


Role of Cell-Cell Junctions in Oesophageal Squamous Cell Carcinoma

 and
and
Abstract
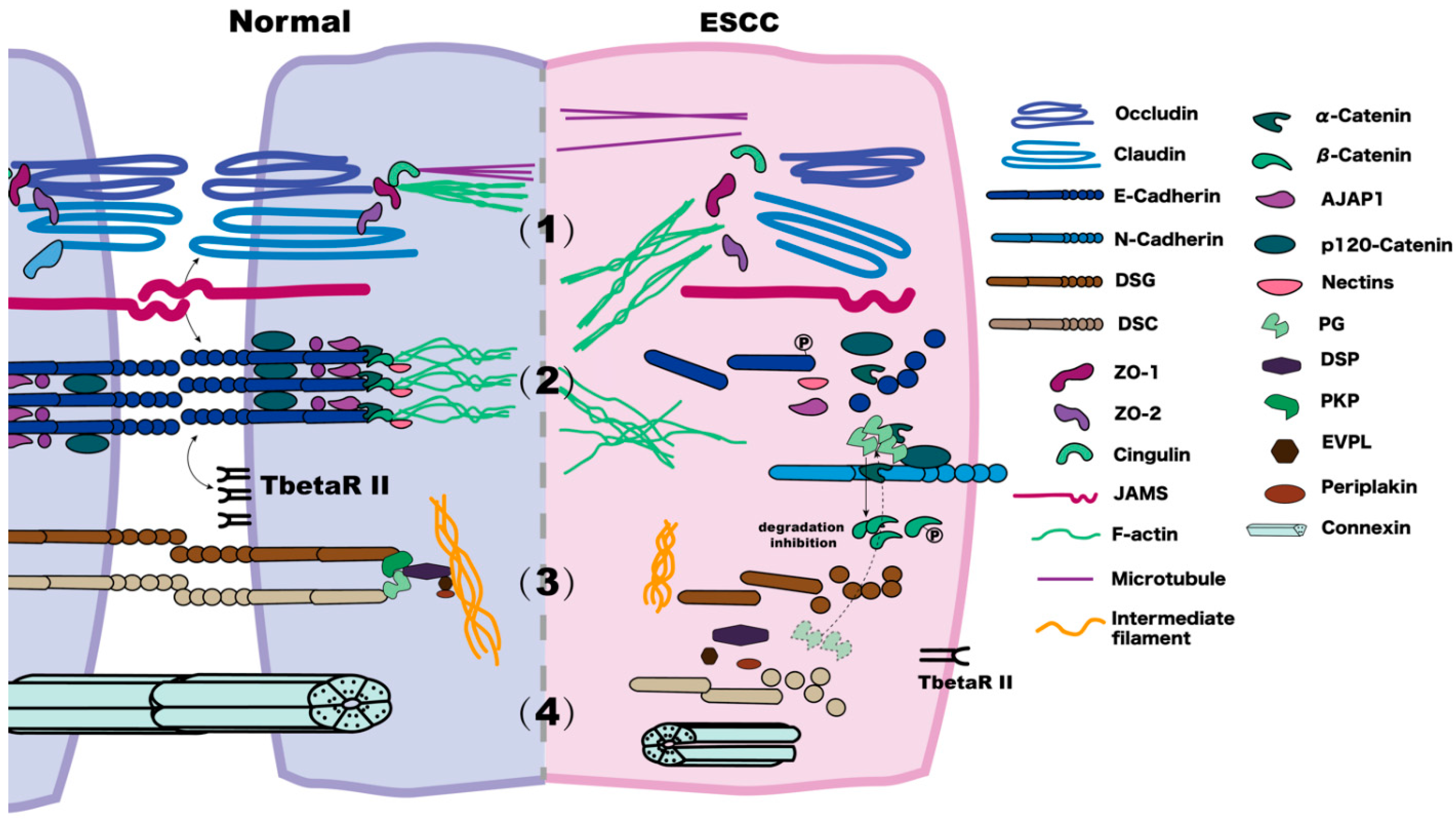
1. Overview
2. Tight Junctions
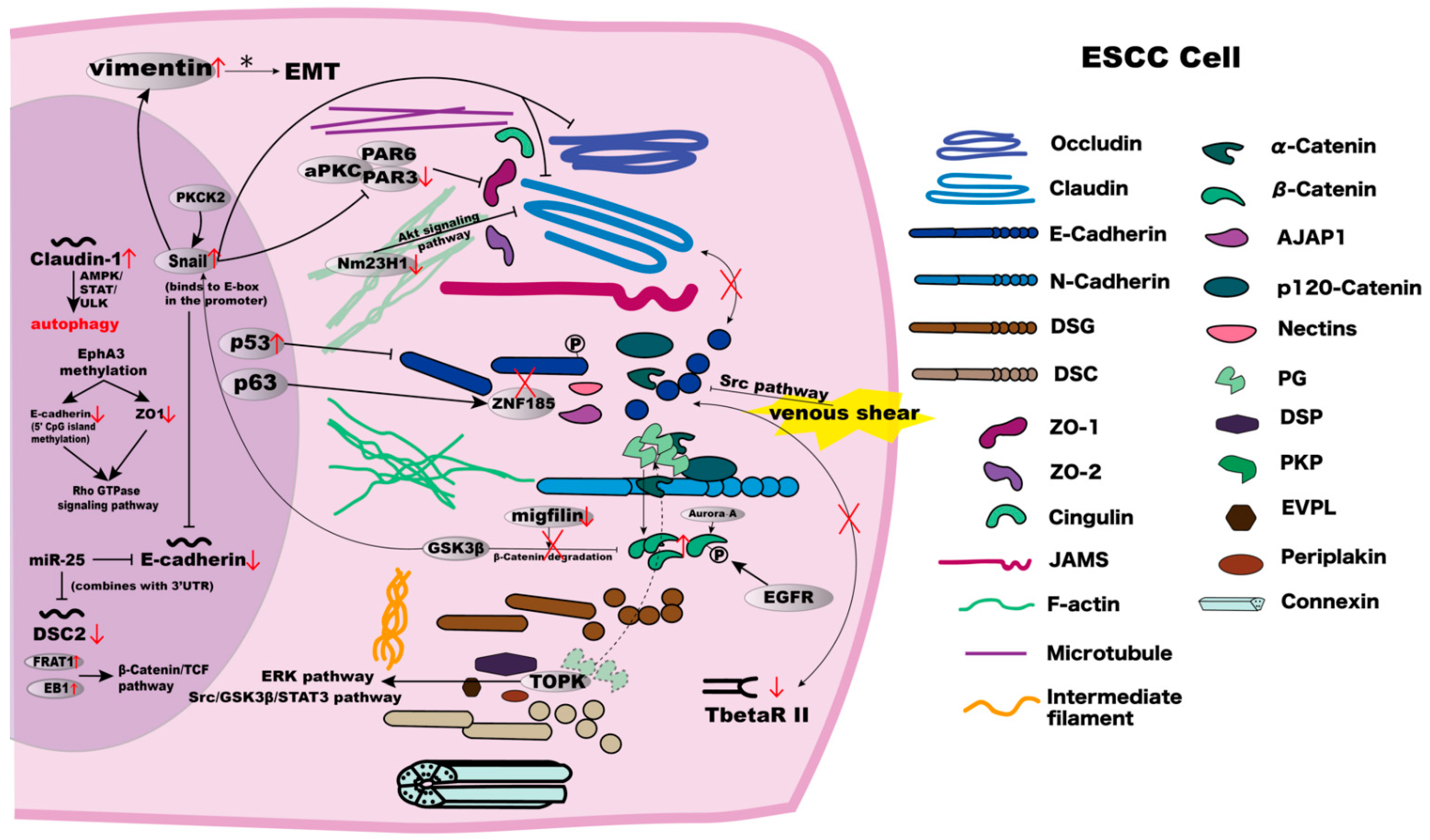
2.1. Effects and Possible Mechanisms of the Abnormal Expression of Claudins and Occludins in ESCC
2.2. ZO Family Roles and Alterations in ESCC
3. Adherens Junctions
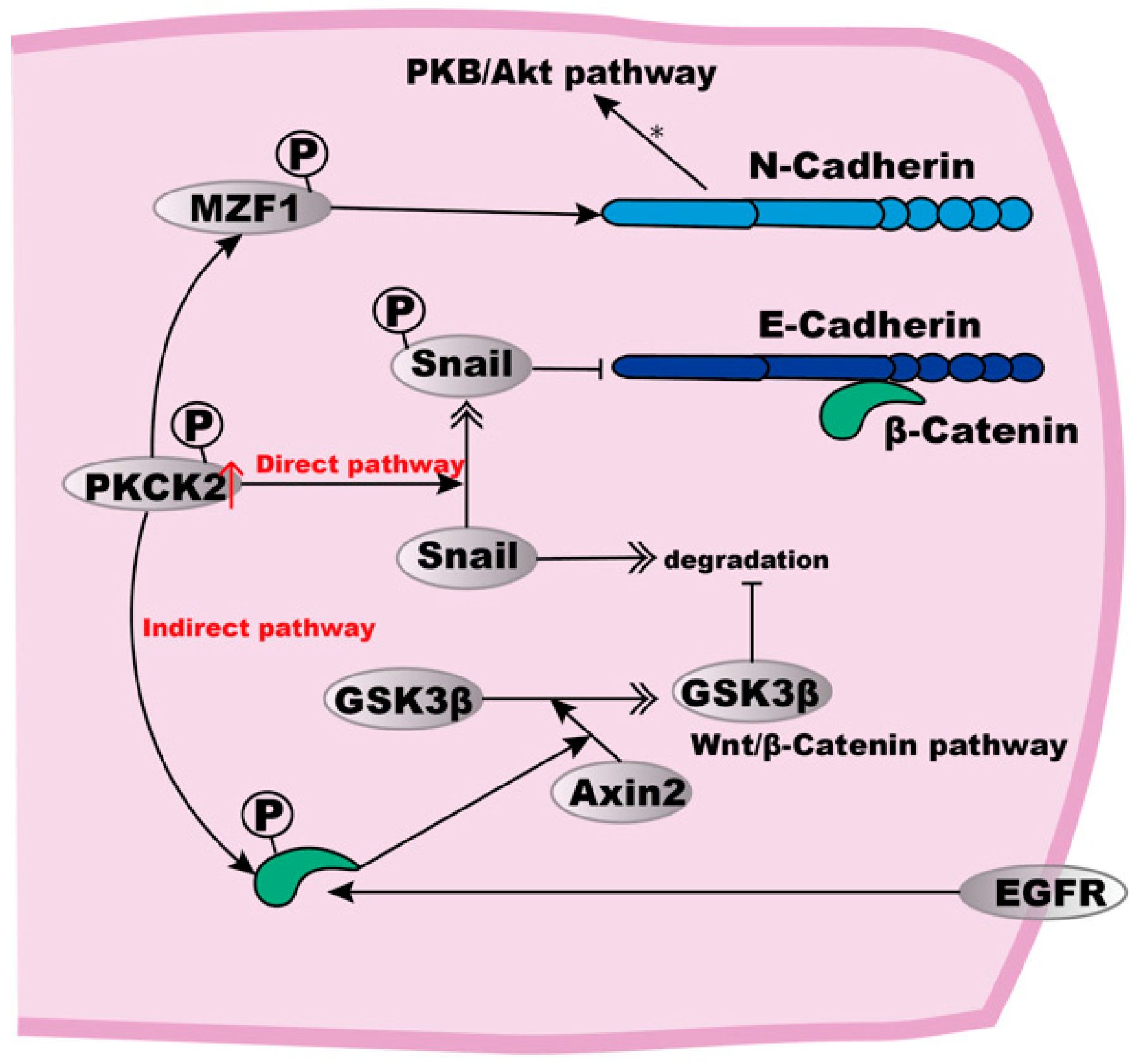
3.1. Abnormal Expression of E-Cadherin in ESCC Cells and Possible Mechanisms
3.2. Possible Effects and Mechanisms of α- and β-Catenin on the Invasiveness and Metastasis of ESCC
3.3. Potential of N-Cadherin as a Biomarker of ESCC
3.4. Two Possible Adhesion Proteins Affecting Migration and Invasion of ESCC Cells
3.5. ESCC and AJAP1
4. Desmosomes
4.1. DSC2 Deficiency in ESCC and Its Mechanism
4.2. Expression of DSG1 and DSG3 in ESCC
4.3. PG Loss and β-Catenin Accumulation in the Cross-Talk between Adherens Junctions and Desmosomes
5. Gap Junctions
6. Therapeutic Implication
7. Summary
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ZO | zonula occludens |
| TAMP | tight junction-associated MARVEL proteins |
| AJAP1 | Adherens junctions-associated protein-1 |
| Cdh23 | cadherin-23 |
| circAMOTL1L | angiomotin-like 1-derived circRNA |
| CTC | circulating tumour cells |
| Cx | connexin |
| DSC | desmocollin |
| DSG | desmoglein |
| DSP | desmoplakin |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-to-mesenchymal transition |
| ER | endoplasmic reticulum |
| ESCC | Oesophageal squamous cell carcinoma |
| EVPL | envoplakin |
| GSK-3β | glycogen synthase kinase-3β |
| GUK | guanylate kinase |
| HDACs | histone deacetylases |
| HSP70RY | heat shock 70kD protein 4 |
| JAMs | junction adhesion molecules |
| LPS | lipopolysaccharide |
| miR-25 | microRNA25 |
| Nm23H1 | non-metastatic protein 23-H1 |
| PCDH10 | protocadherin 10 |
| PDZ | PSD95, DlgA and ZO-1 |
| PG | plakoglobin |
| PG | plakoglobin |
| Pkp | plakophilin |
| RBM25 | RNA Binding Motif Protein 25 |
| SCC | squamous cell carcinoma |
| SH3 | SRC homology 3 |
| TOPK | T-LAK cell-originated protein kinase |
| UPR | unfolded protein response |
| ZNF185 | zinc finger protein 185 |
| ZONAB | ZO-1-associated nucleic acid binding protein |
References
- Garcia, M.A.; Nelson, W.J.; Chavez, N. Cell-Cell Junctions Organize Structural and Signaling Networks. Cold Spring Harb. Perspect. Biol. 2018, 10, a029181. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K. Organization of multiprotein complexes at cell-cell junctions. Histochem. Cell Biol. 2008, 130, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.H.; Chen, M.C.; Tsai, Y.P.; Tan, G.Y.T.; Hsu, P.H.; Jeng, Y.M.; Tsai, Y.F.; Yang, M.H.; Hwang-Verslues, W.W. Interplay between desmoglein2 and hypoxia controls metastasis in breast cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2014408118. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Wang, C.; Wang, J.; Chen, Z.; Gao, Y.; He, J. Immunohistochemical prognostic markers of esophageal squamous cell carcinoma: A systematic review. Chin. J. Cancer 2017, 36, 65. [Google Scholar] [CrossRef]
- Takeno, S.; Noguchi, T.; Fumoto, S.; Kimura, Y.; Shibata, T.; Kawahara, K. E-cadherin expression in patients with esophageal squamous cell carcinoma: Promoter hypermethylation, Snail overexpression, and clinicopathologic implications. Am. J. Clin. Pathol. 2004, 122, 78–84. [Google Scholar] [CrossRef]
- Zhai, J.W.; Yang, X.G.; Yang, F.S.; Hu, J.G.; Hua, W.X. Expression and clinical significance of Ezrin and E-cadherin in esophageal squamous cell carcinoma. Chin. J. Cancer 2010, 29, 317–320. [Google Scholar] [CrossRef][Green Version]
- Fang, W.K.; Liao, L.D.; Li, L.Y.; Xie, Y.M.; Xu, X.E.; Zhao, W.J.; Wu, J.Y.; Zhu, M.X.; Wu, Z.Y.; Du, Z.P.; et al. Down-regulated desmocollin-2 promotes cell aggressiveness through redistributing adherens junctions and activating beta-catenin signalling in oesophageal squamous cell carcinoma. J. Pathol. 2013, 231, 257–270. [Google Scholar] [CrossRef]
- Sung, C.O.; Han, S.Y.; Kim, S.H. Low expression of claudin-4 is associated with poor prognosis in esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2011, 18, 273–281. [Google Scholar] [CrossRef]
- Rusu, A.D.; Georgiou, M. The multifarious regulation of the apical junctional complex. Open Biol. 2020, 10, 190278. [Google Scholar] [CrossRef]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Lioni, M.; Brafford, P.; Andl, C.; Rustgi, A.; El-Deiry, W.; Herlyn, M.; Smalley, K.S. Dysregulation of claudin-7 leads to loss of E-cadherin expression and the increased invasion of esophageal squamous cell carcinoma cells. Am. J. Pathol. 2007, 170, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Cowin, P.; Kapprell, H.P.; Franke, W.W.; Tamkun, J.; Hynes, R.O. Plakoglobin: A protein common to different kinds of intercellular adhering junctions. Cell 1986, 46, 1063–1073. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, X.K.; Xu, H.Y.; Shan, Z.Z.; Wang, T.; Yang, Z.C.; He, W.; Wang, L.X.; Fan, Q.X. N-cadherin participated in invasion and metastasis of human esophageal squamous cell carcinoma via taking part in the formation of vasculogenic mimicry. Med. Oncol. 2015, 32, 480. [Google Scholar] [CrossRef]
- Li, K.; He, W.; Lin, N.; Wang, X.; Fan, Q.X. N-cadherin knock-down decreases invasiveness of esophageal squamous cell carcinoma in vitro. World J. Gastroenterol. 2009, 15, 697–704. [Google Scholar] [CrossRef]
