
SARS-CoV-2 Delta Variant: Interplay between Individual Mutations and Their Allosteric Synergy


Abstract
1. Introduction
2. Materials and Methods
3. Results
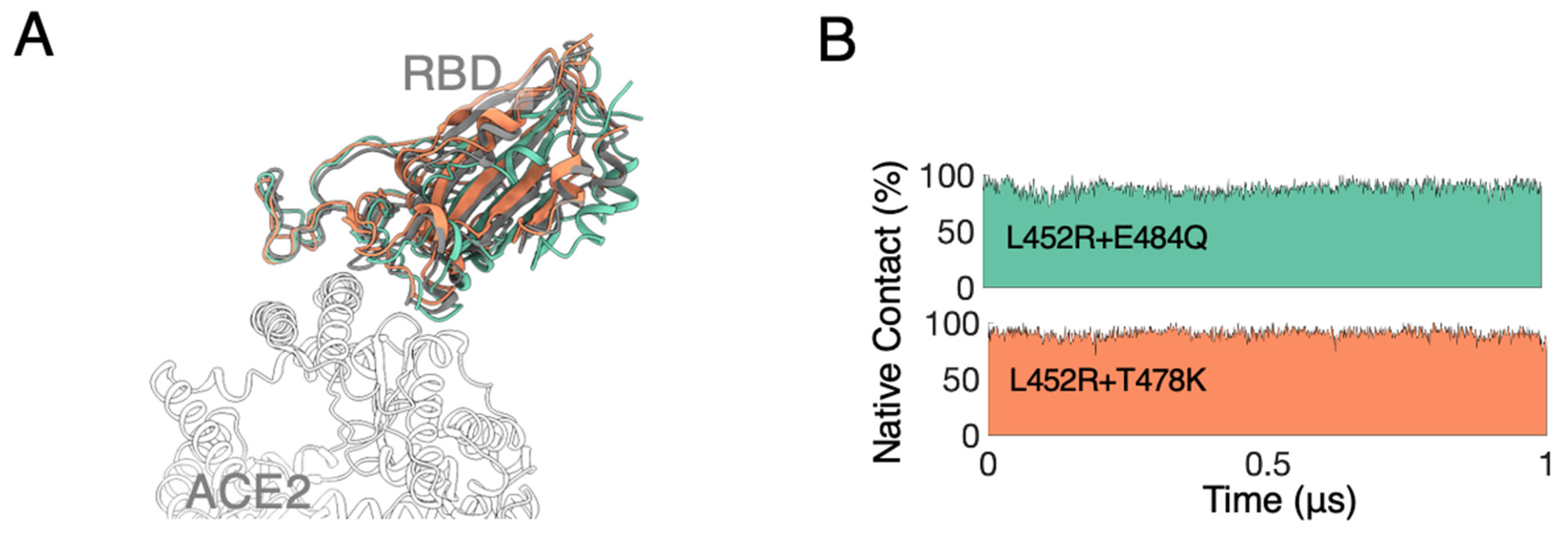
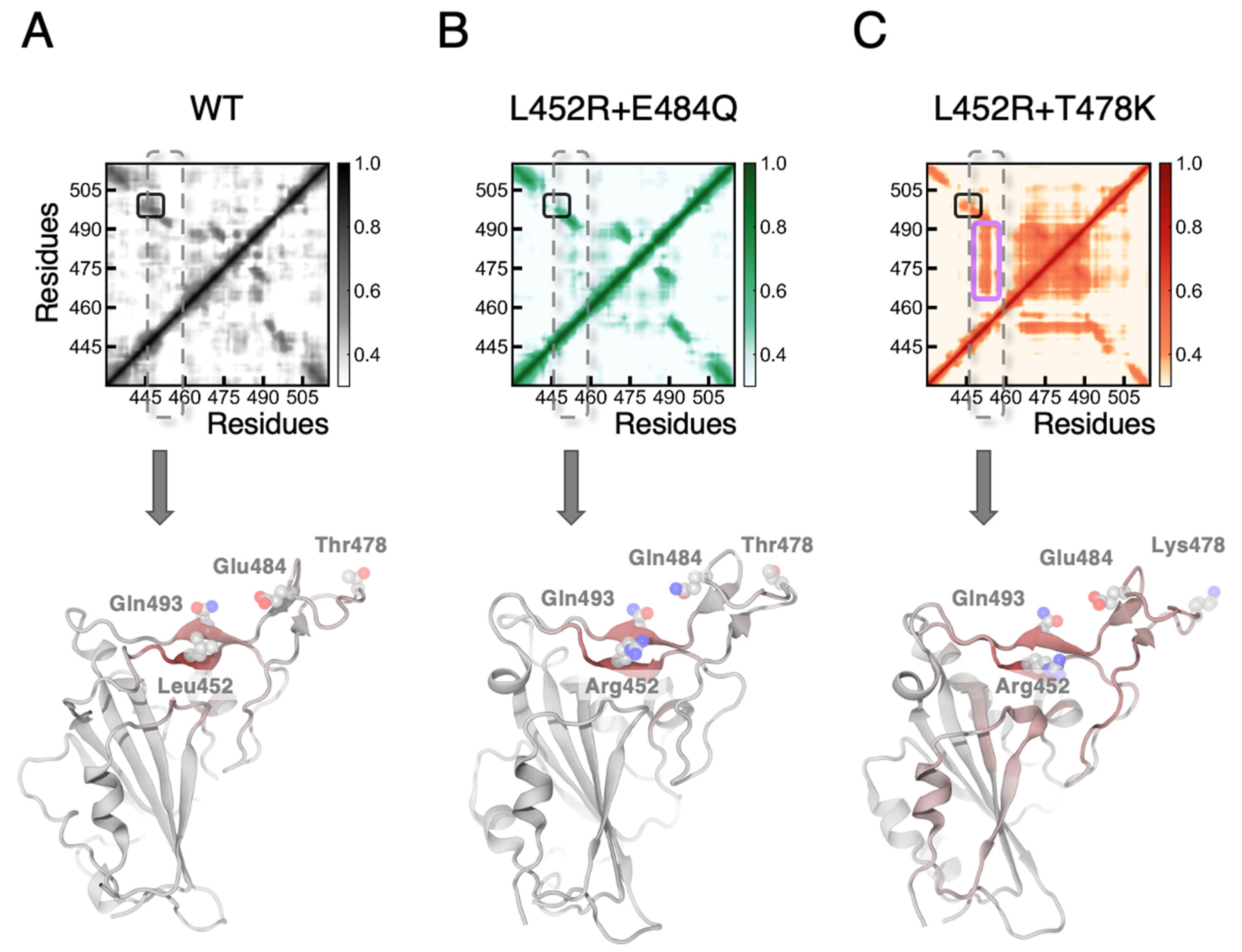
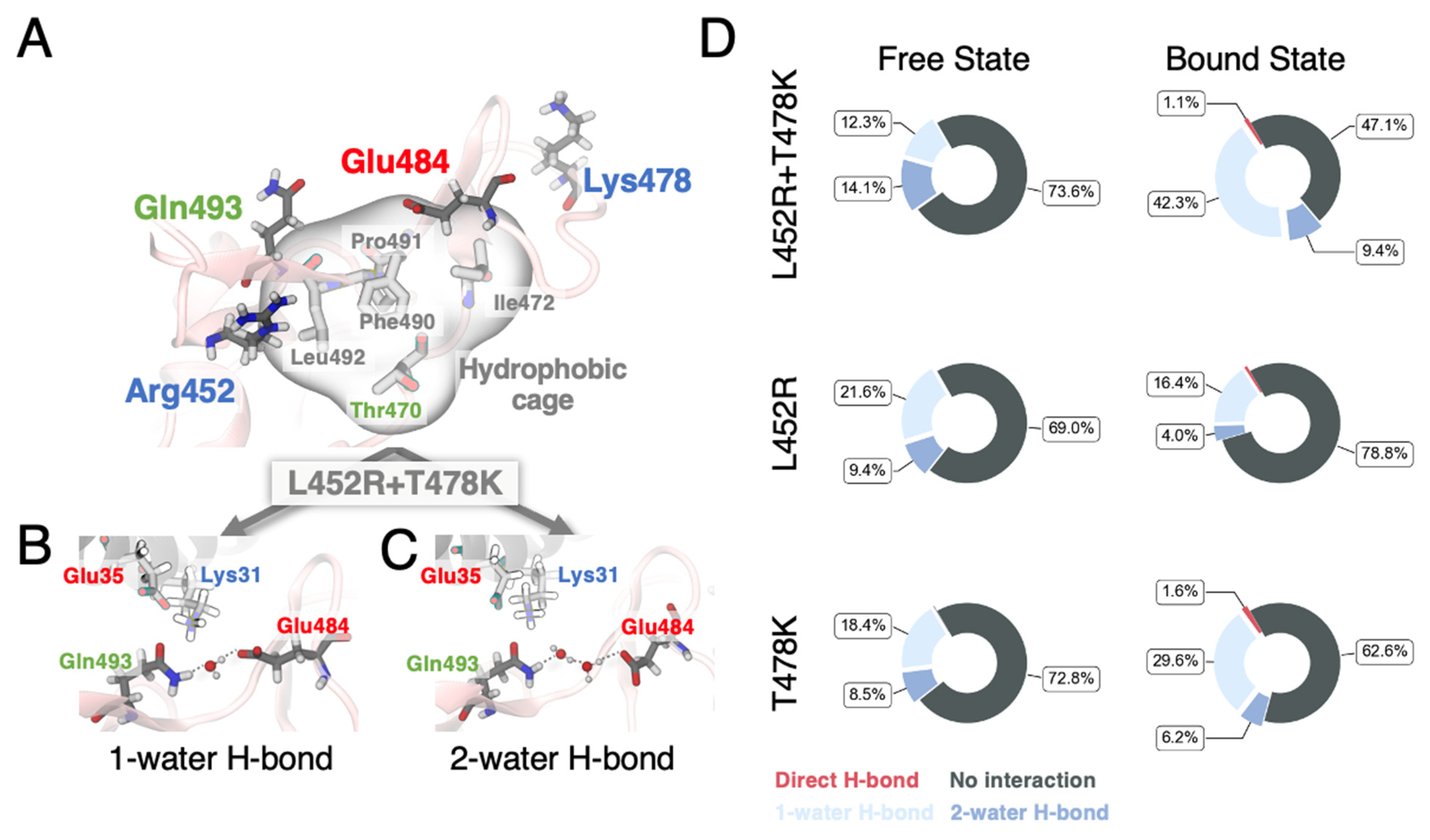
3.1. MD Simulations Revealed a Tighter Binding to ACE2 by Mutated RBDs
3.2. FEP Calculations Revealed Synergy between Mutations Enhancing RBD-ACE2 Binding
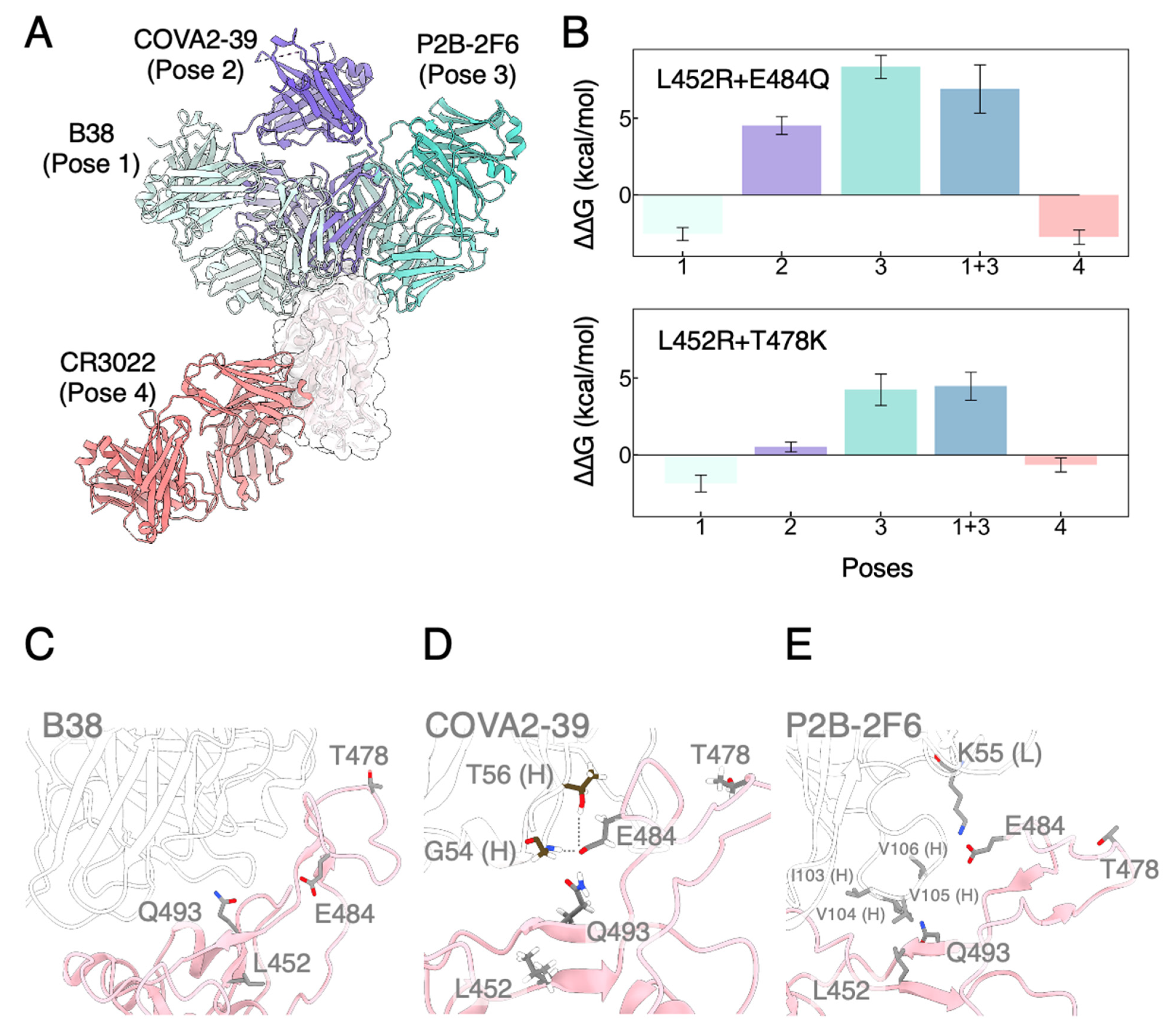
3.3. Mutated RBDs Show Immunoevasion to Several mAbs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and Functional Characterizations of Infectivity and Immune Evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor Binding and Complex Structures of Human ACE2 to Spike RBD from Omicron and Delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, L.; Mo, M.; Liu, T.; Wu, C.; Gong, C.; Lu, K.; Gong, L.; Zhu, W.; Xu, Z. SARS-CoV-2 Omicron RBD Shows Weaker Binding Affinity than the Currently Dominant Delta Variant to Human ACE2. Signal Transduct. Target. 2022, 7, 8. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Sprouse, K.R.; Bowen, J.E.; Rosen Laura, E.; Dang Ha, V.; De Marco, A.; Nicholas, F.; Tilles, S.W.; Logue, J.; et al. Molecular Basis of Immune Evasion by the Delta and Kappa SARS-CoV-2 Variants. Science 2021, 374, 1621–1626. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta Variant Replication and Immune Evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 Spike D614G Change Enhances Replication and Transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Washington, N.L.; Gangavarapu, K.; Zeller, M.; Bolze, A.; Cirulli, E.T.; Schiabor Barrett, K.M.; Larsen, B.B.; Anderson, C.; White, S.; Cassens, T.; et al. Emergence and Rapid Transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell 2021, 184, 2587–2594.e7. