; Zalesak, M.; Hendrix, S.; et al. The Alzheimer’s Prevention Initiative Generation Program: Study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimer’s Dement. 2019, 5, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Janelidze, S.; Palmqvist, S.; Cullen, N.; Svenningsson, A.L.; Strandberg, O.; Mengel, D.; Walsh, D.M.; Stomrud, E.; Dage, J.L.; et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain 2020, 143, 3234–3241. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef] [PubMed]
- McDade, E.; Llibre-Guerra, J.J.; Holtzman, D.M.; Morris, J.C.; Bateman, R.J. The informed road map to prevention of Alzheimer Disease: A call to arms. Mol. Neurodegener. 2021, 16, 49. [Google Scholar] [CrossRef]
- Kurkinen, M. The amyloid hypothesis is too good to be true. Alzheimer’s Dement. Cogn. Neurol. 2017, 1, 1–9. [Google Scholar] [CrossRef]
- Mullard, A. BACE failures lower AD expectations, again. Nat. Rev. Drug Discov. 2018, 17, 385. [Google Scholar] [CrossRef]
- Henley, D.; Raghavan, N.; Sperling, R.; Aisen, P.; Raman, R.; Romano, G. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1483–1485. [Google Scholar] [CrossRef]
- Egan, M.F.; Mukai, Y.; Voss, T.; Kost, J.; Stone, J.; Furtek, C.; Mahoney, E.; Cummings, J.L.; Tariot, P.N.; Aisen, P.S.; et al. Further analyses of the safety of verubecestat in the phase 3 EPOCH trial of mild-to-moderate Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.-H. Reasons for Failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicines 2019, 7, 97. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Lucca, U.; Watling, M. Can Anti-β-amyloid Monoclonal Antibodies Work in Autosomal Dominant Alzheimer Disease? Neurol. Genet. 2021, 7, e535. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 18 February 2023).
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Kelleher, R.J. 3rd The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Zoltowska, K.M.; Maesako, M.; Meier, J.; Berezovska, O. Novel interaction between Alzheimer’s disease-related protein presenilin 1 and glutamate transporter 1. Sci. Rep. 2018, 8, 8718. [Google Scholar] [CrossRef]
- Güner, G.; Lichtenthaler, S.F. The substrate repertoire of γ-secretase/presenilin. Semin. Cell Dev. Biol. 2020, 105, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, F.A.C.; Carvalho, L.R.B.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.L.; Leite, R.E.P.; Jacob Filho, W.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef]
- Alonso-Nanclares, L.; Gonzalez-Soriano, J.; Rodriguez, J.R.; DeFelipe, J. Gender differences in human cortical synaptic density. Proc. Natl. Acad. Sci. USA 2008, 105, 14615–14619. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef]
- Xu, W.; Südhof, T.C. A neural circuit for memory specificity and generalization. Science 2013, 339, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.P.; Rusakov, D.A. Morphological plasticity of astroglia: Understanding synaptic microenvironment. Glia 2015, 63, 2133–2151. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Dunphy, J.; Tolman, M.; Foley, J.C.; Haydon, P.G. Astrocytic control of synaptic function. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. Towards an understanding of synapse formation. Neuron 2018, 100, 276–293. [Google Scholar] [CrossRef] [PubMed]
- Froemke, R.C. Plasticity of cortical excitatory-inhibitory balance. Annu. Rev. Neurosci. 2015, 38, 195–219. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Babiloni, C.; Blinowska, K.; Bonanni, L.; Cichocki, A.; De Haan, W.; Del Percio, C.; Dubois, B.; Escudero, J.; Fernández, A.; Frisoni, G.; et al. What electrophysiology tells us about Alzheimer’s disease: A window into the synchronization and connectivity of brain neurons. Neurobiol. Aging 2020, 85, 58–73. [Google Scholar] [CrossRef]
- Wisch, J.K.; Roe, C.M.; Babulal, G.M.; Schindler, S.E.; Fagan, A.M.; Benzinger, T.L.; Morris, J.C.; Ances, B.M. Resting State Functional Connectivity Signature Differentiates Cognitively Normal from Individuals Who Convert to Symptomatic Alzheimer’s Disease. J. Alzheimers Dis. 2020, 74, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; Salat, D.H.; Greve, D.N.; Chua, E.F.; Rand-Giovannetti, E.; Rentz, D.M.; Bertram, L.; Mullin, K.; Tanzi, R.E.; Blacker, D.; et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 2005, 65, 404–411. [Google Scholar] [CrossRef]