- Liu, D.; Liu, Y.; Qi, B.; Gu, C.; Huo, S.; Zhao, B. Trichostatin A promotes esophageal squamous cell carcinoma cell migration and EMT through BRD4/ERK1/2-dependent pathway. Cancer Med. 2021, 10, 5235–5245. [Google Scholar] [CrossRef]
- Chen, Z.; Che, D.; Gu, X.; Lin, J.; Deng, J.; Jiang, P.; Xu, K.; Xu, B.; Zhang, T. Upregulation of PEDF Predicts a Poor Prognosis and Promotes Esophageal Squamous Cell Carcinoma Progression by Modulating the MAPK/ERK Signaling Pathway. Front. Oncol. 2021, 11, 625612. [Google Scholar] [CrossRef]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Otani, T.; Furuse, M. Tight Junction Structure and Function Revisited. Trends Cell Biol. 2020, 30, 805–817. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Tokumasu, R.; Kimura, H.; Tsukita, S. Role of claudin species-specific dynamics in reconstitution and remodeling of the zonula occludens. Mol. Biol. Cell 2011, 22, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tani, K.; Tamura, A.; Tsukita, S.; Fujiyoshi, Y. Model for the architecture of claudin-based paracellular ion channels through tight junctions. J. Mol. Biol. 2015, 427, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Choi, W.; Kuo, W.T.; Singh, G.; Sailer, A.; Wang, Y.; Shen, L.; Fanning, A.S.; Turner, J.R. The scaffolding protein ZO-1 coordinates actomyosin and epithelial apical specializations in vitro and in vivo. J. Biol. Chem. 2018, 293, 17317–17335. [Google Scholar] [CrossRef]
- Usami, Y.; Chiba, H.; Nakayama, F.; Ueda, J.; Matsuda, Y.; Sawada, N.; Komori, T.; Ito, A.; Yokozaki, H. Reduced expression of claudin-7 correlates with invasion and metastasis in squamous cell carcinoma of the esophagus. Hum. Pathol. 2006, 37, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Kusumi, T.; Sato, F.; Kawasaki, H.; Shibata, S.; Ohashi, M.; Hakamada, K.; Sasaki, M.; Kijima, H. Decreased expression of claudin-1 is correlated with recurrence status in esophageal squamous cell carcinoma. Biomed. Res. 2008, 29, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.T.; Chen, C.L.; Chou, T.Y.; Yeh, C.T.; Lee, W.H.; Wang, L.S. Nm23H1 mediates tumor invasion in esophageal squamous cell carcinoma by regulation of CLDN1 through the AKT signaling. Oncogenesis 2016, 5, e239. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, J.; Gao, F.; Xu, T.; Li, J.; Hu, Z.; Wang, C.; Long, Y.; He, X.; Deng, X.; Ren, D.; et al. CLDN1 induces autophagy to promote proliferation and metastasis of esophageal squamous carcinoma through AMPK/STAT1/ULK1 signaling. J. Cell Physiol. 2020, 235, 2245–2259. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Wang, Z.; Song, L.; Wang, D.; Sun, Z. Low expression of claudin-4: An indicator of recurrence in esophageal squamous cell carcinoma after Ivor Lewis esophagectomy? Med. Oncol. 2014, 31, 951. [Google Scholar] [CrossRef]
- Suzuki, K.; Sentani, K.; Tanaka, H.; Yano, T.; Suzuki, K.; Oshima, M.; Yasui, W.; Tamura, A.; Tsukita, S. Deficiency of Stomach-Type Claudin-18 in Mice Induces Gastric Tumor Formation Independent of H pylori Infection. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 119–142. [Google Scholar] [CrossRef]
- Usami, Y.; Satake, S.; Nakayama, F.; Matsumoto, M.; Ohnuma, K.; Komori, T.; Semba, S.; Ito, A.; Yokozaki, H. Snail-associated epithelial-mesenchymal transition promotes oesophageal squamous cell carcinoma motility and progression. J. Pathol. 2008, 215, 330–339. [Google Scholar] [CrossRef]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Estrada, O.M.; Cullerés, A.; Soriano, F.X.; Peinado, H.; Bolós, V.; Martínez, F.O.; Reina, M.; Cano, A.; Fabre, M.; Vilaró, S. The transcription factors Slug and Snail act as repressors of Claudin-1 expression in epithelial cells. Biochem. J. 2006, 394, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Li, C.F.; Chen, J.Y.; Ho, Y.H.; Hsu, W.H.; Wu, L.C.; Lan, H.Y.; Hsu, D.S.; Tai, S.K.; Chang, Y.C.; Yang, M.H. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Ahmad, R.; Chaturvedi, R.; Smith, J.J.; Midha, R.; Mittal, M.K.; Krishnan, M.; Chen, X.; Eschrich, S.; Yeatman, T.J.; et al. Claudin-2 expression increases tumorigenicity of colon cancer cells: Role of epidermal growth factor receptor activation. Oncogene 2011, 30, 3234–3247. [Google Scholar] [CrossRef]
- Macara, I.G. Parsing the polarity code. Nat. Rev. Mol. Cell Biol. 2004, 5, 220–231. [Google Scholar] [CrossRef]
- Suzuki, A.; Ohno, S. The PAR-aPKC system: Lessons in polarity. J. Cell Sci. 2006, 119, 979–987. [Google Scholar] [CrossRef]
- Zen, K.; Yasui, K.; Gen, Y.; Dohi, O.; Wakabayashi, N.; Mitsufuji, S.; Itoh, Y.; Zen, Y.; Nakanuma, Y.; Taniwaki, M.; et al. Defective expression of polarity protein PAR-3 gene (PARD3) in esophageal squamous cell carcinoma. Oncogene 2009, 28, 2910–2918. [Google Scholar] [CrossRef]
- Hirokawa, N.; Heuser, J.E. Quick-freeze, deep-etch visualization of the cytoskeleton beneath surface differentiations of intestinal epithelial cells. J. Cell Biol. 1981, 91, 399–409. [Google Scholar] [CrossRef]
- Miyaguchi, K. Ultrastructure of the zonula adherens revealed by rapid-freeze deep-etching. J. Struct. Biol. 2000, 132, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.; Weis, W.I. Structure and biochemistry of cadherins and catenins. Cold Spring Harb. Perspect. Biol. 2009, 1, a003053. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Tan, J.; Anderson, K.L.; Hanein, D.; Volkmann, N.; Weis, W.I.; Nelson, W.J.; Dunn, A.R. Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science 2014, 346, 1254211. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.A.; Ireton, R.C.; Reynolds, A.B. A core function for p120-catenin in cadherin turnover. J. Cell Biol. 2003, 163, 525–534. [Google Scholar] [CrossRef]