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 Immune Evasion by the B.1.427/B.1.429 Variant of Concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-Binding Domain That Affect Recognition by Polyclonal Human Plasma Antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Jensen, B.; Luebke, N.; Feldt, T.; Keitel, V.; Brandenburger, T.; Kindgen-Milles, D.; Lutterbeck, M.; Freise, N.F.; Schoeler, D.; Haas, R.; et al. Emergence of the E484K Mutation in SARS-CoV-2-Infected Immunocompromised Patients Treated with Bamlanivimab in Germany. Lancet Reg. Health—Eur. 2021, 8, 100164. [Google Scholar] [CrossRef]
- Yan, R.; Wang, R.; Ju, B.; Yu, J.; Zhang, Y.; Liu, N.; Wang, J.; Zhang, Q.; Chen, P.; Zhou, B.; et al. Structural Basis for Bivalent Binding and Inhibition of SARS-CoV-2 Infection by Human Potent Neutralizing Antibodies. Cell Res 2021, 31, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A Noncompeting Pair of Human Neutralizing Antibodies Block COVID-19 Virus Binding to Its Receptor ACE2. Science 2020, 368, 1274. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.-W.; Sahi, V.; Figueroa, A.; et al. Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human Neutralizing Antibodies Elicited by SARS-CoV-2 Infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Prévost, J.; Finzi, A. The Great Escape? SARS-CoV-2 Variants Evading Neutralizing Responses. Cell Host Microbe 2021, 29, 322–324. [Google Scholar] [CrossRef]
- Starr, T.N.; Czudnochowski, N.; Liu, Z.; Zatta, F.; Park, Y.-J.; Addetia, A.; Pinto, D.; Beltramello, M.; Hernandez, P.; Greaney, A.J.; et al. SARS-CoV-2 RBD Antibodies That Maximize Breadth and Resistance to Escape. Nature 2021, 597, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. MRNA Vaccine-Elicited Antibodies to SARS-CoV-2 and Circulating Variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Xia, H.; Zou, J.; Weaver, S.C.; Swanson, K.A.; Cai, H.; Cutler, M.; Cooper, D.; Muik, A.; et al. BNT162b2-Elicited Neutralization of B.1.617 and Other SARS-CoV-2 Variants. Nature 2021, 596, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Pegu, A.; O’Connell, S.; Schmidt, S.D.; O’Dell, S.; Talana, C.A.; Lai, L.; Albert, J.; Anderson, E.; Bennett, H.; Corbett, K.S.; et al. Durability of MRNA-1273 Vaccine–Induced Antibodies against SARS-CoV-2 Variants. Science 2021, 373, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y.; et al. Reduced Neutralization of SARS-CoV-2 B.1.617 by Vaccine and Convalescent Serum. Cell 2021, 184, 4220–4236.