- Quiroz, Y.T.; Budson, A.E.; Celone, K.; Ruiz, A.; Newmark, R.; Castrillón, G.; Lopera, F.; Stern, C.E. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann. Neurol. 2010, 68, 865–875. [Google Scholar] [CrossRef]
- Busche, M.A.; Konnerth, A. Neuronal hyperactivity--A key defect in Alzheimer’s disease? Bioessays 2015, 37, 624–632. [Google Scholar] [CrossRef]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What Happens with the Circuit in Alzheimer’s Disease in Mice and Humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef]
- Bronner, F. Extracellular and intracellular regulation of calcium homeostasis. ScientificWorldJournal. 2001, 1, 919–925. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Südhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef]
- Zheng, K.; Jensen, T.P.; Savtchenko, L.P.; Levitt, J.A.; Suhling, K.; Rusakov, D.A. Nanoscale diffusion in the synaptic cleft and beyond measured with time-resolved fluorescence anisotropy imaging. Sci. Rep. 2017, 7, 42022. [Google Scholar] [CrossRef]
- Clements, J.D.; Lester, R.A.; Tong, G.; Jahr, C.E.; Westbrook, G.L. The time course of glutamate in the synaptic cleft. Science 1992, 258, 1498–1501. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Jahr, C.E. Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 2007, 27, 9736–9741. [Google Scholar] [CrossRef]
- Scimemi, A.; Beato, M. Determining the neurotransmitter concentration profile at active synapses. Mol. Neurobiol. 2009, 40, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, R.J.; Ryan, R.M. Mechanisms of glutamate transport. Physiol. Rev. 2013, 93, 1621–1657. [Google Scholar] [CrossRef]
- Murphy-Royal, C.; Dupuis, J.; Groc, L.; Oliet, S.H.R. Astroglial glutamate transporters in the brain: Regulating neurotransmitter homeostasis and synaptic transmission. J. Neurosci. Res. 2017, 95, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Bañuelos, T.N.; Chí-Castañeda, D.; Ortega, A. Glutamate transporters: Gene expression regulation and signaling properties. Neuropharmacology 2019, 161, 107550. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Roberts, R.C.; Roche, J.K.; McCullumsmith, R.E. Localization of excitatory amino acid transporters EAAT1 and EAAT2 in human postmortem cortex: A light and electron microscopic study. Neuroscience 2014, 277, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Kurkinen, M. Glutamate and Neuropsychiatric Disorders—Current and Emerging Treatments; Pavlovic, Z.M., Ed.; Springer Nature: Berlin, Germany, 2022; pp. 229–260. [Google Scholar]
- Lin, C.-L.G.; Kong, Q.; Cuny, G.D.; Glicksman, M.A. Glutamate transporter EAAT2: A new target for the treatment of neurodegenerative diseases. Future Med. Chem. 2012, 4, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Chang, L.-C.; Takahashi, K.; Liu, Q.; Schulte, D.A.; Lai, L.; Ibabao, B.; Lin, Y.; Stouffer, N.; Das Mukhopadhyay, C.; et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J. Clin. Investig. 2014, 124, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, P.; Green, P.S.; Watson, G.S.; Marques, M.A.; Tanaka, K.; Meeker, K.D.; Meabon, J.S.; Li, N.; Zhu, P.; Olson, V.G.; et al. GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J. Alzheimers Dis. 2011, 26, 447–455. [Google Scholar] [CrossRef]
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef]
- Jacob, C.P.; Koutsilieri, E.; Bartl, J.; Neuen-Jacob, E.; Arzberger, T.; Zander, N.; Ravid, R.; Roggendorf, W.; Riederer, P.; Grünblatt, E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimers Dis. 2007, 11, 97–116. [Google Scholar] [CrossRef]
- Takahashi, K.; Foster, J.B.; Lin, C.-L.G. Glutamate transporter EAAT2: Regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell. Mol. Life Sci. 2015, 72, 3489–3506. [Google Scholar] [CrossRef]
- Fontana, A.C.K. Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 2015, 134, 982–1007. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Falcucci, R.M.; Wertz, R.; Green, J.L.; Meucci, O.; Salvino, J.; Fontana, A.C.K. Novel Positive Allosteric Modulators of Glutamate Transport Have Neuroprotective Properties in an in Vitro Excitotoxic Model. ACS Chem. Neurosci. 2019, 10, 3437–3453. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval for Alzheimer’s Drug. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug?utm_medium=email&utm_source=govdelivery (accessed on 18 February 2023).