- Takai, Y.; Nakanishi, H. Nectin and afadin: Novel organizers of intercellular junctions. J. Cell Sci. 2003, 116, 17–27. [Google Scholar] [CrossRef]
- Weis, W.I.; Nelson, W.J. Re-solving the cadherin-catenin-actin conundrum. J. Biol. Chem. 2006, 281, 35593–35597. [Google Scholar] [CrossRef]
- Yamamoto, T.; Harada, N.; Kano, K.; Taya, S.; Canaani, E.; Matsuura, Y.; Mizoguchi, A.; Ide, C.; Kaibuchi, K. The Ras target AF-6 interacts with ZO-1 and serves as a peripheral component of tight junctions in epithelial cells. J. Cell Biol. 1997, 139, 785–795. [Google Scholar] [CrossRef]
- Shiozaki, H.; Doki, Y.; Oka, H.; Iihara, K.; Miyata, M.; Kadowaki, T.; Matsui, S.; Tamura, S.; Inoue, M.; Mori, T. E-cadherin expression and cancer invasion and metastasis. Hum. Cell 1993, 6, 94–99. [Google Scholar]
- Kaihara, T.; Kusaka, T.; Kawamata, H.; Oda, Y.; Fujii, S.; Morita, K.; Imura, J.; Fujimori, T. Decreased expression of E-cadherin and Yamamoto-Kohama’s mode of invasion highly correlates with lymph node metastasis in esophageal squamous cell carcinoma. Pathobiology 2001, 69, 172–178. [Google Scholar] [CrossRef]
- Jankowski, J.; Newham, P.; Kandemir, O.; Hirano, S.; Takeichi, M.; Pignatelli, M. Differential expression of e-cadherin in normal, metaplastic and dysplastic esophageal mucosa—A putative biomarker. Int. J. Oncol. 1994, 4, 441–448. [Google Scholar] [CrossRef]
- Nair, K.S.; Naidoo, R.; Chetty, R. Microsatellite analysis of the APC gene and immunoexpression of E-cadherin, catenin, and tubulin in esophageal squamous cell carcinoma. Hum. Pathol. 2006, 37, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yang, R.; Gu, J.; Jiang, H.; Li, H. The expression of AGGF1, FOXC2, and E-cadherin in esophageal carcinoma and their clinical significance. Medicine 2020, 99, e22173. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lotan, R.; Menter, D.; Lippman, S.M.; Xu, X.C. Expression of E-cadherin is associated with squamous differentiation in squamous cell carcinomas. Anticancer Res. 2000, 20, 1385–1390. [Google Scholar]
- Nishimura, T.; Tamaoki, M.; Komatsuzaki, R.; Oue, N.; Taniguchi, H.; Komatsu, M.; Aoyagi, K.; Minashi, K.; Chiwaki, F.; Shinohara, H.; et al. SIX1 maintains tumor basal cells via transforming growth factor-β pathway and associates with poor prognosis in esophageal cancer. Cancer Sci. 2017, 108, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Andl, C.D.; Fargnoli, B.B.; Okawa, T.; Bowser, M.; Takaoka, M.; Nakagawa, H.; Klein-Szanto, A.; Hua, X.; Herlyn, M.; Rustgi, A.K. Coordinated functions of E-cadherin and transforming growth factor beta receptor II in vitro and in vivo. Cancer Res. 2006, 66, 9878–9885. [Google Scholar] [CrossRef]
- Si, H.X.; Tsao, S.W.; Lam, K.Y.; Srivastava, G.; Liu, Y.; Wong, Y.C.; Shen, Z.Y.; Cheung, A.L. E-cadherin expression is commonly downregulated by CpG island hypermethylation in esophageal carcinoma cells. Cancer Lett. 2001, 173, 71–78. [Google Scholar] [CrossRef]
- Ling, Z.Q.; Li, P.; Ge, M.H.; Zhao, X.; Hu, F.J.; Fang, X.H.; Dong, Z.M.; Mao, W.M. Hypermethylation-modulated down-regulation of CDH1 expression contributes to the progression of esophageal cancer. Int. J. Mol. Med. 2011, 27, 625–635. [Google Scholar] [CrossRef]
- Chen, X.; Lu, B.; Ma, Q.; Ji, C.D.; Li, J.Z. EphA3 inhibits migration and invasion of esophageal cancer cells by activating the mesenchymal-epithelial transition process. Int. J. Oncol. 2019, 54, 722–732. [Google Scholar] [CrossRef]
- Liu, B.; Li, X.; Li, C.; Xu, R.; Sun, X. miR-25 mediates metastasis and epithelial-mesenchymal-transition in human esophageal squamous cell carcinoma via regulation of E-cadherin signaling. Bioengineered 2019, 10, 679–688. [Google Scholar] [CrossRef]
- Handra-Luca, A.; Terris, B.; Couvelard, A.; Molas, G.; Degott, C.; Flejou, J.F. Spindle cell squamous carcinoma of the oesophagus: An analysis of 17 cases, with new immunohistochemical evidence for a clonal origin. Histopathology 2001, 39, 125–132. [Google Scholar] [CrossRef]
- Ren, D.; Wang, M.; Guo, W.; Zhao, X.; Tu, X.; Huang, S.; Zou, X.; Peng, X. Wild-type p53 suppresses the epithelial-mesenchymal transition and stemness in PC-3 prostate cancer cells by modulating miR-145. Int. J. Oncol. 2013, 42, 1473–1481. [Google Scholar] [CrossRef]
- Yang, Z.; Qu, C.B.; Zhang, Y.; Zhang, W.F.; Wang, D.D.; Gao, C.C.; Ma, L.; Chen, J.S.; Liu, K.L.; Zheng, B.; et al. Dysregulation of p53-RBM25-mediated circAMOTL1L biogenesis contributes to prostate cancer progression through the circAMOTL1L-miR-193a-5p-Pcdha pathway. Oncogene 2019, 38, 2516–2532. [Google Scholar] [CrossRef]
- Smirnov, A.; Lena, A.M.; Cappello, A.; Panatta, E.; Anemona, L.; Bischetti, S.; Annicchiarico-Petruzzelli, M.; Mauriello, A.; Melino, G.; Candi, E. ZNF185 is a p63 target gene critical for epidermal differentiation and squamous cell carcinoma development. Oncogene 2019, 38, 1625–1638. [Google Scholar] [CrossRef]
- Lawler, K.; O’Sullivan, G.; Long, A.; Kenny, D. Shear stress induces internalization of E-cadherin and invasiveness in metastatic oesophageal cancer cells by a Src-dependent pathway. Cancer Sci. 2009, 100, 1082–1087. [Google Scholar] [CrossRef]