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Mannar, D.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.-P.; Saville, J.W.; Leopold, K.; Li, W.; Dimitrov, D.S.; Tuttle, K.S.; et al. Cryo-Electron Microscopy Structures of the N501Y SARS-CoV-2 Spike Protein in Complex with ACE2 and 2 Potent Neutralizing Antibodies. PLoS Biol. 2021, 19, e3001237. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; West, A.P.; Huey-Tubman, K.E.; Hoffmann, M.A.G.; Sharaf, N.G.; Hoffman, P.R.; Koranda, N.; Gristick, H.B.; Gaebler, C.; Muecksch, F.; et al. Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell 2020, 182, 828–842.e16. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, C.; Wang, Y.; Hong, Q.; Zhang, C.; Li, Z.; Xu, S.; Zuo, Q.; Liu, C.; Huang, Z.; et al. Conformational Dynamics of the Beta and Kappa SARS-CoV-2 Spike Proteins and Their Complexes with ACE2 Receptor Revealed by Cryo-EM. Nat. Commun. 2021, 12, 7345. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural Basis for SARS-CoV-2 Delta Variant Recognition of ACE2 Receptor and Broadly Neutralizing Antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef]
- Padhi, A.K.; Rath, S.L.; Tripathi, T. Accelerating COVID-19 Research Using Molecular Dynamics Simulation. J. Phys. Chem. B 2021, 125, 9078–9091. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Huynh, T. Insights into SARS-CoV-2’s Mutations for Evading Human Antibodies: Sacrifice and Survival. J. Med. Chem. 2022, 65, 2820–2826. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.; Zhang, Z.; Acharya, A.; Lynch, D.L.; Pang, Y.T.; Mou, Z.; Parks, J.M.; Chipot, C.; Gumbart, J.C. Machine Learning Reveals the Critical Interactions for SARS-CoV-2 Spike Protein Binding to ACE2. J. Phys. Chem. Lett. 2021, 12, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Wang, H.; Huynh, T. Enhanced Binding of the N501Y-Mutated SARS-CoV-2 