- Deyts, C.; Clutter, M.; Pierce, N.; Chakrabarty, P.; Ladd, T.B.; Goddi, A.; Rosario, A.M.; Cruz, P.; Vetrivel, K.; Wagner, S.L.; et al. APP-Mediated Signaling Prevents Memory Decline in Alzheimer’s Disease Mouse Model. Cell Rep. 2019, 27, 1345–1355. [Google Scholar] [CrossRef]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Davis, J.; Hoos, M.; Van Nostrand, W.E. Mutation of the Kunitz-type proteinase inhibitor domain in the amyloid β-protein precursor abolishes its anti-thrombotic properties in vivo. Thromb. Res. 2017, 155, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, W.E. The influence of the amyloid ß-protein and its precursor in modulating cerebral hemostasis. Biochim. Biophys. Acta 2016, 1862, 1018–1026. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365. [Google Scholar] [CrossRef]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an antimicrobial peptide: A review of the evidence. J. Alzheimers Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Vélez, J.I.; Lopera, F.; Silva, C.T.; Villegas, A.; Espinosa, L.G.; Vidal, O.M.; Mastronardi, C.A.; Arcos-Burgos, M. Familial Alzheimer’s Disease and recessive modifiers. Mol. Neurobiol. 2020, 57, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Vélez, J.I.; Lopera, F.; Sepulveda-Falla, D.; Patel, H.R.; Johar, A.S.; Chuah, A.; Tobón, C.; Rivera, D.; Villegas, A.; Cai, Y.; et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol. Psychiatry 2016, 21, 916–924. [Google Scholar] [CrossRef]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurkinen, M.; Fułek, M.; Fułek, K.; Beszłej, J.A.; Kurpas, D.; Leszek, J. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: Should We Change Our Thinking? Biomolecules 2023, 13, 453. https://doi.org/10.3390/biom13030453
Kurkinen M, Fułek M, Fułek K, Beszłej JA, Kurpas D, Leszek J. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: Should We Change Our Thinking? Biomolecules. 2023; 13(3):453. https://doi.org/10.3390/biom13030453
Chicago/Turabian StyleKurkinen, Markku, Michał Fułek, Katarzyna Fułek, Jan Aleksander Beszłej, Donata Kurpas, and Jerzy Leszek. 2023. "The Amyloid Cascade Hypothesis in Alzheimer’s Disease: Should We Change Our Thinking?" Biomolecules 13, no. 3: 453. https://doi.org/10.3390/biom13030453
APA StyleKurkinen, M., Fułek, M., Fułek, K., Beszłej, J. A., Kurpas, D., & Leszek, J. (2023). The Amyloid Cascade Hypothesis in Alzheimer’s Disease: Should We Change Our Thinking? Biomolecules, 13(3), 453. https://doi.org/10.3390/biom13030453