- Reynolds, A.B.; Roesel, D.J.; Kanner, S.B.; Parsons, J.T. Transformation-specific tyrosine phosphorylation of a novel cellular protein in chicken cells expressing oncogenic variants of the avian cellular src gene. Mol. Cell Biol. 1989, 9, 629–638. [Google Scholar] [CrossRef]
- Ireton, R.C.; Davis, M.A.; van Hengel, J.; Mariner, D.J.; Barnes, K.; Thoreson, M.A.; Anastasiadis, P.Z.; Matrisian, L.; Bundy, L.M.; Sealy, L.; et al. A novel role for p120 catenin in E-cadherin function. J. Cell Biol. 2002, 159, 465–476. [Google Scholar] [CrossRef]
- Chung, Y.; Lam, A.K.; Luk, J.M.; Law, S.; Chan, K.W.; Lee, P.Y.; Wong, J. Altered E-cadherin expression and p120 catenin localization in esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2007, 14, 3260–3267. [Google Scholar] [CrossRef]
- Haraguchi, M.; Fukushige, T.; Kanekura, T.; Ozawa, M. E-cadherin loss in RMG-1 cells inhibits cell migration and its regulation by Rho GTPases. Biochem. Biophys. Rep. 2019, 18, 100650. [Google Scholar] [CrossRef]
- Cai, D.; Chen, S.C.; Prasad, M.; He, L.; Wang, X.; Choesmel-Cadamuro, V.; Sawyer, J.K.; Danuser, G.; Montell, D.J. Mechanical feedback through E-cadherin promotes direction sensing during collective cell migration. Cell 2014, 157, 1146–1159. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Ochiai, A.; Akimoto, S.; Kato, H.; Watanabe, H.; Tachimori, Y.; Yamamoto, S.; Hirohashi, S. Expression of E-cadherin, alpha-catenin, beta-catenin and plakoglobin in esophageal carcinomas and its prognostic significance: Immunohistochemical analysis of 96 lesions. Oncology 1997, 54, 158–165. [Google Scholar] [CrossRef]
- Kadowaki, T.; Shiozaki, H.; Inoue, M.; Tamura, S.; Oka, H.; Doki, Y.; Iihara, K.; Matsui, S.; Iwazawa, T.; Nagafuchi, A.; et al. E-cadherin and alpha-catenin expression in human esophageal cancer. Cancer Res. 1994, 54, 291–296. [Google Scholar]
- Khare, L.; Sabourin, C.L.; De Young, B.R.; Jamasbi, R.J.; Stoner, G.D. Altered localization of E-cadherin and alpha-catenin in rat esophageal tumors. Int. J. Oncol. 1999, 14, 33–40. [Google Scholar] [CrossRef]
- Zhang, G.; Zhou, X.; Xue, L.; Quan, L.; Wang, Y.; Zhou, C.; Lu, N.; Wang, Q.; Zhu, H.; Xu, N. Accumulation of cytoplasmic beta-catenin correlates with reduced expression of E-cadherin, but not with phosphorylated Akt in esophageal squamous cell carcinoma: Immunohistochemical study. Pathol. Int. 2005, 55, 310–317. [Google Scholar] [CrossRef]
- Deng, Y.Z.; Chen, P.P.; Wang, Y.; Yin, D.; Koeffler, H.P.; Li, B.; Tong, X.J.; Xie, D. Connective tissue growth factor is overexpressed in esophageal squamous cell carcinoma and promotes tumorigenicity through beta-catenin-T-cell factor/Lef signaling. J. Biol. Chem. 2007, 282, 36571–36581. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Zhu, H.; Zhang, W.; Zhang, G.; Zhou, X.; Zhou, C.; Quan, L.; Bai, J.; Xue, L.; et al. FRAT1 overexpression leads to aberrant activation of beta-catenin/TCF pathway in esophageal squamous cell carcinoma. Int. J. Cancer 2008, 123, 561–568. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, X.; Zhu, H.; Liu, S.; Zhou, C.; Zhang, G.; Xue, L.; Lu, N.; Quan, L.; Bai, J.; et al. Overexpression of EB1 in human esophageal squamous cell carcinoma (ESCC) may promote cellular growth by activating beta-catenin/TCF pathway. Oncogene 2005, 24, 6637–6645. [Google Scholar] [CrossRef]
- He, H.; Ding, F.; Li, Y.; Luo, A.; Chen, H.; Wu, C.; Liu, Z. Migfilin regulates esophageal cancer cell motility through promoting GSK-3β-mediated degradation of β-catenin. Mol. Cancer Res. 2012, 10, 273–281. [Google Scholar] [CrossRef]
- Wang, S.; Cai, J.; Zhang, S.; Dong, M.; Zhang, L.; Xu, Y.; Shen, B.; Chen, S. Loss of polarity protein Par3, via transcription factor Snail, promotes bladder cancer metastasis. Cancer Sci. 2021, 112, 2625–2641. [Google Scholar] [CrossRef]
- Jin, S.; Wang, X.; Tong, T.; Zhang, D.; Shi, J.; Chen, J.; Zhan, Q. Aurora-A enhances malignant development of esophageal squamous cell carcinoma (ESCC) by phosphorylating β-catenin. Mol. Oncol. 2015, 9, 249–259. [Google Scholar] [CrossRef]
- Shiozaki, H.; Kadowaki, T.; Doki, Y.; Inoue, M.; Tamura, S.; Oka, H.; Iwazawa, T.; Matsui, S.; Shimaya, K.; Takeichi, M.; et al. Effect of epidermal growth factor on cadherin-mediated adhesion in a human oesophageal cancer cell line. Br. J. Cancer 1995, 71, 250–258. [Google Scholar] [CrossRef]
- Hoschuetzky, H.; Aberle, H.; Kemler, R. Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J. Cell Biol. 1994, 127, 1375–1380. [Google Scholar] [CrossRef]
- Hazan, R.B.; Norton, L. The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J. Biol. Chem. 1998, 273, 9078–9084. [Google Scholar] [CrossRef]
- Jones, L.J.; Veale, R.B. Redistribution of β-catenin in response to EGF and lithium signalling in human oesophageal squamous carcinoma cell lines. Cancer Cell Int. 2003, 3, 13. [Google Scholar] [CrossRef][Green Version]
- Radice, G.L. N-cadherin-mediated adhesion and signaling from development to disease: Lessons from mice. Prog. Mol. Biol. Transl. Sci. 2013, 116, 263–289. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef]
- Zisis, T.; Brückner, D.B.; Brandstätter, T.; Siow, W.X.; d’Alessandro, J.; Vollmar, A.M.; Broedersz, C.P.; Zahler, S. Disentangling cadherin-mediated cell-cell interactions in collective cancer cell migration. Biophys. J. 2022, 121, 44–60. [Google Scholar] [CrossRef]
- Ko, H.; Kim, S.; Jin, C.H.; Lee, E.; Ham, S.; Yook, J.I.; Kim, K. Protein kinase casein kinase 2-mediated upregulation of N-cadherin confers anoikis resistance on esophageal carcinoma cells. Mol. Cancer Res. 2012, 10, 1032–1038. [Google Scholar] [CrossRef]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef]