Spike Protein to the Human ACE2 Receptor: Insights from Molecular Dynamics Simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y Mutation of Spike Protein in SARS-CoV-2 Strengthens Its Binding to Receptor ACE2. eLife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Socher, E.; Conrad, M.; Heger, L.; Paulsen, F.; Sticht, H.; Zunke, F.; Arnold, P. Computational Decomposition Reveals Reshaping of the SARS-CoV-2 ACE2 Interface among Viral Variants Expressing the N501Y Mutation. J. Cell. Biochem. 2021, 122, 1863–1872. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, Z.; Gao, J.; Wang, Y. SARS-CoV-2 Spike Protein N501Y Mutation Causes Differential Species Transmissibility and Antibody Sensitivity: A Molecular Dynamics and Alchemical Free Energy Study. Mol. Syst. Des. Eng. 2021, 6, 964–974. [Google Scholar] [CrossRef]
- Antony, P.; Vijayan, R. Molecular Dynamics Simulation Study of the Interaction between Human Angiotensin Converting Enzyme 2 and Spike Protein Receptor Binding Domain of the SARS-CoV-2 B.1.617 Variant. Biomolecules 2021, 11, 1244. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.-W. Omicron Variant (B.1.1.529): Infectivity, Vaccine Breakthrough, and Antibody Resistance. J. Chem. Inf. Model. 2022, 62, 412–422. [Google Scholar] [CrossRef]
- Aggarwal, A.; Naskar, S.; Maroli, N.; Gorai, B.; Dixit, N.M.; Maiti, P.K. Mechanistic Insights into the Effects of Key Mutations on SARS-CoV-2 RBD–ACE2 Binding. Phys. Chem. Chem. Phys. 2021, 23, 26451–26458. [Google Scholar] [CrossRef]
- Cheng, M.H.; Krieger, J.M.; Banerjee, A.; Xiang, Y.; Kaynak, B.; Shi, Y.; Arditi, M.; Bahar, I. Impact of New Variants on SARS-CoV-2 Infectivity and Neutralization: A Molecular Assessment of the Alterations in the Spike-Host Protein Interactions. iScience 2022, 25, 103939. [Google Scholar] [CrossRef] [PubMed]