- Yook, J.I.; Li, X.Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef]
- Calvert, M.E.; Keck, K.M.; Ptak, C.; Shabanowitz, J.; Hunt, D.F.; Pemberton, L.F. Phosphorylation by casein kinase 2 regulates Nap1 localization and function. Mol. Cell Biol. 2008, 28, 1313–1325. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gross, W.; Hong, S.H.; Privalsky, M.L. The SMRT corepressor is a target of phosphorylation by protein kinase CK2 (casein kinase II). Mol. Cell Biochem. 2001, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Le Mée, S.; Fromigué, O.; Marie, P.J. Sp1/Sp3 and the myeloid zinc finger gene MZF1 regulate the human N-cadherin promoter in osteoblasts. Exp. Cell Res. 2005, 302, 129–142. [Google Scholar] [CrossRef]
- Vanniya, S.P.; Srisailapathy, C.R.S.; Kunka Mohanram, R. The tip link protein Cadherin-23: From Hearing Loss to Cancer. Pharmacol. Res. 2018, 130, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Sannigrahi, M.K.; Srinivas, C.S.; Deokate, N.; Rakshit, S. The strong propensity of Cadherin-23 for aggregation inhibits cell migration. Mol. Oncol. 2019, 13, 1092–1109. [Google Scholar] [CrossRef]
- Ying, J.; Li, H.; Seng, T.J.; Langford, C.; Srivastava, G.; Tsao, S.W.; Putti, T.; Murray, P.; Chan, A.T.; Tao, Q. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene 2006, 25, 1070–1080. [Google Scholar] [CrossRef]
- Tanaka, H.; Kanda, M.; Koike, M.; Iwata, N.; Shimizu, D.; Ezaka, K.; Sueoka, S.; Tanaka, Y.; Takami, H.; Hashimoto, R.; et al. Adherens junctions associated protein 1 serves as a predictor of recurrence of squamous cell carcinoma of the esophagus. Int. J. Oncol. 2015, 47, 1811–1818. [Google Scholar] [CrossRef]
- Hötte, K.; Smyrek, I.; Starzinski-Powitz, A.; Stelzer, E.H.K. Endogenous AJAP1 associates with the cytoskeleton and attenuates angiogenesis in endothelial cells. Biol. Open 2017, 6, 723–731. [Google Scholar] [CrossRef]
- Green, K.J.; Gaudry, C.A. Are desmosomes more than tethers for intermediate filaments? Nat. Rev. Mol. Cell Biol. 2000, 1, 208–216. [Google Scholar] [CrossRef]
- Chidgey, M.; Dawson, C. Desmosomes: A role in cancer? Br. J. Cancer 2007, 96, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M. Cell adhesion: The molecular basis of tissue architecture and morphogenesis. Cell 1996, 84, 345–357. [Google Scholar] [CrossRef]
- Getsios, S.; Huen, A.C.; Green, K.J. Working out the strength and flexibility of desmosomes. Nat. Rev. Mol. Cell Biol. 2004, 5, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Tucker, D.K.; Kohlhorst, D.; Niessen, C.M.; Kowalczyk, A.P. Classical and desmosomal cadherins at a glance. J. Cell Sci. 2012, 125, 2547–2552. [Google Scholar] [CrossRef]
- Khan, K.; Hardy, R.; Haq, A.; Ogunbiyi, O.; Morton, D.; Chidgey, M. Desmocollin switching in colorectal cancer. Br. J. Cancer 2006, 95, 1367–1370. [Google Scholar] [CrossRef]
- Kolegraff, K.; Nava, P.; Helms, M.N.; Parkos, C.A.; Nusrat, A. Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/β-catenin signaling. Mol. Biol. Cell 2011, 22, 1121–1134. [Google Scholar] [CrossRef]
- Nuber, U.A.; Schäfer, S.; Schmidt, A.; Koch, P.J.; Franke, W.W. The widespread human desmocollin Dsc2 and tissue-specific patterns of synthesis of various desmocollin subtypes. Eur. J. Cell Biol. 1995, 66, 69–74. [Google Scholar]
- Fang, W.K.; Gu, W.; Li, E.M.; Wu, Z.Y.; Shen, Z.Y.; Shen, J.H.; Wu, J.Y.; Pan, F.; Lv, Z.; Xu, X.E.; et al. Reduced membranous and ectopic cytoplasmic expression of DSC2 in esophageal squamous cell carcinoma: An independent prognostic factor. Hum. Pathol. 2010, 41, 1456–1465. [Google Scholar] [CrossRef]
- Fang, W.K.; Liao, L.D.; Zeng, F.M.; Zhang, P.X.; Wu, J.Y.; Shen, J.; Xu, L.Y.; Li, E.M. Desmocollin-2 affects the adhesive strength and cytoskeletal arrangement in esophageal squamous cell carcinoma cells. Mol. Med. Rep. 2014, 10, 2358–2364. [Google Scholar] [CrossRef]
- Fang, W.K.; Liao, L.D.; Gu, W.; Chen, B.; Wu, Z.Y.; Wu, J.Y.; Shen, J.; Xu, L.Y.; Li, E.M. Down-regulated γ-catenin expression is associated with tumor aggressiveness in esophageal cancer. World J. Gastroenterol. 2014, 20, 5839–5848. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.E.; Ben-Yedidia, T.; Shtutman, M.; Feinstein, E.; Einat, P.; Ben-Ze’ev, A. Nr-CAM is a target gene of the beta-catenin/LEF-1 pathway in melanoma and colon cancer and its expression enhances motility and confers tumorigenesis. Genes Dev. 2002, 16, 2058–2072. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Ong, C.W.; Salto-Tellez, M. Overexpression of neurone glial-related cell adhesion molecule is an independent predictor of poor prognosis in advanced colorectal cancer. Cancer Sci. 2011, 102, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.D.; Li, J.Y.; Li, M.; Gu, J.; Shi, X.T.; Ke, Y.; Chen, K.N. Matrix metalloproteinases expression correlates with survival in patients with esophageal squamous cell carcinoma. Am. J. Gastroenterol. 2005, 100, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Pittella, F.; Katsube, K.; Takemura, T.; Hashimoto, T.; Kawano, T.; Garrod, D.; Takagi, M. Perinuclear and cytoplasmic distribution of desmoglein in esophageal squamous cell carcinomas. Pathol. Res. Pract. 2001, 197, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Natsugoe, S.; Aikou, T.; Shimada, M.; Kumanohoso, T.; Tezuka, Y.; Sagara, M.; Yoshinaka, H.; Baba, M.; Fukumoto, T. Expression of desmoglein I in squamous cell carcinoma of the esophagus. J. Surg. Oncol. 1994, 57, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.K.; Chen, B.; Xu, X.E.; Liao, L.D.; Wu, Z.Y.; Wu, J.Y.; Shen, J.; Xu, L.Y.; Li, E.M. Altered expression and localization of desmoglein 3 in esophageal squamous cell carcinoma. Acta Histochem. 2014, 116, 803–809. [Google Scholar] [CrossRef]