- Socher, E.; Heger, L.; Paulsen, F.; Zunke, F.; Arnold, P. Molecular Dynamics Simulations of the Delta and Omicron SARS-CoV-2 Spike—ACE2 Complexes Reveal Distinct Changes between Both Variants. Comput. Struct. Biotechnol. J. 2022, 20, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Baek, S.-H.; Park, D. Interaction Analysis of the Spike Protein of Delta and Omicron Variants of SARS-CoV-2 with HACE2 and Eight Monoclonal Antibodies Using the Fragment Molecular Orbital Method. J. Chem. Inf. Model. 2022, 62, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Gheeraert, A.; Vuillon, L.; Chaloin, L.; Moncorgé, O.; Very, T.; Perez, S.; Leroux, V.; Chauvot de Beauchêne, I.; Mias-Lucquin, D.; Devignes, M.-D.; et al. Singular Interface Dynamics of the SARS-CoV-2 Delta Variant Explained with Contact Perturbation Analysis. J. Chem. Inf. Model. 2022, 62, 3107–3122. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, T.; Mo, M.; Shi, Y.; Wu, L.; Li, Y.; Qin, Q.; Zhu, W.; Wu, C.; Gong, L.; et al. Exploring the Binding Affinity and Mechanism between ACE2 and the Trimers of Delta and Omicron Spike Proteins by Molecular Dynamics Simulation and Bioassay. J. Chem. Inf. Model. 2022, 62, 4512–4522. [Google Scholar] [CrossRef]
- Mahmood, T.B.; Hossan, M.I.; Mahmud, S.; Shimu, M.S.S.; Alam, M.J.; Bhuyan, M.M.R.; Emran, T.B. Missense Mutations in Spike Protein of SARS-CoV-2 Delta Variant Contribute to the Alteration in Viral Structure and Interaction with HACE2 Receptor. Immun. Inflamm. Dis. 2022, 10, e683. [Google Scholar] [CrossRef]
- Pipitò, L.; Rujan, R.-M.; Reynolds, C.A.; Deganutti, G. Molecular Dynamics Studies Reveal Structural and Functional Features of the SARS-CoV-2 Spike Protein. Bioessays 2022, 44, e2200060. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.Z.H. Mutational Effect of Some Major COVID-19 Variants on Binding of the S Protein to ACE2. Biomolecules 2022, 12, 572. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron Variant: Antibody Evasion and Cryo-EM Structure of Spike Protein–ACE2 Complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Liu, S.; Huynh, T.; Stauft, C.B.; Wang, T.T.; Luan, B. Structure–Function Analysis of Resistance to Bamlanivimab by SARS-CoV-2 Variants Kappa, Delta, and Lambda. J. Chem. Inf. Model. 2021, 61, 5133–5140. [Google Scholar] [CrossRef]
- Chen, J.; Gao, K.; Wang, R.; Wei, G.-W. Prediction and Mitigation of Mutation Threats to COVID-19 Vaccines and Antibody Therapies. Chem. Sci. 2021, 12, 6929–6948. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ 1and χ 2Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Humphrey, W.; Humphrey, W.; Dalke, A.; Dalke, A.; Schulten, K.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Gao, J. Enhanced Receptor Binding of SARS-CoV-2 through Networks of Hydrogen-Bonding and Hydrophobic Interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Li, F. Structural Analysis of Major Species Barriers between Humans and Palm Civets for Severe Acute Respiratory Syndrome Coronavirus Infections. J. Virol. 2008, 82, 6984–6991. [Google Scholar] [CrossRef]
- Luan, B.; Huynh, T. In Silico Antibody Mutagenesis for Optimizing Its Binding to Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2. J. Phys. Chem. Lett. 2020, 11, 9781–9787. [Google Scholar] [CrossRef]
- Brooijmans, N.; Sharp, K.A.; Kuntz, I.D. Stability of Macromolecular Complexes. Proteins Struct. Funct. Bioinform. 2002, 48, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key Residues of the Receptor Binding Motif in the Spike Protein of SARS-CoV-2 That Interact with ACE2 and Neutralizing Antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.V.F.; Moreira, E.G.; Coêlho, D.F.; Wallau, G.L.; Lins, R.D. Immune Evasion of SARS-CoV-2 Variants of Concern Is Driven by Low Affinity to Neutralizing Antibodies. Chem. Commun. 2021, 57, 6094–6097. [Google Scholar] [CrossRef]
- Lange, O.F.; Grubmüller, H. Generalized Correlation for Biomolecular Dynamics. Proteins 2005, 62, 1053–1061. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Ma, A.K.; Fonseca, R.; Latorraca, N.R.; Kelly, B.; Betz, R.M.; Asawa, C.; Kobilka, B.K.; Dror, R.O. Diverse GPCRs Exhibit Conserved Water Networks for Stabilization and Activation. Proc. Natl. Acad. Sci. USA 2019, 116, 3288–3293. [Google Scholar] [CrossRef] [PubMed]