- Fang, W.K.; Gu, W.; Liao, L.D.; Chen, B.; Wu, Z.Y.; Wu, J.Y.; Shen, J.; Xu, L.Y.; Li, E.M. Prognostic significance of desmoglein 2 and desmoglein 3 in esophageal squamous cell carcinoma. Asian Pac. J. Cancer Prev. 2014, 15, 871–876. [Google Scholar] [CrossRef]
- Zhurinsky, J.; Shtutman, M.; Ben-Ze’ev, A. Plakoglobin and beta-catenin: Protein interactions, regulation and biological roles. J. Cell Sci. 2000, 113 Pt 18, 3127–3139. [Google Scholar] [CrossRef]
- Lewis, J.E.; Wahl, J.K., III; Sass, K.M.; Jensen, P.J.; Johnson, K.R.; Wheelock, M.J. Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J. Cell Biol. 1997, 136, 919–934. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, J.; Zhao, J.; Li, Z.; Chen, H.; Qiao, Y.; Chen, X.; Liu, K.; Dong, Z. TOPK promotes metastasis of esophageal squamous cell carcinoma by activating the Src/GSK3β/STAT3 signaling pathway via γ-catenin. BMC Cancer 2019, 19, 1264. [Google Scholar] [CrossRef]
- Iwaya, T.; Maesawa, C.; Kimura, T.; Ogasawara, S.; Ikeda, K.; Kimura, Y.; Noda, Y.; Ishida, K.; Sato, N.; Saito, K.; et al. Infrequent mutation of the human envoplakin gene is closely linked to the tylosis oesophageal cancer locus in sporadic oesophageal squamous cell carcinomas. Oncol. Rep. 2005, 13, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Chun, M.G.; Hanahan, D. Genetic deletion of the desmosomal component desmoplakin promotes tumor microinvasion in a mouse model of pancreatic neuroendocrine carcinogenesis. PLoS Genet. 2010, 6, e1001120. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, V.; Koetsier, J.L.; Godsel, L.M.; Green, K.J. Plakophilin 3 mediates Rap1-dependent desmosome assembly and adherens junction maturation. Mol. Biol. Cell 2014, 25, 3749–3764. [Google Scholar] [CrossRef] [PubMed]
- Meşe, G.; Richard, G.; White, T.W. Gap junctions: Basic structure and function. J. Investig. Dermatol. 2007, 127, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R. Junctional intercellular communication and the control of growth. Biochim. Biophys. Acta 1979, 560, 1–65. [Google Scholar] [CrossRef]
- Pitts, J.D.; Finbow, M.E. The gap junction. J. Cell Sci. Suppl. 1986, 4, 239–266. [Google Scholar] [CrossRef]
- Kamibayashi, Y.; Oyamada, Y.; Mori, M.; Oyamada, M. Aberrant expression of gap junction proteins (connexins) is associated with tumor progression during multistage mouse skin carcinogenesis in vivo. Carcinogenesis 1995, 16, 1287–1297. [Google Scholar] [CrossRef]
- Tsai, H.; Werber, J.; Davia, M.O.; Edelman, M.; Tanaka, K.E.; Melman, A.; Christ, G.J.; Geliebter, J. Reduced connexin 43 expression in high grade, human prostatic adenocarcinoma cells. Biochem. Biophys. Res. Commun. 1996, 227, 64–69. [Google Scholar] [CrossRef]
- Wilgenbus, K.K.; Kirkpatrick, C.J.; Knuechel, R.; Willecke, K.; Traub, O. Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues. Int. J. Cancer 1992, 51, 522–529. [Google Scholar] [CrossRef]
- Loncarek, J.; Yamasaki, H.; Levillain, P.; Milinkevitch, S.; Mesnil, M. The expression of the tumor suppressor gene connexin 26 is not mediated by methylation in human esophageal cancer cells. Mol. Carcinog. 2003, 36, 74–81. [Google Scholar] [CrossRef]
- Matono, S.; Tanaka, T.; Sueyoshi, S.; Yamana, H.; Fujita, H.; Shirouzu, K. Bystander effect in suicide gene therapy is directly proportional to the degree of gap junctional intercellular communication in esophageal cancer. Int. J. Oncol. 2003, 23, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Li, Q.; Nishikawa, Y.; Yoshioka, T.; Yoshida, M.; Nishimura, T.; Enomoto, K. Pathological significance of intracytoplasmic connexin proteins: Implication in tumor progression. J. Membr. Biol. 2007, 218, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Iikawa, N.; Yamamoto, Y.; Kawasaki, Y.; Nishijima-Matsunobu, A.; Suzuki, M.; Yamada, T.; Omori, Y. Intrinsic Oncogenic Function of Intracellular Connexin26 Protein in Head and Neck Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2018, 19, 2134. [Google Scholar] [CrossRef]
- Tanaka, T.; Kimura, M.; Ishiguro, H.; Mizoguchi, K.; Takeyama, H. Connexin 43 expression is associated with poor survival in patients with esophageal squamous cell carcinoma. Mol. Clin. Oncol. 2016, 4, 989–993. [Google Scholar] [CrossRef]
- Inose, T.; Kato, H.; Kimura, H.; Faried, A.; Tanaka, N.; Sakai, M.; Sano, A.; Sohda, M.; Nakajima, M.; Fukai, Y.; et al. Correlation between connexin 26 expression and poor prognosis of esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2009, 16, 1704–1710. [Google Scholar] [CrossRef] [PubMed]
- Di Conza, G.; Ho, P.C. ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Battaglia-Hsu, S.F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef]
- Hou, X.; Khan, M.R.A.; Turmaine, M.; Thrasivoulou, C.; Becker, D.L.; Ahmed, A. Wnt signaling regulates cytosolic translocation of connexin 43. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R248–R261. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, L.; Liu, J.; Goldstein, A.; Bado, I.; Zhang, W.; Arenkiel, B.R.; Li, Z.; Yang, M.; Du, S.; et al. The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell 2018, 34, 823–839.e827. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, X.; Luo, W. Advances in the application of claudins to tumor therapy. Sheng Wu Gong Cheng Xue Bao 2019, 35, 931–941. [Google Scholar] [CrossRef]