- Alaofi, A.L.; Shahid, M. Mutations of SARS-CoV-2 RBD May Alter Its Molecular Structure to Improve Its Infection Efficiency. Biomolecules 2021, 11, 1273. [Google Scholar] [CrossRef] [PubMed]
- Hastie, K.M.; Li, H.; Bedinger, D.; Schendel, S.L.; Dennison, S.M.; Li, K.; Rayaprolu, V.; Yu, X.; Mann, C.; Zandonatti, M.; et al. Defining Variant-Resistant Epitopes Targeted by SARS-CoV-2 Antibodies: A Global Consortium Study. Science 2021, 374, 472–478. [Google Scholar] [CrossRef]
- Du, S.; Cao, Y.; Zhu, Q.; Yu, P.; Qi, F.; Wang, G.; Du, X.; Bao, L.; Deng, W.; Zhu, H.; et al. Structurally Resolved SARS-CoV-2 Antibody Shows High Efficacy in Severely Infected Hamsters and Provides a Potent Cocktail Pairing Strategy. Cell 2020, 183, 1013–1023.e13. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete Map of SARS-CoV-2 RBD Mutations That Escape the Monoclonal Antibody LY-CoV555 and Its Cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants, B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Bell, D.R.; Weber, J.K.; Yin, W.; Huynh, T.; Duan, W.; Zhou, R. In Silico Design and Validation of High-Affinity RNA Aptamers Targeting Epithelial Cellular Adhesion Molecule Dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 8486–8493. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Abel, R.; Clark, A.; Friesner, R. Free Energy Perturbation Calculations of the Thermodynamics of Protein Side-Chain Mutations. J. Mol. Biol. 2017, 429, 923–929. [Google Scholar] [CrossRef]
- Davis, C.; Logan, N.; Tyson, G.; Orton, R.; Harvey, W.T.; Perkins, J.S.; Mollett, G.; Blacow, R.M.; The COVID-19 Genomics UK (COG-UK) Consortium; Peacock, T.P.; et al. Reduced Neutralisation of the Delta (B.1.617.2) SARS-CoV-2 Variant of Concern Following Vaccination. PLoS Pathog. 2021, 17, e1010022. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, T.; Zhang, Y.; Yang, E.S.; Schramm, C.A.; Shi, W.; Pegu, A.; Oloniniyi, O.K.; Henry, A.R.; Darko, S.; et al. Ultrapotent Antibodies against Diverse and Highly Transmissible SARS-CoV-2 Variants. Science 2021, 373, eabh1766. [Google Scholar] [CrossRef]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.-Y.; Sali, A. Protein Structure Modeling with MODELLER. In Structural Proteomics: High-Throughput Methods; Kobe, B., Guss, M., Huber, T., Eds.; Humana Press: Totowa, NJ, USA, 2008; pp. 145–159. [Google Scholar]