- Li, H.; Li, H.; Waresijiang, Y.; Chen, Y.; Li, Y.; Yu, L.; Li, Y.; Liu, L. Clinical significance of HDAC1, -2 and -3 expression levels in esophageal squamous cell carcinoma. Exp. Ther. Med. 2020, 20, 315–324. [Google Scholar] [CrossRef]
- Zeng, L.S.; Yang, X.Z.; Wen, Y.F.; Mail, S.J.; Wang, M.H.; Zhang, M.Y.; Zheng, X.F.; Wang, H.Y. Overexpressed HDAC4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Aging 2016, 8, 1236–1249. [Google Scholar] [CrossRef]
- Ahmad, R.; Kumar, B.; Pan, K.; Dhawan, P.; Singh, A.B. HDAC-4 regulates claudin-2 expression in EGFR-ERK1/2 dependent manner to regulate colonic epithelial cell differentiation. Oncotarget 2017, 8, 87718–87736. [Google Scholar] [CrossRef][Green Version]
- Kakiuchi, A.; Kakuki, T.; Ohwada, K.; Kurose, M.; Kondoh, A.; Obata, K.; Nomura, K.; Miyata, R.; Kaneko, Y.; Konno, T.; et al. HDAC inhibitors suppress the proliferation, migration and invasiveness of human head and neck squamous cell carcinoma cells via p63-mediated tight junction molecules and p21-mediated growth arrest. Oncol. Rep. 2021, 45, 46. [Google Scholar] [CrossRef]
- Ma, J.; Guo, X.; Zhang, S.; Liu, H.; Lu, J.; Dong, Z.; Liu, K.; Ming, L. Trichostatin A, a histone deacetylase inhibitor, suppresses proliferation and promotes apoptosis of esophageal squamous cell lines. Mol. Med. Rep. 2015, 11, 4525–4531. [Google Scholar] [CrossRef]
- Wang, Z.; Mandell, K.J.; Parkos, C.A.; Mrsny, R.J.; Nusrat, A. The second loop of occludin is required for suppression of Raf1-induced tumor growth. Oncogene 2005, 24, 4412–4420. [Google Scholar] [CrossRef]
- Song, Z.H.; Ke, Y.L.; Xiao, K.; Jiao, L.F.; Hong, Q.H.; Hu, C.H. Diosmectite-zinc oxide composite improves intestinal barrier restoration and modulates TGF-β1, ERK1/2, and Akt in piglets after acetic acid challenge. J. Anim. Sci. 2015, 93, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Guo, Q.; Chen, L.; Liu, S. Clinicopathological significance and potential drug targeting of CDH1 in lung cancer: A meta-analysis and literature review. Drug Des. Dev. Ther. 2015, 9, 2171–2178. [Google Scholar] [CrossRef]
- Huang, R.; Ding, P.; Yang, F. Clinicopathological significance and potential drug target of CDH1 in breast cancer: A meta-analysis and literature review. Drug Des. Dev. Ther. 2015, 9, 5277–5285. [Google Scholar] [CrossRef]
- Geißler, A.L.; Geißler, M.; Kottmann, D.; Lutz, L.; Fichter, C.D.; Fritsch, R.; Weddeling, B.; Makowiec, F.; Werner, M.; Lassmann, S. ATM mutations and E-cadherin expression define sensitivity to EGFR-targeted therapy in colorectal cancer. Oncotarget 2017, 8, 17164–17190. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, D.A.; Asangani, I.A.; Nelson, L.D.; Hughes, D.P.; Siwak, D.R.; Mills, G.B.; Harms, A.; Buchholz, E.; Pilz, L.R.; Manegold, C.; et al. Cetuximab attenuates metastasis and u-PAR expression in non-small cell lung cancer: U-PAR and E-cadherin are novel biomarkers of cetuximab sensitivity. Cancer Res. 2009, 69, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Zhou, X.; Zhang, W.; Qu, Y.; Ke, X. Is β-Catenin a Druggable Target for Cancer Therapy? Trends Biochem. Sci. 2018, 43, 623–634. [Google Scholar] [CrossRef]
- Erez, N.; Zamir, E.; Gour, B.J.; Blaschuk, O.W.; Geiger, B. Induction of apoptosis in cultured endothelial cells by a cadherin antagonist peptide: Involvement of fibroblast growth factor receptor-mediated signalling. Exp. Cell Res. 2004, 294, 366–378. [Google Scholar] [CrossRef]
- Perotti, A.; Sessa, C.; Mancuso, A.; Noberasco, C.; Cresta, S.; Locatelli, A.; Carcangiu, M.L.; Passera, K.; Braghetti, A.; Scaramuzza, D.; et al. Clinical and pharmacological phase I evaluation of Exherin (ADH-1), a selective anti-N-cadherin peptide in patients with N-cadherin-expressing solid tumours. Ann. Oncol. 2009, 20, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Beasley, G.M.; McMahon, N.; Sanders, G.; Augustine, C.K.; Selim, M.A.; Peterson, B.; Norris, R.; Peters, W.P.; Ross, M.I.; Tyler, D.S. A phase 1 study of systemic ADH-1 in combination with melphalan via isolated limb infusion in patients with locally advanced in-transit malignant melanoma. Cancer 2009, 115, 4766–4774. [Google Scholar] [CrossRef]
- Caiazza, C.; Mallardo, M. The Roles of miR-25 and its Targeted Genes in Development of Human Cancer. Microrna 2016, 5, 113–119. [Google Scholar] [CrossRef]
- Brink, P.R.; Valiunas, V.; Gordon, C.; Rosen, M.R.; Cohen, I.S. Can gap junctions deliver? Biochim. Biophys. Acta 2012, 1818, 2076–2081. [Google Scholar] [CrossRef] [PubMed]
- Bonacquisti, E.E.; Nguyen, J. Connexin 43 (Cx43) in cancer: Implications for therapeutic approaches via gap junctions. Cancer Lett. 2019, 442, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Alpha, K.M.; Zehrbach, N.M.; Turner, C.E. Paxillin promotes breast tumor collective cell invasion through maintenance of adherens junction integrity. Mol. Biol. Cell 2022, 33, ar14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Structure | Transmembrane Proteins | Adaptor Proteins | Expression | Consequence | Mechanism | Reference | |
|---|---|---|---|---|---|---|---|
| Tight junction | Claudin-1 | / | Membrane (↓) | Associated with recurrence and poor prognosis | Enhanced invasiveness | AKT signalling pathway, activated by reduction of Nm23H1; binding of Snail to E-box elements | [26,30] |
| Nucleus (↑) | Enhanced proliferation and metastasis | Autophagy triggered by the AMPK/STAT1/ULK1 signalling pathway | [27] | ||||
| Claudin-4 | / | Membrane (↓) | Associated with poor differentiation, deeper invasion, positive lymph node metastasis, low disease-free survival, and overall survival; independent risk factor for recurrence | Promoter methylation | [9,28] | ||
| Claudin-7 | / | Membrane (↓) | Associated with deeper invasion, advanced tumour stage, positive lymph node metastasis, and lymphatic invasion | Binding of Snail to E-box elements; loss of synergistic effect between claudin-7 and E-Cadherin | [12,24,30] | ||
| Occludin | / | Membrane (↓) | / | Binding of Snail to E-box elements | [30] | ||
| / | ZO1 | Membrane (↓) | Associated with positive lymph node metastasis and poor differentiation | Decrease of PAR-3 (GSK-3β/Snail/Par3/ZO-1 axis); EphA3 methylation | [39,58] | ||
| Adherens junction | E-Cadherin | / | Membrane (↓) | Associated with the differentiation and prognosis | / | Methylation of CpG island; p53 mutation | [53,55,56,57,60] |
| Promote proliferation and invasion | Down-regulation of claudin-7 causes loss of synergistic effect between claudin-7 and E-Cadherin | [12] | |||||
| Promote migration and invasion | EphA3 methylation, activation of the Rho GTPase signalling pathway; absence of TbetaRII expression | [49,50,59] | |||||
| High expression of miR-25 inhibits E-Cadherin expression which is crucial for mediating EMT | [59] | ||||||
| Acquire resistance to anoikis and anoxia | High-activity of PKCK2, stabilisation of Snail, upregulation of N-Cadherin | [88,89,90,94] | |||||
| Destroy the integrity of the epithelium | Mutation of p63 and absence of the expression of ZNF185 | [63] | |||||
| Cytoplasm (↑) | Associated with poor differentiation | Reduce the glycosylation of E-Cadherin or truncate the E-Cadherin | [50] | ||||
| N-Cadherin | / | Membrane (↑) | Induce a migratory phenotype, influence the ability of tumour cells by altering their cadherin expression profiles | High activity of PKCK2, directly and indirectly stabilises Snail (see Figure 2) | [88] | ||
| / | α-Catenin | Membrane (↓) | Associated with an aggressive phenotype and lymph node metastasis | / | [71] | ||
| Associated with tumour progression and poor prognosis | / | [70,72] | |||||
| β-Catenin | Cytoplasm (↑) Nucleus (↑) | Associated with poor prognosis, tumour cell invasion and metastasis | Dissociation from cell–cell contacts, intracellular accumulation, inhibition of E-Cadherin mediated cell adhesion | Phosphorylation of β-Catenin by EGFR, disrupting the binding between β-Catenin and α-Catenin | [80,83] | ||
| FRAT1 and EB1 activating the β-Catenin/TCF pathway, promoting transcriptional activity of β-Catenin | [75,76] | ||||||
| Down-regulation of migfilin, inhibiting the GSK-3β-dependent pathway, β-Catenin degradation and β-Catenin TCF-mediated transcriptional activity | [77] | ||||||
| Overexpression of Aurora-A inhibiting degradation, increasing phosphorylated β-Catenin at Ser552 and Ser675 | [79] | ||||||
| p120-Catenin | Membrane (↓) | Associated with lymph node metastasis | Positively correlated with differentiation | / | [67] | ||
| Desmosome | DSG1 | / | ↓ | Associated with tumour invasion, lymph node metastasis and lymphatic invasion | / | [115] | |
| DSG3 | / | Associated with lymph node metastasis, but negative DSG3 expression contributed to a poor survival rate | / | [114] | |||
| DSC2 | / | Membrane (↓), Cytoplasm (↑) | Promote the invasion and metastasis | High expression of miR-25; binding of miR-25 to the DSC2 mRNA 3′UTR | [8,108] | ||
| ↓ | Increased free PG (γ-Catenin), which competes with the β-Catenin in the E-Cadherin/β-Catenin complex and causes an increase in β-Catenin cytoplasmic delivery, nuclear localisation, and transcriptional activity; activating β-Catenin/TCF signalling pathway | [8] | |||||
| PG | ↓ | Cause reduced cell-cell adhesion and enhance cell migration | A tumour metastasis suppressor; lower DSC2 levels cause PG competes with β-Catenin to bind to E-Cadherin | [8,110] | |||
| Gap junction | Cx26; Cx43 | / | Membrane (↓) Cytoplasm (↑) | Associated with an aggressive phenotype and lymph node metastasis | / | [131,133,134,135] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Q.-R.; Du, X.-H.; Huang, T.-T.; Zheng, Y.-C.; Li, Y.-L.; Huang, D.-Y.; Dai, H.-Q.; Li, E.-M.; Fang, W.-K. Role of Cell-Cell Junctions in Oesophageal Squamous Cell Carcinoma. Biomolecules 2022, 12, 1378. https://doi.org/10.3390/biom12101378
Xu Q-R, Du X-H, Huang T-T, Zheng Y-C, Li Y-L, Huang D-Y, Dai H-Q, Li E-M, Fang W-K. Role of Cell-Cell Junctions in Oesophageal Squamous Cell Carcinoma. Biomolecules. 2022; 12(10):1378. https://doi.org/10.3390/biom12101378
Chicago/Turabian StyleXu, Qian-Rui, Xiao-Hui Du, Ting-Ting Huang, Yu-Chun Zheng, Yu-Ling Li, Dan-Yi Huang, Hao-Qiang Dai, En-Min Li, and Wang-Kai Fang. 2022. "Role of Cell-Cell Junctions in Oesophageal Squamous Cell Carcinoma" Biomolecules 12, no. 10: 1378. https://doi.org/10.3390/biom12101378
APA StyleXu, Q.-R., Du, X.-H., Huang, T.-T., Zheng, Y.-C., Li, Y.-L., Huang, D.-Y., Dai, H.-Q., Li, E.-M., & Fang, W.-K. (2022). Role of Cell-Cell Junctions in Oesophageal Squamous Cell Carcinoma. Biomolecules, 12(10), 1378. https://doi.org/10.3390/biom12101378