- Miyamoto, S.; Kollman, P.A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101-10. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hansmann, U.; Okamoto, Y.; Eisenmenger, F. Molecular dynamics, Langevin and hydrid Monte Carlo simulations in a multicanonical ensemble. Chem. Phys. Lett. 1996, 259, 321–330. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L452R + E484Q | L452R + T478K | L452R | E484Q | T478K | ||
|---|---|---|---|---|---|---|
| ACE2 | −2.27 ± 0.27 | −6.22 ± 0.28 | −0.87 ± 0.29 | 0.51 ± 0.42 | −1.18 ± 0.33 | |
| mAbs | Pose 1 | −2.54 ± 0.42 | −1.86 ± 0.55 | −1.29 ± 0.30 | −1.68 ± 0.26 | −0.71 ± 0.43 |
| Pose 2 | 4.53 ± 0.59 | 0.52 ± 0.33 | −1.04 ± 0.24 | 4.92 ± 0.48 | 1.38 ± 0.38 | |
| Pose 3 | 8.35 ± 0.75 | 4.25 ± 1.03 | 7.46 ± 0.57 | 0.75 ± 0.34 | −0.61 ± 0.34 | |
| Pose 1 + 3 | 6.92 ± 1.57 | 4.47 ± 0.91 | ||||
| Pose 4 | −2.75 ± 0.47 | −0.65 ± 0.46 | −0.95 ± 0.24 | −0.74 ± 0.38 | −0.88 ± 0.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, K.C.; Song, Y.; Xu, Z.; Shang, C.; Zhou, R. SARS-CoV-2 Delta Variant: Interplay between Individual Mutations and Their Allosteric Synergy. Biomolecules 2022, 12, 1742. https://doi.org/10.3390/biom12121742
Chan KC, Song Y, Xu Z, Shang C, Zhou R. SARS-CoV-2 Delta Variant: Interplay between Individual Mutations and Their Allosteric Synergy. Biomolecules. 2022; 12(12):1742. https://doi.org/10.3390/biom12121742
Chicago/Turabian StyleChan, Kevin C., Yi Song, Zheng Xu, Chun Shang, and Ruhong Zhou. 2022. "SARS-CoV-2 Delta Variant: Interplay between Individual Mutations and Their Allosteric Synergy" Biomolecules 12, no. 12: 1742. https://doi.org/10.3390/biom12121742
APA StyleChan, K. C., Song, Y., Xu, Z., Shang, C., & Zhou, R. (2022). SARS-CoV-2 Delta Variant: Interplay between Individual Mutations and Their Allosteric Synergy. Biomolecules, 12(12), 1742. https://doi.org/10.3390/biom12121742